Conformational Investigations in Flexible Molecules Using Orientational NMR Constraints in Combination with 3J-Couplings and NOE Distances


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
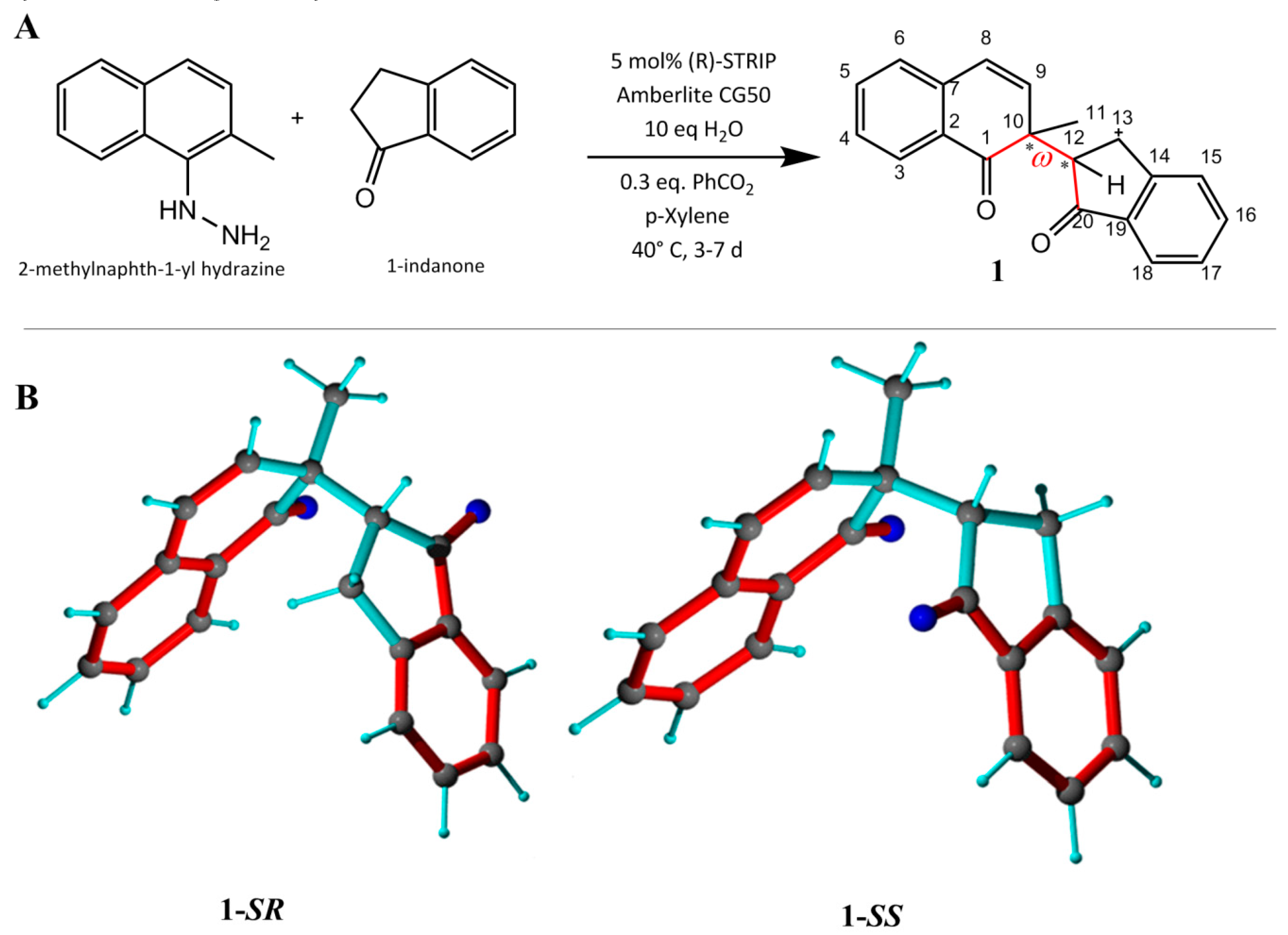
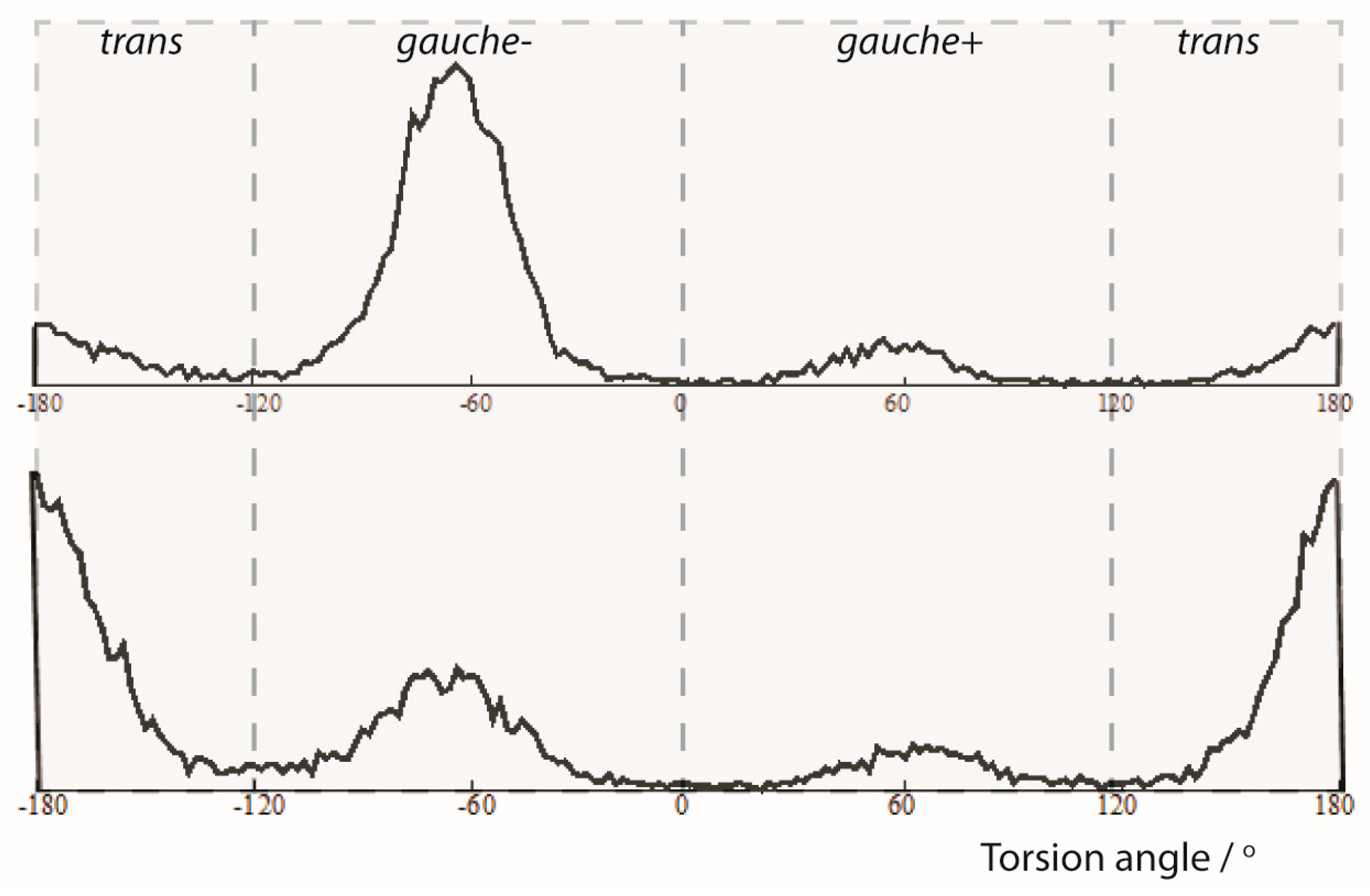
2.1. Single-Bond Dynamics in Diketone 1
2.1.1. NMR Datasets
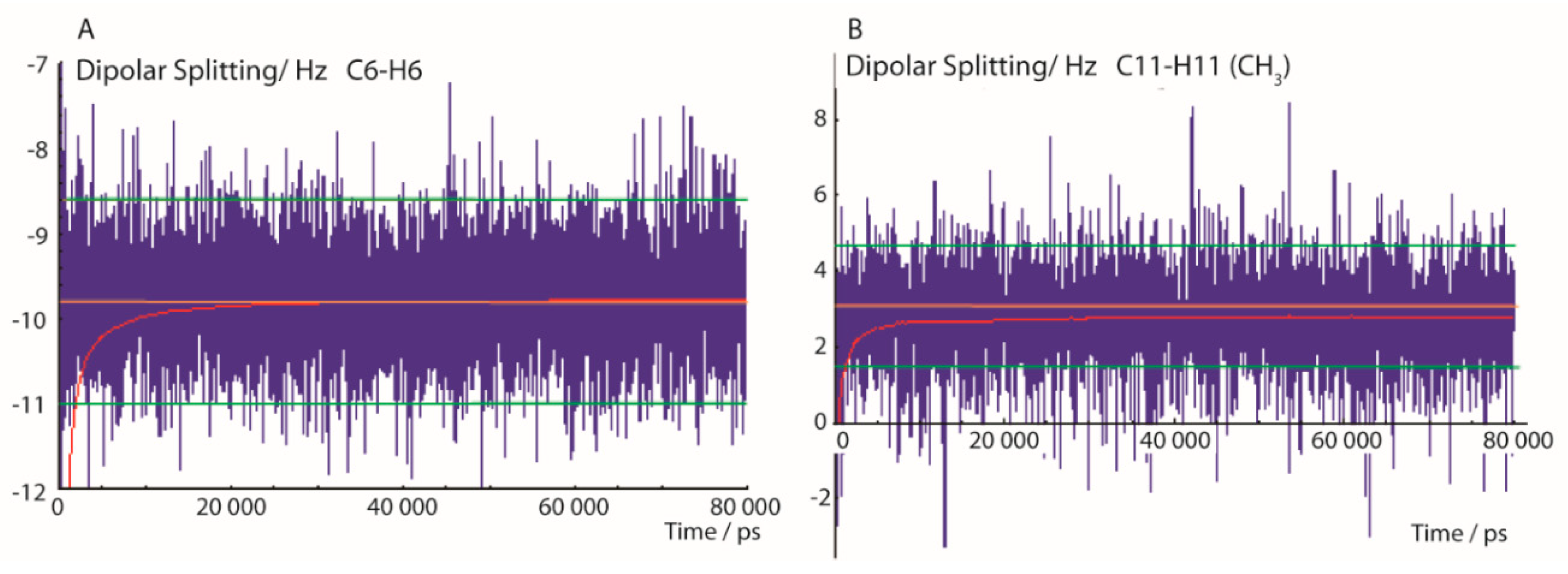
2.1.2. MDOC Simulation
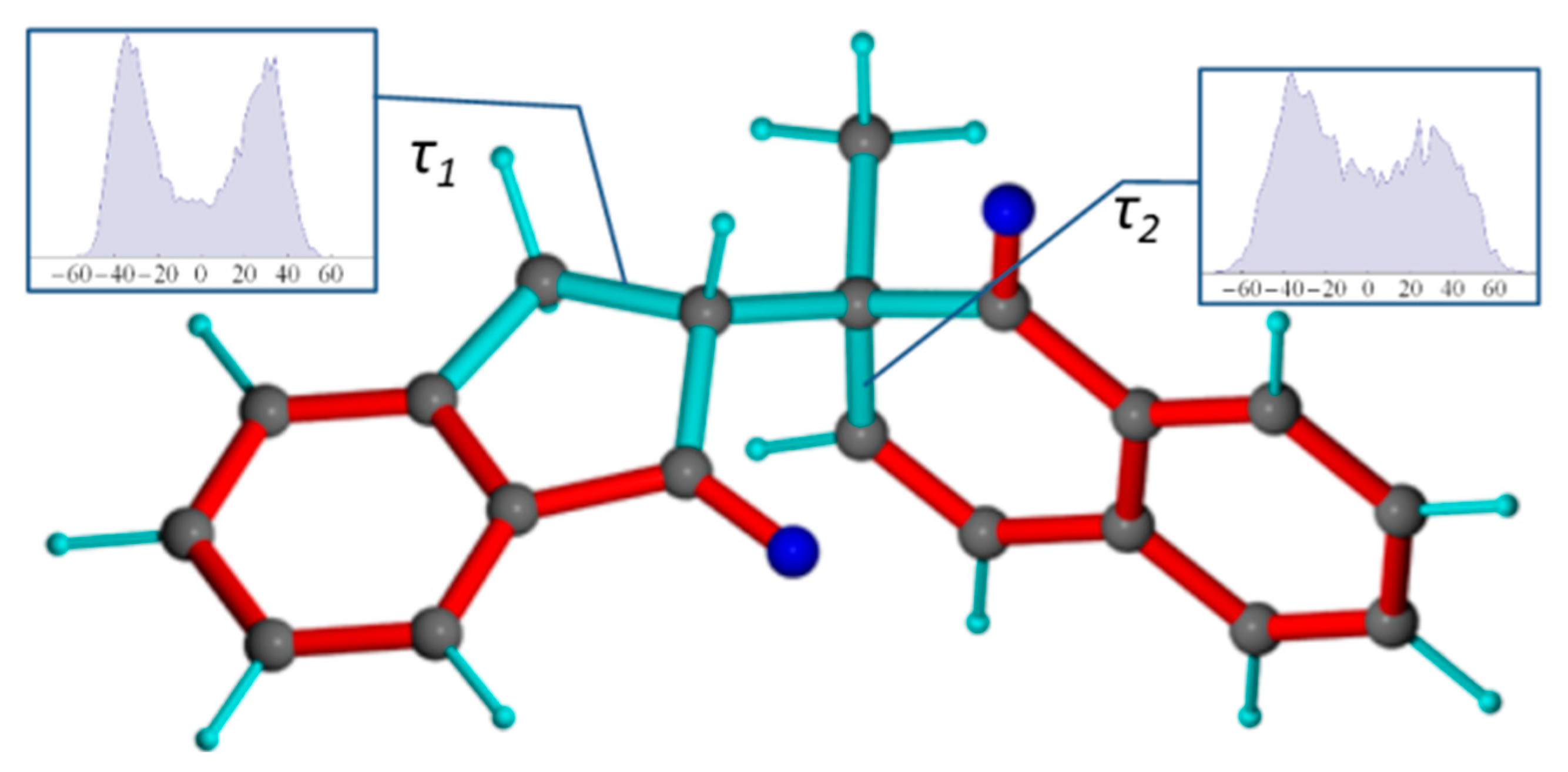
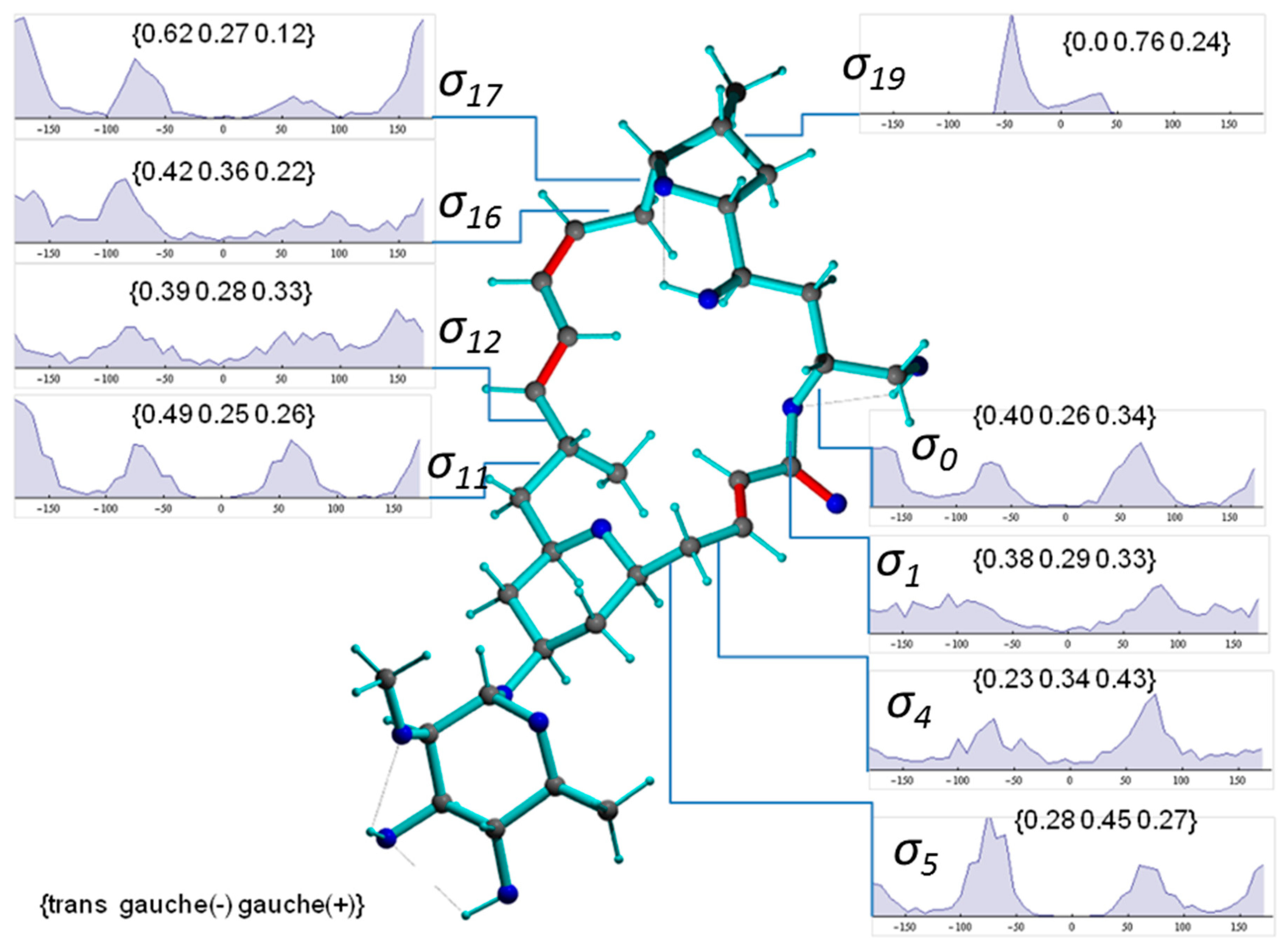
2.2. Mandelalide A
2.2.1. NMR Datasets
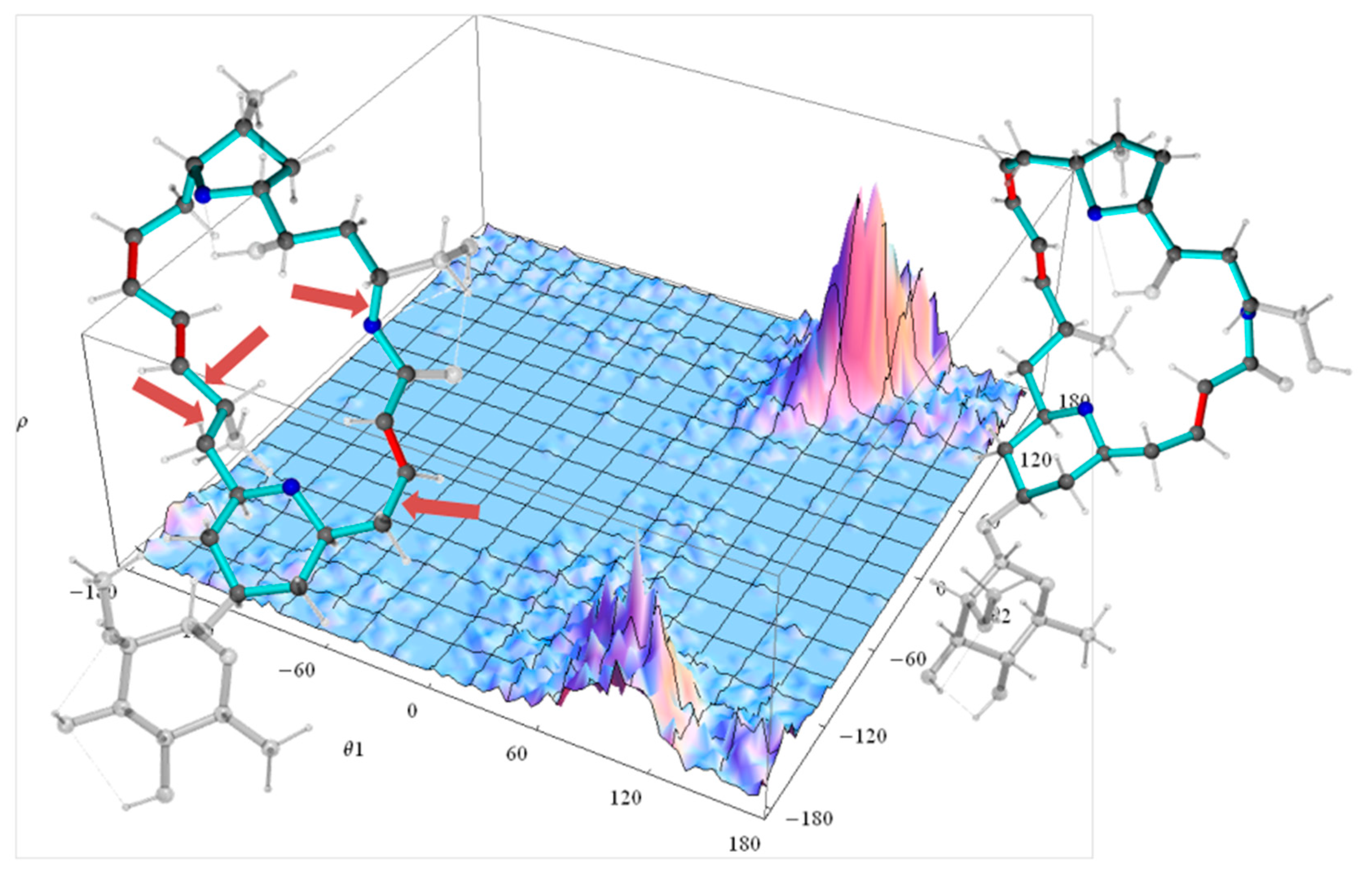
2.2.2. Torsion Angle Distributions
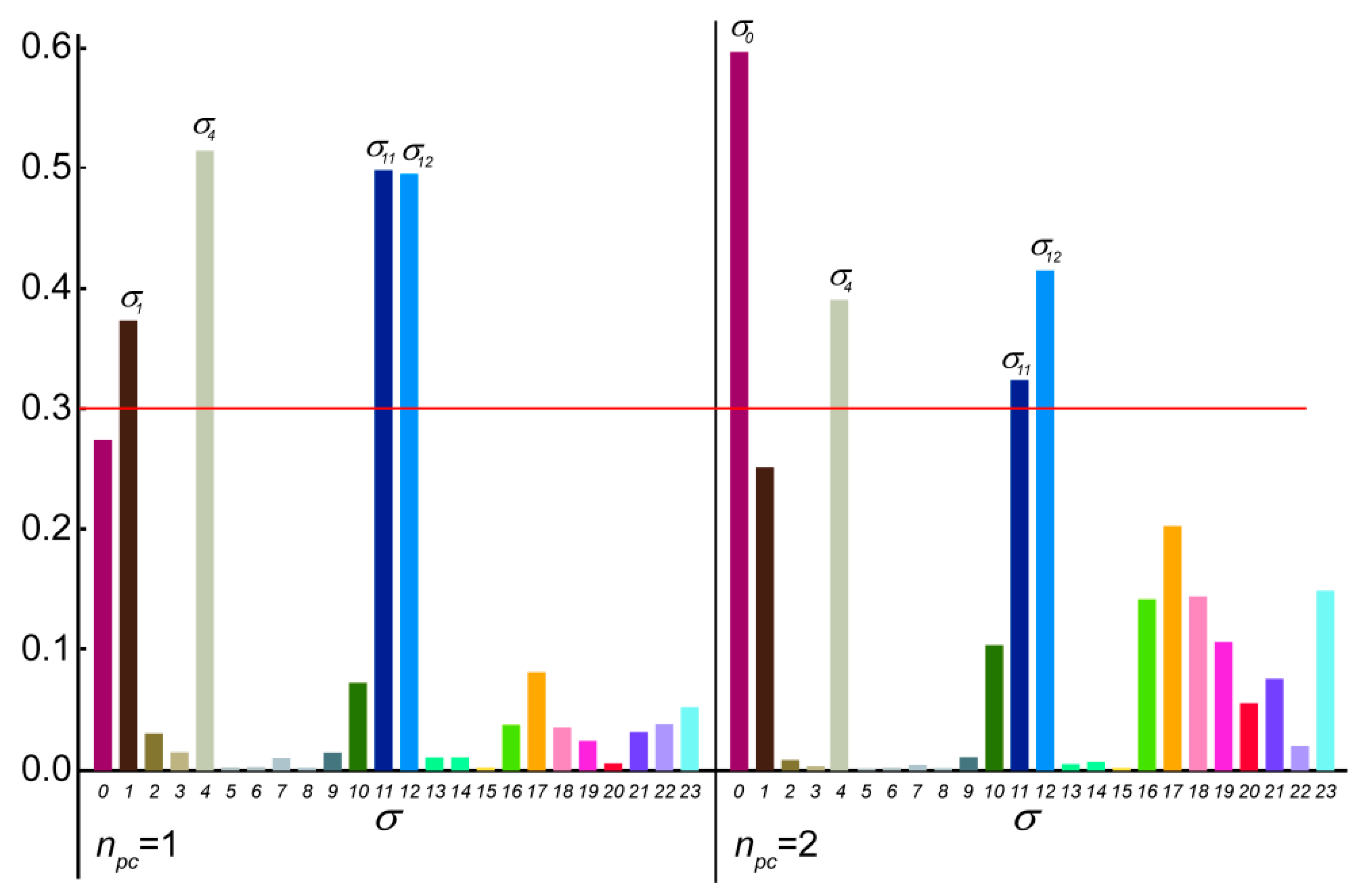
2.2.3. Principal Component Analysis
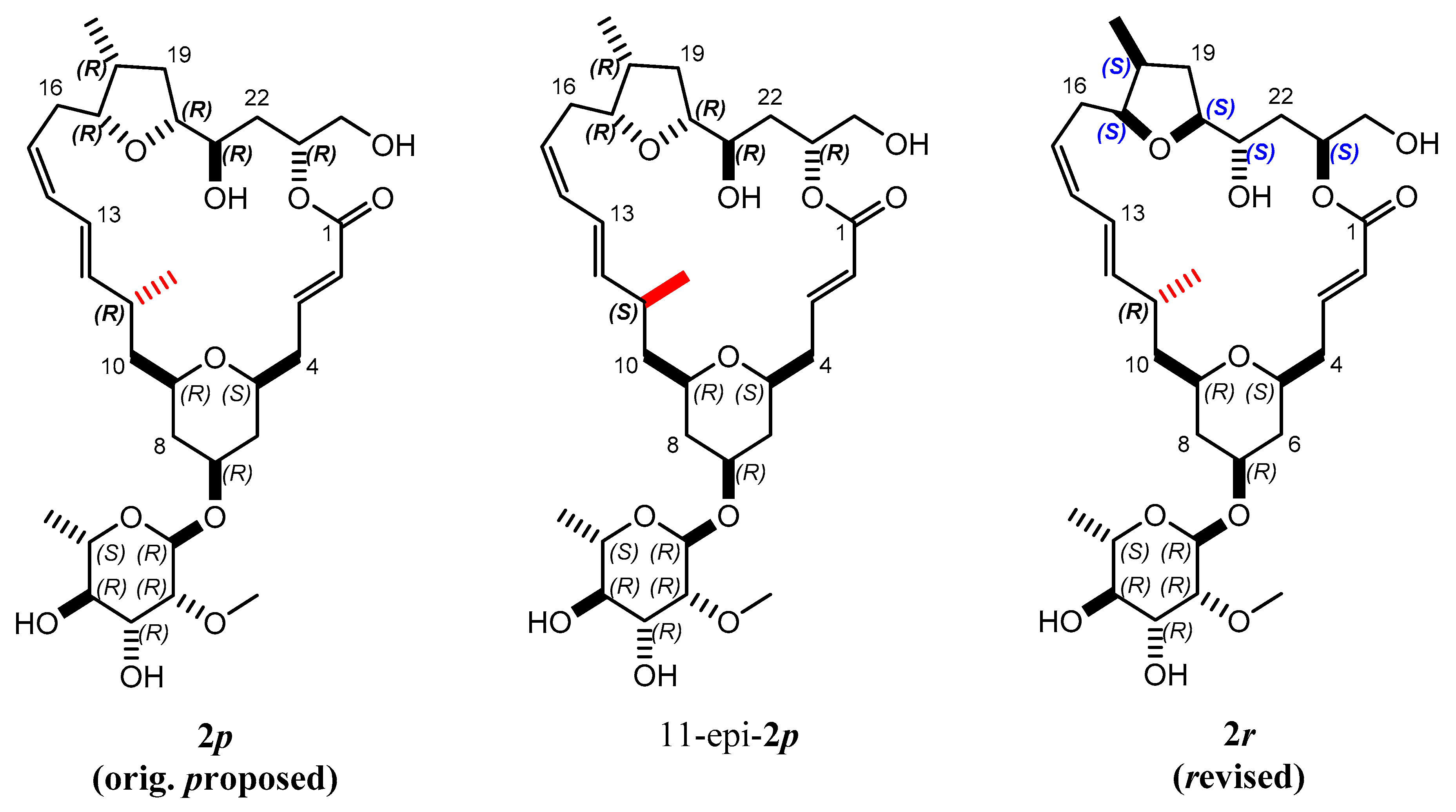
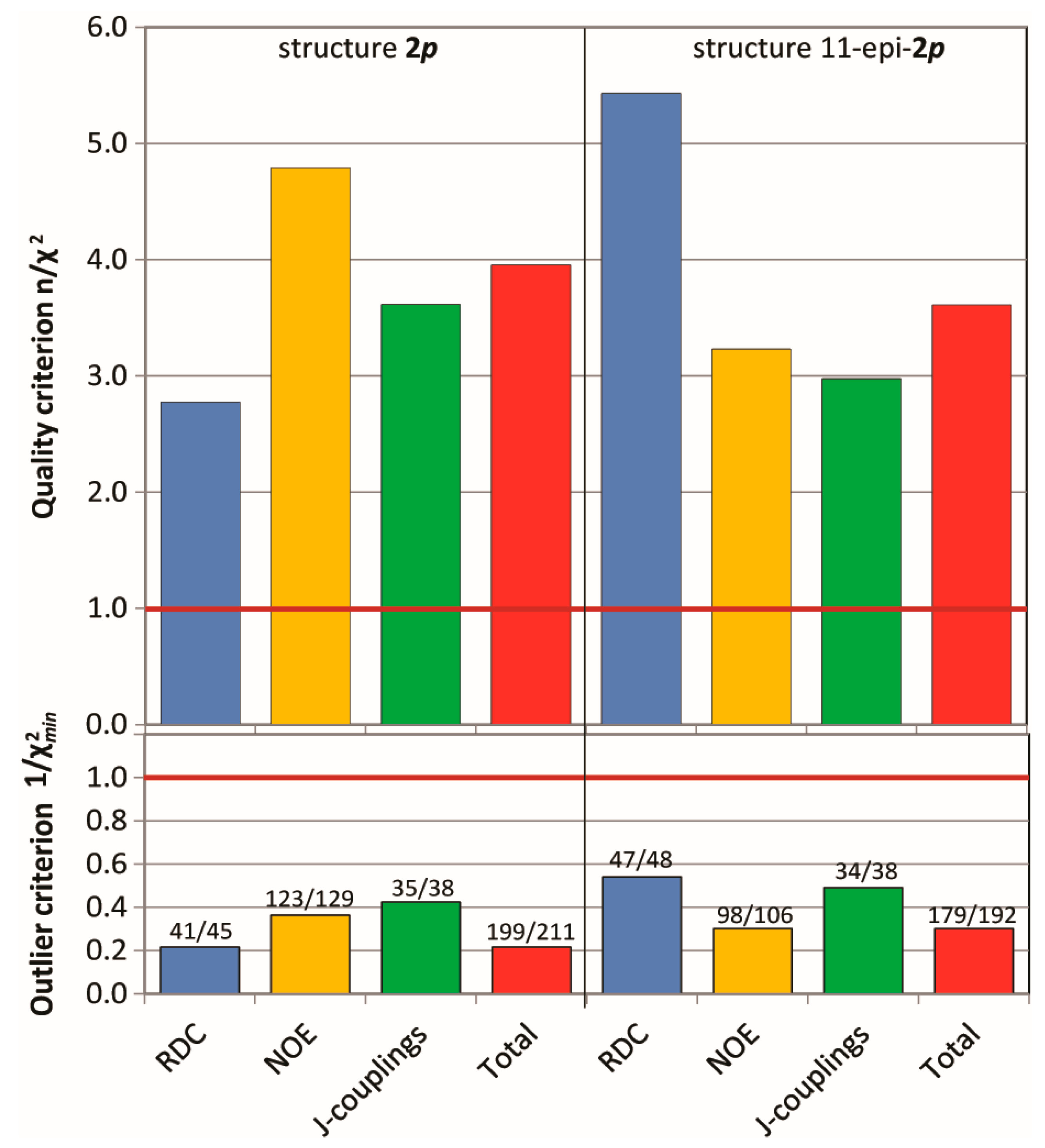
2.3. Configuration Determination with MDOC
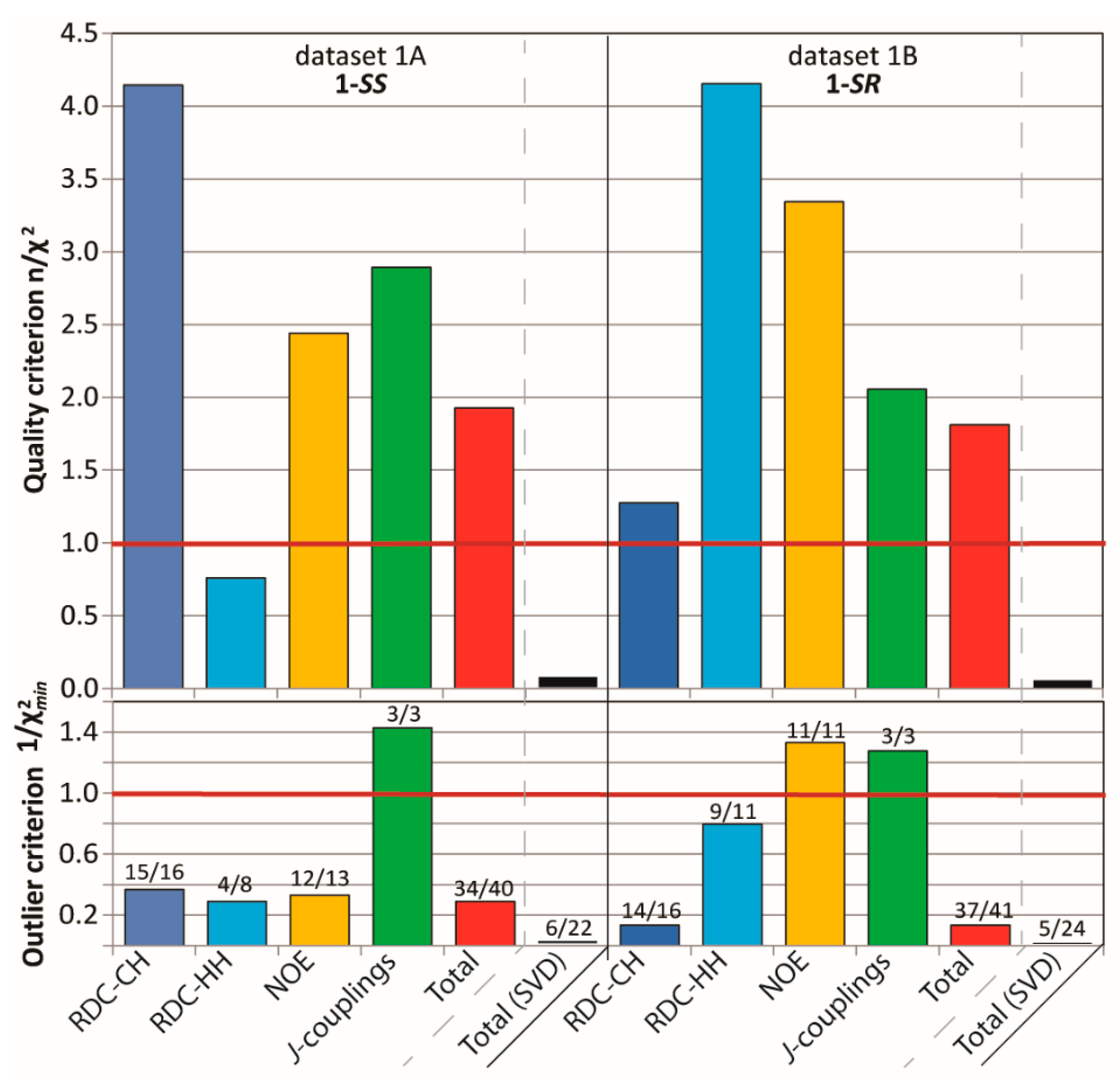
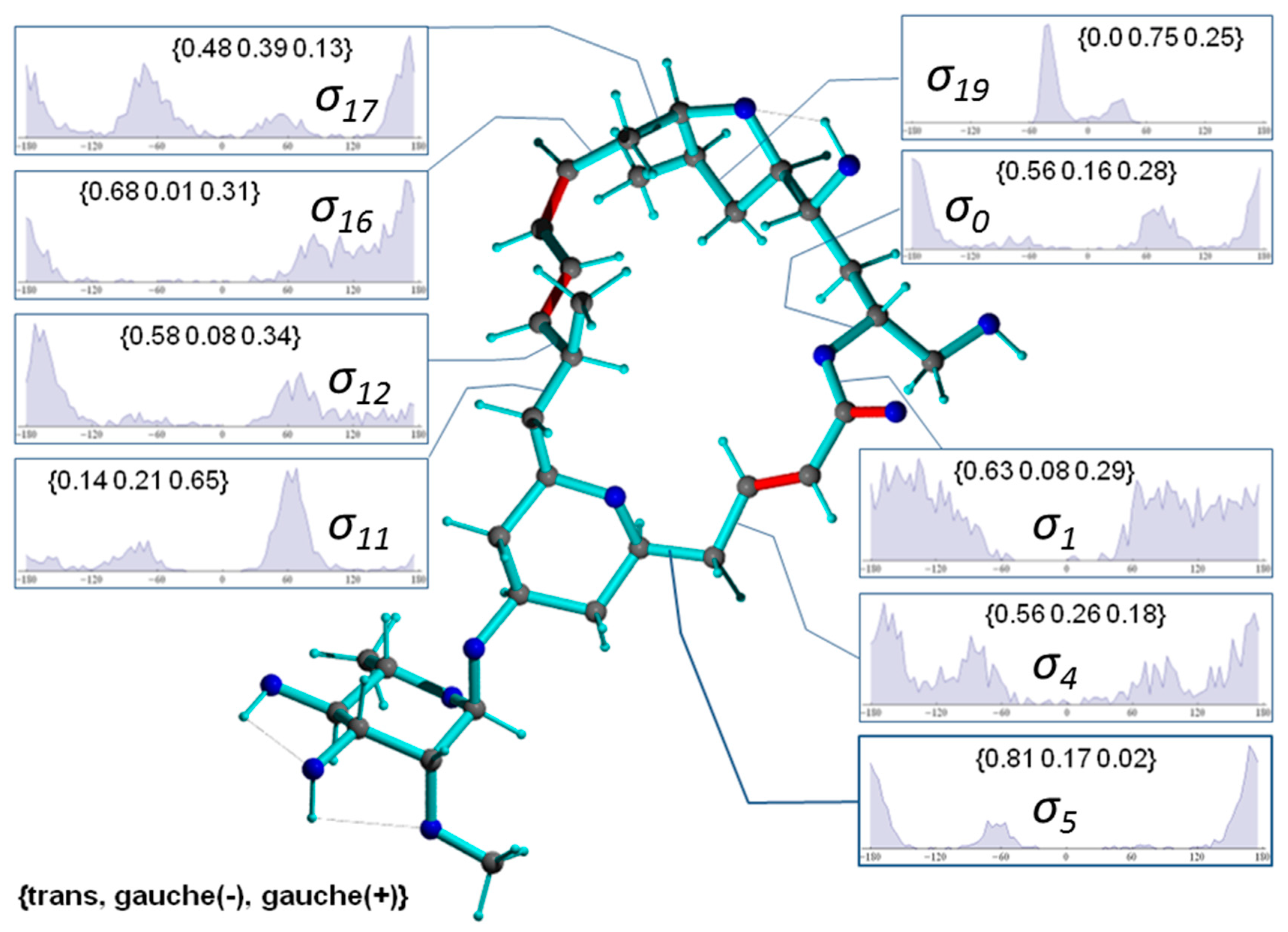
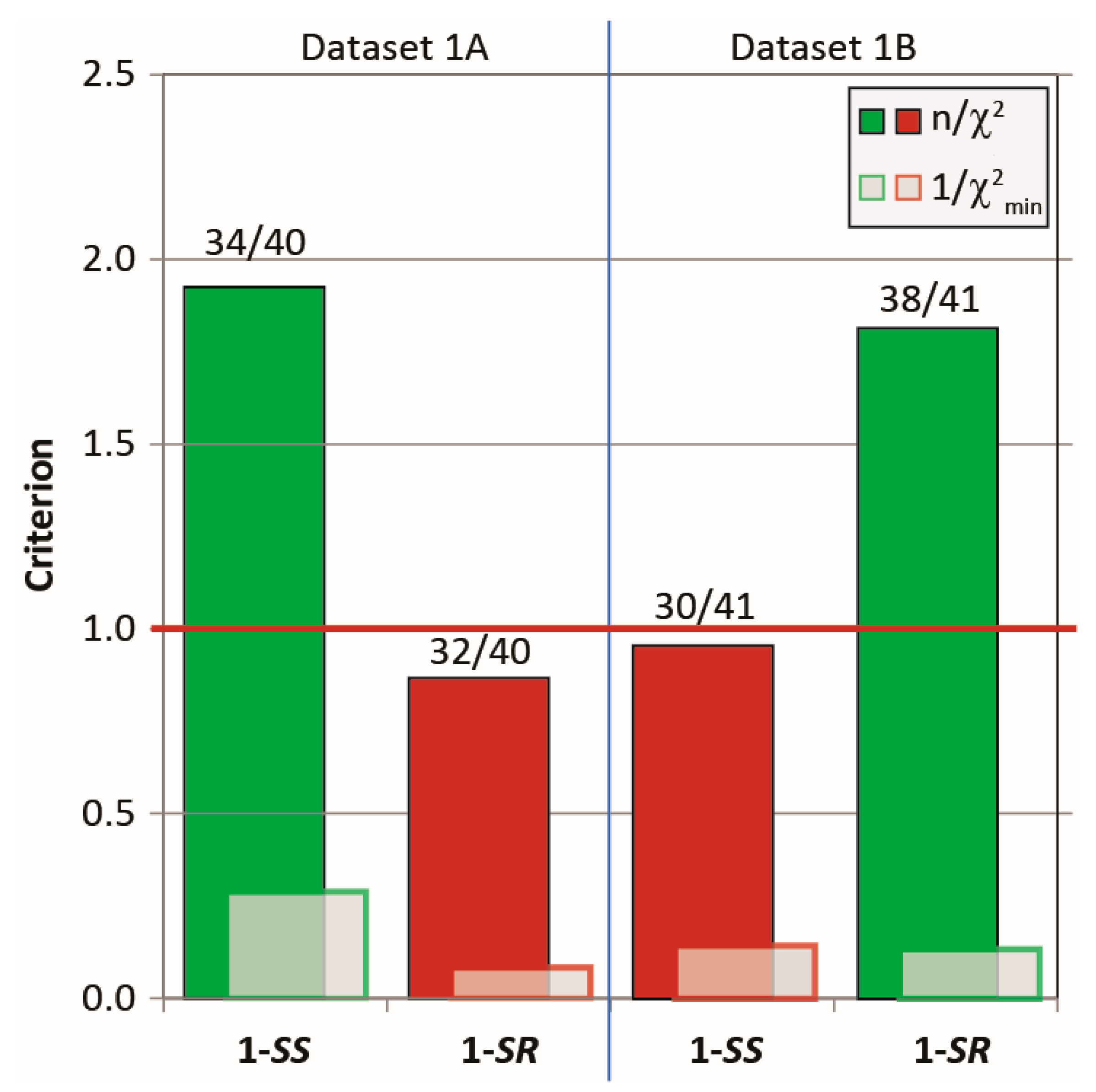
2.3.1. Discriminating 1,4-Diketone 1-SS vs. 1-SR
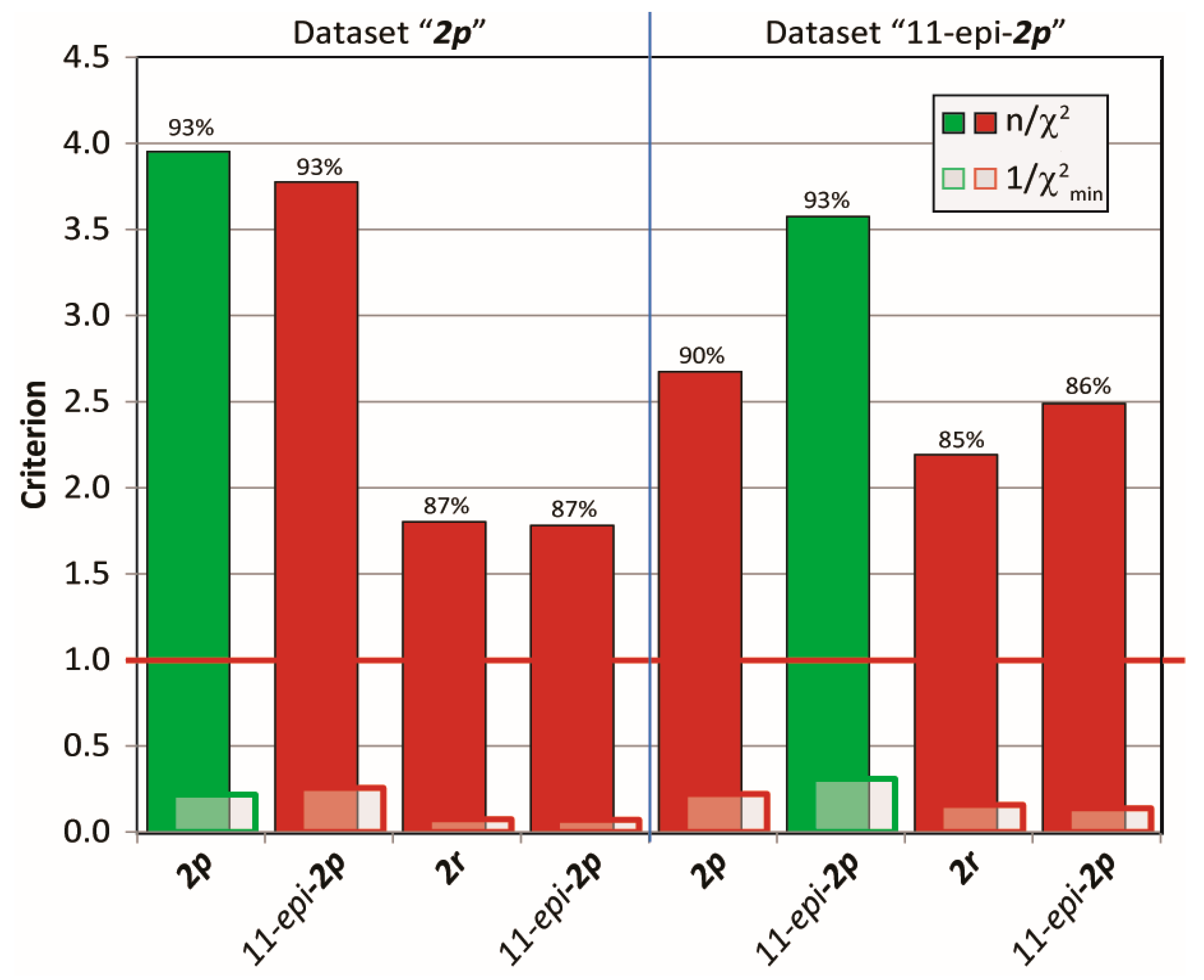
2.3.2. Discriminating between 4 Stereoisomers of Mandelalide A 2
3. Conclusions
4. Materials and Methods
4.1. NMR
4.1.1. Materials
4.1.2. Sample Alignment
4.1.3. NMR Experiments
4.1.4. Conformational Analysis
4.2. MDOC Simulations
4.2.1. Theory: RDC-Based Orientation Constraints
4.2.2. Theory: Scalar Constraints
4.2.3. Evaluation of Quality
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Englert, G.; Saupe, A. Hochaufgeloste Protonenresonanzspektren Mit Direkter Magnetischer Dipol-Dipol-Wechselwirkung, Teil II. Z Nat. Pt. A 1964, 19a, 172–177. [Google Scholar]
- Burnell, E.E.; De Lange, C.A. NMR of Ordered Liquids; Kluwer Academic Publishers: Dordrecht, The Netherlands; Boston, MA, USA, 2003; p. 455. [Google Scholar]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Haasnoot, C.A.G.; Deleeuw, F.A.A.M.; Altona, C. The Relationship between Proton-Proton Nmr Coupling-Constants and Substituent Electronegativities.1. An Empirical Generalization of the Karplus Equation. Tetrahedron 1980, 36, 2783–2792. [Google Scholar] [CrossRef]
- Chen, K.; Tjandra, N. The Use of Residual Dipolar Coupling in Studying Proteins by NMR. Nmr. Proteins. Small. Biomol. 2012, 326, 47–67. [Google Scholar] [CrossRef]
- Lange, O.F.; Lakomek, N.A.; Farès, C.; Schroder, G.F.; Walter, K.F.A.; Becker, S.; Meiler, J.; Grubmuller, H.; Griesinger, C.; de Groot, B.L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. [Google Scholar] [CrossRef]
- Farès, C.; Lakomek, N.A.; Walter, K.F.A.; Frank, B.T.C.; Meiler, J.; Becker, S.; Griesinger, C. Accessing ns-mu s side chain dynamics in ubiquitin with methyl RDCs. J. Biomol. Nmr. 2009, 45, 23–44. [Google Scholar] [CrossRef]
- Thiele, C.M. Use of RDCs in rigid organic compounds and some practical considerations concerning alignment media. Concepts Magn. Reson. Part. A 2007, 30A, 65–80. [Google Scholar] [CrossRef]
- Hallwass, F.; Schmidt, M.; Sun, H.; Mazur, A.; Kummerlowe, G.; Luy, B.; Navarro-Vazquez, A.; Griesinger, C.; Reinscheid, U.M. Residual Chemical Shift Anisotropy (RCSA): A Tool for the Analysis of the Configuration of Small Molecules. Angew. Chem. Int. Ed. 2011, 50, 9487–9490. [Google Scholar] [CrossRef]
- Navarro-Vazquez, A.; Gil, R.R.; Blinov, K. Computer-Assisted 3D Structure Elucidation (CASE-3D) of Natural Products Combining Isotropic and Anisotropic NMR Parameters. J. Nat. Prod. 2018, 81, 203–210. [Google Scholar] [CrossRef]
- Trigo-Mourino, P.; Sifuentes, R.; Navarro-Vazquez, A.; Gayathri, C.; Maruenda, H.; Gil, R.R. Determination of the Absolute Configuration of 19-OH-(-)-eburnamonine Using a Combination of Residual Dipolar Couplings, DFT Chemical Shift Predictions, and Chiroptics. Nat. Prod. Commun. 2012, 7, 735–738. [Google Scholar] [CrossRef]
- Snider, J.D.; Troche-Pesqueira, E.; Woodruff, S.R.; Gayathri, C.; Tsarevsky, N.V.; Gil, R.R. New strategy for RDC-assisted diastereotopic proton assignment using a combination of J-scaled BIRD HSQC and J-scaled BIRD HMQC/HSQC. Magn. Reson. Chem. 2012, 50, S86–S91. [Google Scholar] [CrossRef]
- Losonczi, J.A.; Andrec, M.; Fischer, M.W.F.; Prestegard, J.H. Order matrix analysis of residual dipolar couplings using singular value decomposition. J. Magn. Reson. 1999, 138, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Saupe, A. Recent Results in Field of Liquid Crystals. Angew. Chem. Int. Ed. 1968, 7, 97–112. [Google Scholar] [CrossRef]
- Freudenberger, J.C.; Spiteller, P.; Bauer, R.; Kessler, H.; Luy, B. Stretched poly(dimethylsiloxane) gels as NMR alignment media for apolar and weakly polar organic solvents: An ideal tool for measuring RDCs at low molecular concentrations. J. Am. Chem. Soc. 2004, 126, 14690–14691. [Google Scholar] [CrossRef]
- Haberz, P.; Farjon, J.; Griesinger, C. A DMSO-Compatible Orienting Medium: Towards the Investigation of the Stereochemistry of Natural Products. Angew. Chem. Int. Ed. 2005, 44, 427–429. [Google Scholar] [CrossRef]
- Kummerlowe, G.; Auernheimer, J.; Lendlein, A.; Luy, B. Stretched poly(acrylonitrile) as a scalable alignment medium for DMSO. J. Am. Chem. Soc. 2007, 129, 6080–6081. [Google Scholar] [CrossRef]
- Sun, H.; Reinscheid, U.M.; Whitson, E.L.; d’Auyergne, E.J.; Ireland, C.M.; Navarro-Vazquez, A.; Griesinger, C. Challenge of Large-Scale Motion for Residual Dipolar Coupling Based Analysis of Configuration: The Case of Fibrosterol Sulfate A. J. Am. Chem. Soc. 2011, 133, 14629–14636. [Google Scholar] [CrossRef]
- Bayer, P.; Varani, L.; Varani, G. Refinement of the structure of protein-RNA complexes by residual dipolar coupling analysis. J. Biomol. Nmr. 1999, 14, 149–155. [Google Scholar] [CrossRef]
- Choy, W.Y.; Tollinger, M.; Mueller, G.A.; Kay, L.E. Direct structure refinement of high molecular weight proteins against residual dipolar couplings and carbonyl chemical shift changes upon alignment: An application to maltose binding protein. J. Biomol. Nmr. 2001, 21, 31–40. [Google Scholar] [CrossRef]
- Lipsitz, R.S.; Tjandra, N. Residual Dipolar Couplings in Nmr Structure Analysis*1. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 387–413. [Google Scholar] [CrossRef]
- Farès, C.; Hassfeld, J.; Menche, D.; Carlomagno, T. Simultaneous determination of the conformation and relative configuration of archazolide A by using nuclear Overhauser effects, J couplings, and residual dipolar couplings. Angew. Chem. Int. Ed. 2008, 47, 3722–3726. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Scheek, R.M. Orientation restraints in molecular dynamics simulations using time and ensemble averaging. J. Magn. Reson. 2003, 164, 19–27. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Best, R.B.; DePristo, M.A.; Dobson, C.M.; Vendruscolo, M. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar] [CrossRef]
- Esteban-Martin, S.; Fenwick, R.B.; Salvatella, X. Refinement of Ensembles Describing Unstructured Proteins Using NMR Residual Dipolar Couplings. J. Am. Chem. Soc. 2010, 132, 4626–4632. [Google Scholar] [CrossRef]
- De Simone, A.; Montalvao, R.W.; Vendruscolo, M. Determination of Conformational Equilibria in Proteins Using Residual Dipolar Couplings. J. Chem. Theory. Comput. 2011, 7, 4189–4195. [Google Scholar] [CrossRef]
- Salmon, L.; Blackledge, M. Investigating protein conformational energy landscapes and atomic resolution dynamics from NMR dipolar couplings: A review. Rep. Prog. Phys. 2015, 78, 12. [Google Scholar] [CrossRef]
- Camilloni, C.; Vendruscolo, M. A Tensor-Free Method for the Structural and Dynamical Refinement of Proteins using Residual Dipolar Couplings. J. Phys. Chem. B 2015, 119, 653–661. [Google Scholar] [CrossRef]
- Tomba, G.; Camilloni, C.; Vendruscolo, M. Determination of the conformational states of strychnine in solution using NMR residual dipolar couplings in a tensor-free approach. Methods 2018, 148, 4–8. [Google Scholar] [CrossRef]
- Sternberg, U.; Witter, R. Molecular dynamics simulations on PGLa using NMR orientational constraints. J. Biomol. Nmr. 2015, 63, 265–274. [Google Scholar] [CrossRef]
- Sternberg, U.; Witter, R.; Ulrich, A.S. All-atom molecular dynamics simulations using orientational constraints from anisotropic NMR samples. J. Biomol. Nmr. 2007, 38, 23–39. [Google Scholar] [CrossRef]
- Tzvetkova, P.; Sternberg, U.; Gloge, T.; Navarro Vazquez, A.; Luy, B. Configuration Determination by Residual Dipolar Couplings: Accessing the Full Conformational Space by Molecular Dynamics with Tensorial Constraints. Chem. Sci. 2019, 10, 8774–8791. [Google Scholar] [CrossRef]
- Di Pietro, M.E.; Sternberg, U.; Luy, B. Molecular Dynamics with Orientational Tensorial Constraints: A New Approach to Probe the Torsional Angle Distributions of Small Rotationally Flexible Molecules. J. Phys. Chem. B 2019. [Google Scholar] [CrossRef]
- Huang, S.L.; Kotzner, L.; De, C.K.; List, B. Catalytic Asymmetric Dearomatizing Redox Cross Coupling of Ketones with Aryl Hydrazines Giving 1,4-Diketones. J. Am. Chem. Soc. 2015, 137, 3446–3449. [Google Scholar] [CrossRef]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A-D, Cytotoxic Macrolides from a New Lissoclinum Species of South African Tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef]
- Willwacher, J.; Furstner, A. Catalysis-Based Total Synthesis of Putative Mandelalide A. Angew. Chem. Int. Ed. 2014, 53, 4217–4221. [Google Scholar] [CrossRef]
- Lei, H.; Yan, J.; Yu, J.; Liu, Y.; Wang, Z.; Xu, Z.; Ye, T. Total synthesis and stereochemical reassignment of mandelalide A. Angew. Chem. Int. Ed. Engl. 2014, 53, 6533–6537. [Google Scholar] [CrossRef]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef]
- Kolmer, A.; Edwards, L.J.; Kuprov, I.; Thiele, C.M. Conformational analysis of small organic molecules using NOE and RDC data: A discussion of strychnine and alpha-methylene-gamma-butyrolactone. J. Magn. Reson. 2015, 261, 101–109. [Google Scholar] [CrossRef]
- Schuetz, A.; Junker, J.; Leonov, A.; Lange, O.F.; Molinski, T.F.; Griesinger, C. Stereochemistry of sagittamide a from residual dipolar coupling enhanced NMR. J. Am. Chem. Soc. 2007, 129, 15114–15115. [Google Scholar] [CrossRef]
- Willwacher, J.; Heggen, B.; Wirtz, C.; Thiel, W.; Furstner, A. Total Synthesis, Stereochemical Revision, and Biological Reassessment of MandelalideA: Chemical Mimicry of Intrafamily Relationships. Chem. -A Eur. J. 2015, 21, 10416–10430. [Google Scholar] [CrossRef]
- Gayathri, C.; Tsarevsky, N.V.; Gil, R.R. Residual Dipolar Couplings (RDCs) Analysis of Small Molecules Made Easy: Fast and Tuneable Alignment by Reversible Compression/Relaxation of Reusable PMMA Gels. Chem. -A Eur. J. 2010, 16, 3622–3626. [Google Scholar] [CrossRef] [PubMed]
- Trigo-Mourino, P.; Merle, C.; Koos, M.R.M.; Luy, B.; Gil, R.R. Probing Spatial Distribution of Alignment by Deuterium NMR Imaging. Chem. Eur. J. 2013, 19, 7013–7019. [Google Scholar] [CrossRef] [PubMed]
- Enthart, A.; Freudenberger, J.C.; Furrer, J.; Kessler, H.; Luy, B. The CLIP/CLAP-HSQC: Pure absorptive spectra for the measurement of one-bond couplings. J. Magn. Reson. 2008, 192, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Reinsperger, T.; Luy, B. Homonuclear BIRD-decoupled spectra for measuring one-bond couplings with highest resolution: CLIP/CLAP-RESET and constant-time-CLIP/CLAP-RESET. J. Magn. Reson. 2014, 239, 110–120. [Google Scholar] [CrossRef]
- Pupier, M.; Nuzillard, J.M.; Wist, J.; Schlorer, N.E.; Kuhn, S.; Erdelyi, M.; Steinbeck, C.; Williams, A.J.; Butts, C.; Claridge, T.D.W.; et al. NMReDATA, a standard to report the NMR assignment and parameters of organic compounds. Magn. Reson. Chem. 2018, 56, 703–715. [Google Scholar] [CrossRef]
- Mollhoff, M.; Sternberg, U. Molecular mechanics with fluctuating atomic charges - a new force field with a semi-empirical charge calculation. J. Mol. Model. 2001, 7, 90–102. [Google Scholar] [CrossRef]
- Sternberg, U.; Koch, F.T.; Brauer, M.; Kunert, M.; Anders, E. Molecular mechanics for zinc complexes with fluctuating atomic charges. J. Mol. Model. 2001, 7, 54–64. [Google Scholar] [CrossRef]
- Sternberg, U.; Koch, F.T.; Mollhoff, M. New Approach to the Semiempirical Calculation of Atomic Charges for Polypeptides and Large Molecular-Systems. J. Comput. Chem. 1994, 15, 524–531. [Google Scholar] [CrossRef]
- Witter, R.; Mollhoff, M.; Koch, F.T.; Sternberg, U. Fast Atomic Charge Calculation for Implementation into a Polarizable Force Field and Application to an Ion Channel Protein. J. Chem-Ny. 2015. [Google Scholar] [CrossRef]
- Torda, A.E.; Scheek, R.M.; Vangunsteren, W.F. Time-Dependent Distance Restraints in Molecular-Dynamics Simulations. Chem. Phys. Lett. 1989, 157, 289–294. [Google Scholar] [CrossRef]
- Limmer, S. Physical Principles Underlying the Experimental Methods for Studying the Orientational Order of Liquid-Crystals. Fortschr. Phys. Prog. Phys. 1989, 37, 879–931. [Google Scholar] [CrossRef]
- Marsan, M.P.; Muller, I.; Ramos, C.; Rodriguez, F.; Dufourc, E.J.; Czaplicki, J.; Milon, A. Cholesterol orientation and dynamics in dimyristoylphosphatidylcholine bilayers: A solid state deuterium NMR analysis. Biophys. J. 1999, 76, 351–359. [Google Scholar] [CrossRef]
- Vogeli, B. The nuclear Overhauser effect from a quantitative perspective. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 78, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Intelmann, D.; Kummerlowe, G.; Haseleu, G.; Desmer, N.; Schulze, K.; Frohlich, R.; Frank, O.; Luy, B.; Hofmann, T. Structures of Storage-Induced Transformation Products of the Beer’s Bitter Principles, Revealed by Sophisticated NMR Spectroscopic and LC-MS Techniques. Chem. Eur. J. 2009, 15, 13047–13058. [Google Scholar] [CrossRef]
Sample Availability: Samples of are no longer available from the authors. |













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farès, C.; Lingnau, J.B.; Wirtz, C.; Sternberg, U. Conformational Investigations in Flexible Molecules Using Orientational NMR Constraints in Combination with 3J-Couplings and NOE Distances. Molecules 2019, 24, 4417. https://doi.org/10.3390/molecules24234417
Farès C, Lingnau JB, Wirtz C, Sternberg U. Conformational Investigations in Flexible Molecules Using Orientational NMR Constraints in Combination with 3J-Couplings and NOE Distances. Molecules. 2019; 24(23):4417. https://doi.org/10.3390/molecules24234417
Chicago/Turabian StyleFarès, Christophe, Julia B. Lingnau, Cornelia Wirtz, and Ulrich Sternberg. 2019. "Conformational Investigations in Flexible Molecules Using Orientational NMR Constraints in Combination with 3J-Couplings and NOE Distances" Molecules 24, no. 23: 4417. https://doi.org/10.3390/molecules24234417
APA StyleFarès, C., Lingnau, J. B., Wirtz, C., & Sternberg, U. (2019). Conformational Investigations in Flexible Molecules Using Orientational NMR Constraints in Combination with 3J-Couplings and NOE Distances. Molecules, 24(23), 4417. https://doi.org/10.3390/molecules24234417