
A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues



Abstract
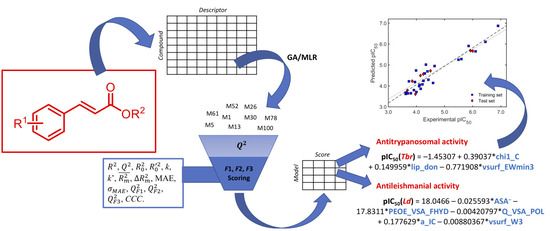
1. Introduction
2. Results and Discussions
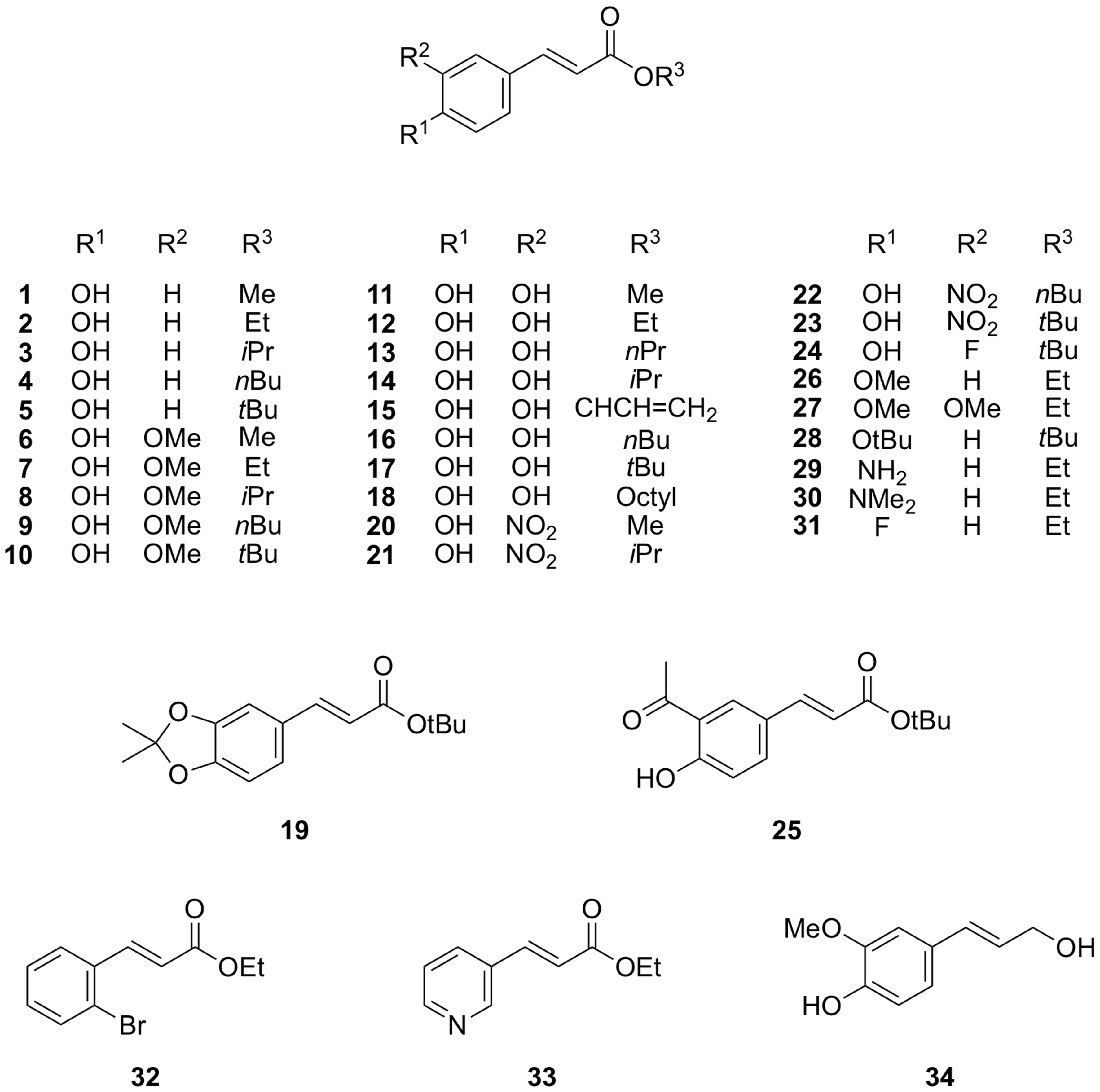
2.1. Cinnamate Ester Analogues and Their Molecular Fingerprints
2.2. QSAR Modeling for Antileishmanial Activity
2.2.1. Building of QSAR Models for Antileishmanial Activity
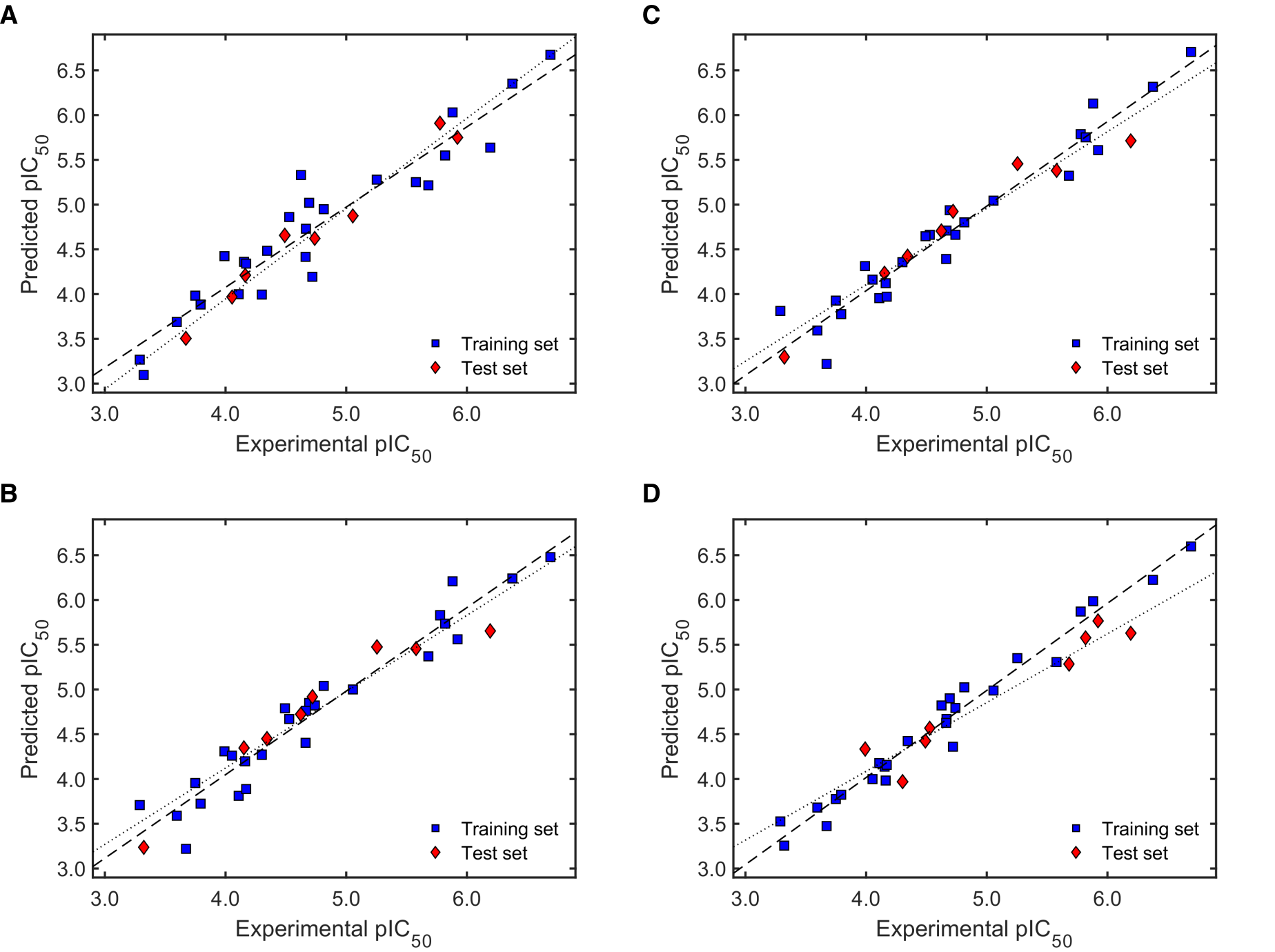
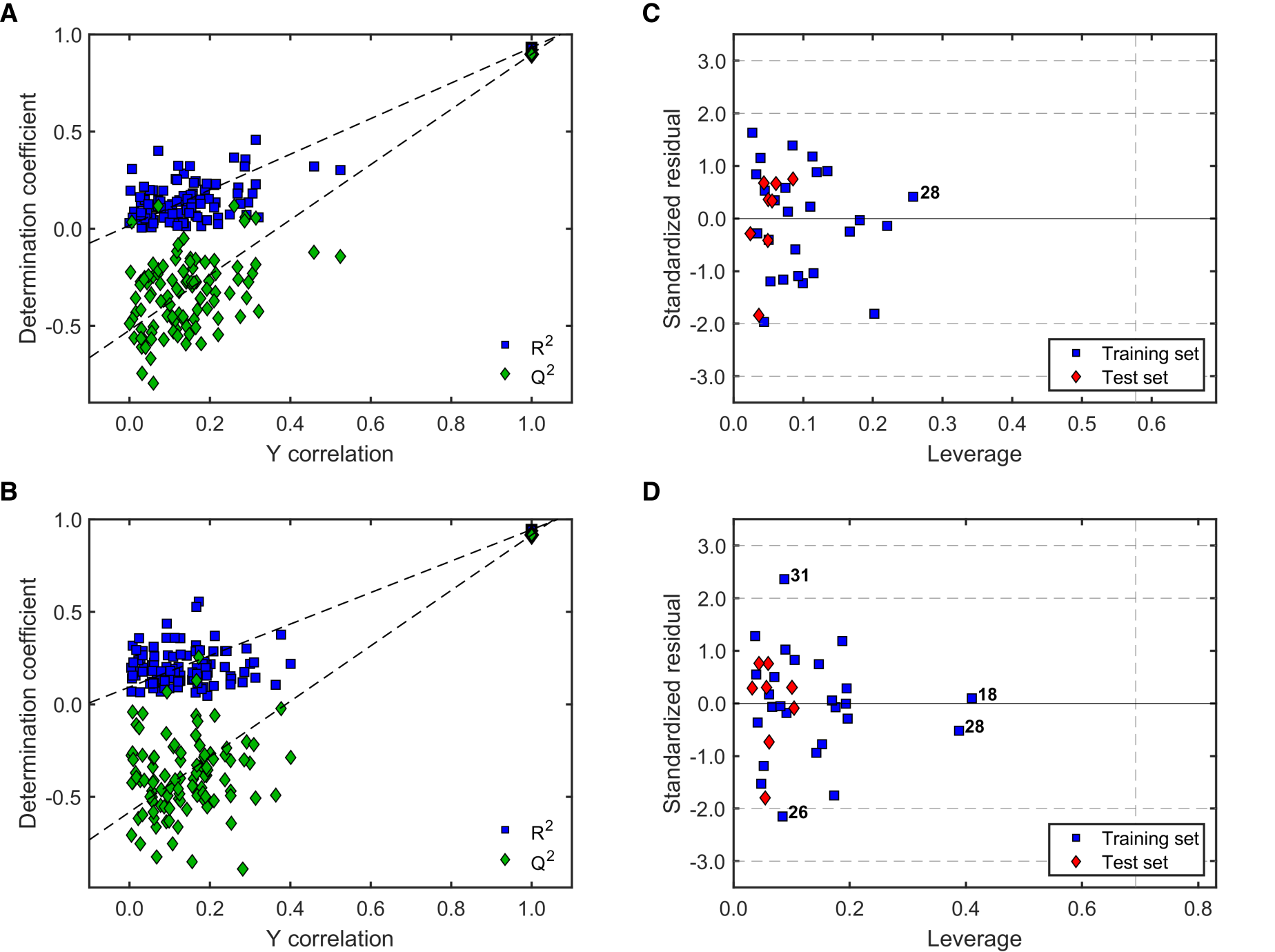
2.2.2. Validation of QSAR Models for Antileishmanial Activity
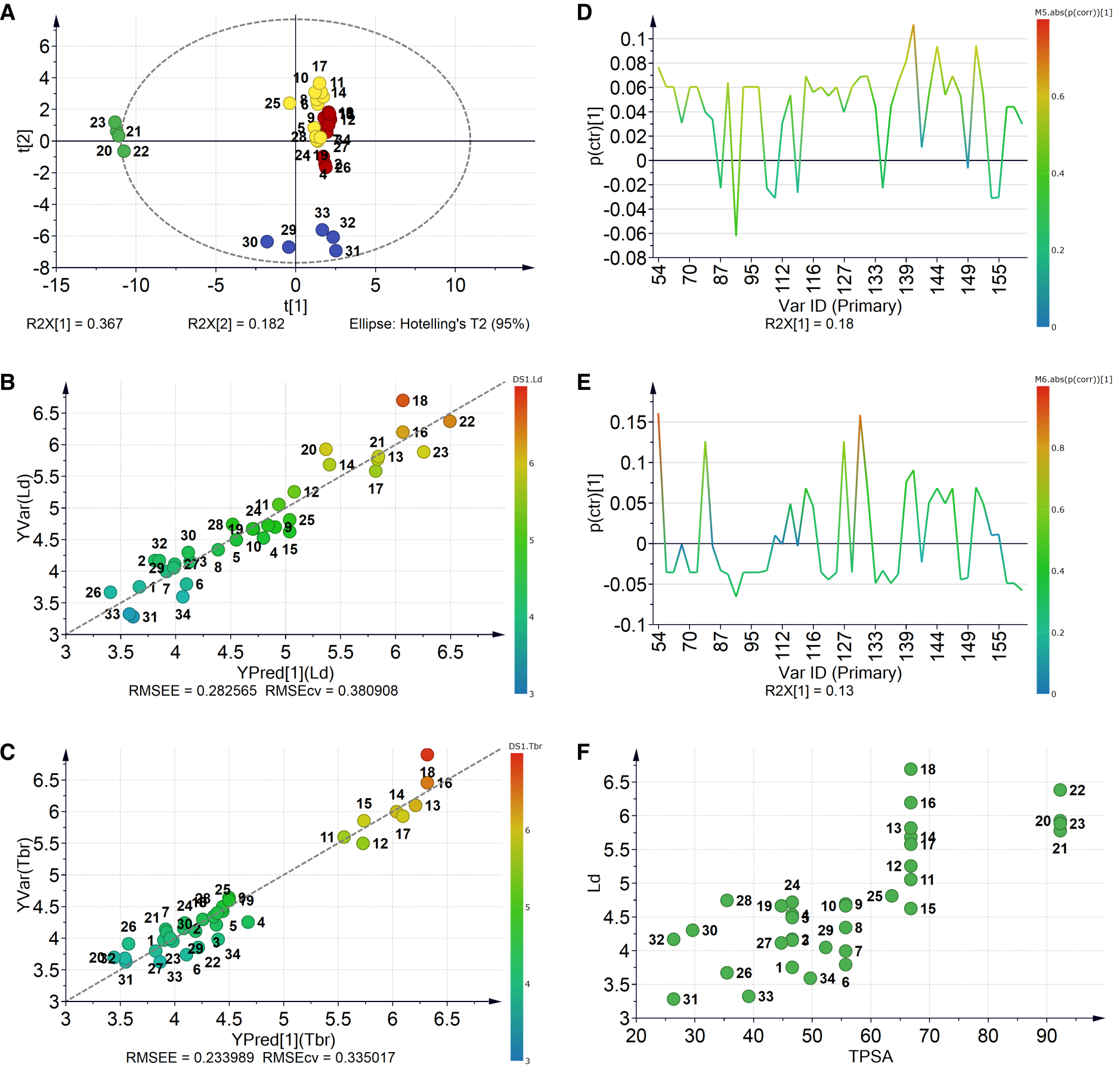
2.2.3. Robustness and Applicability Domain (AD) Definition of the Best QSAR Models for Antileishmanial Activity
2.2.4. Interpretation of the Best QSAR Models for Antileishmanial Activity
2.3. QSAR Modeling for Antitrypanosomal Activity
2.3.1. Building of QSAR Models for Antitrypanosomal Activity
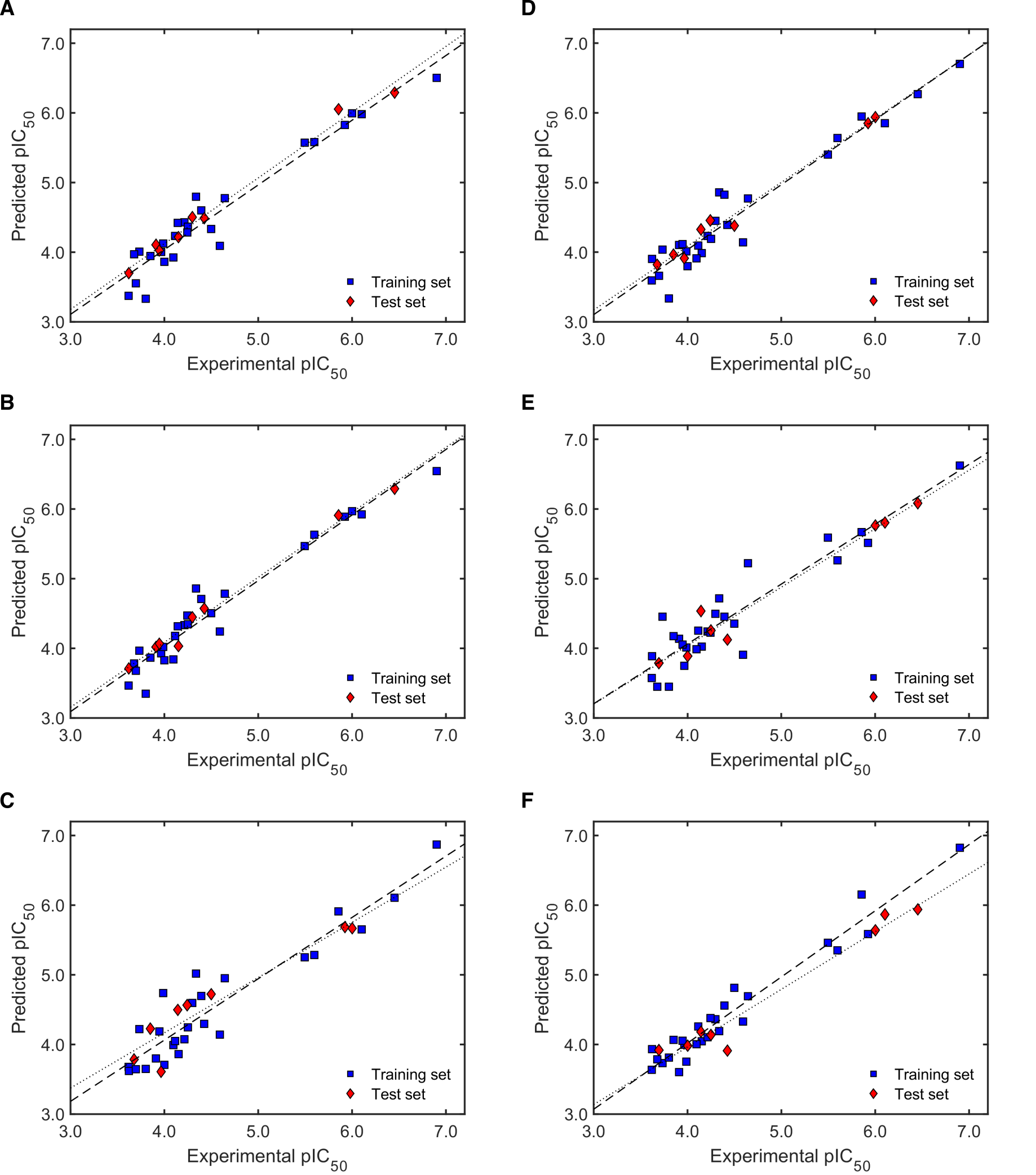
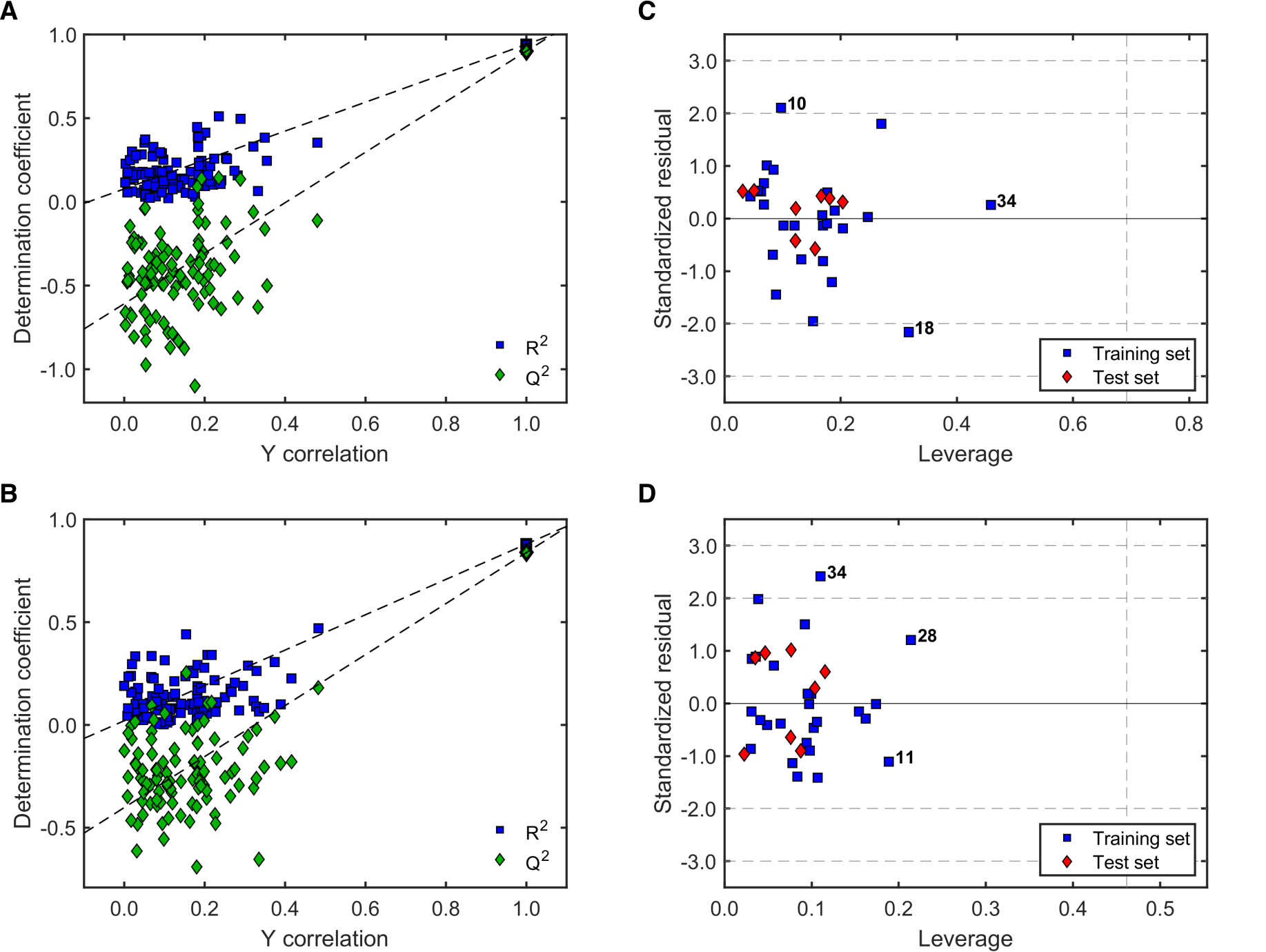
2.3.2. Validation of QSAR Models for Antitrypanosomal Activity
2.3.3. Robustness and AD Definition of the Best QSAR Models for Antitrypanosomal Activity
2.3.4. Interpretation of the Best QSAR Models for Antitrypanosomal Activity
3. Materials and Methods
3.1. Data Preparation
3.2. QSAR Modeling: First Approach Using Molecular Fingerprints
3.3. QSAR Modeling by GA/MLR
3.4. Complete Validation of the QSAR Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization WHO World Health Assembly. Available online: https://www.who.int/neglected_diseases/diseases/en/ (accessed on 31 January 2019).
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- Furuse, Y. Analysis of research intensity on infectious disease by disease burden reveals which infectious diseases are neglected by researchers. Proc. Natl. Acad. Sci. USA. 2019, 116, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Filardy, A.A.; Guimarães-Pinto, K.; Nunes, M.P.; Zukeram, K.; Fliess, L.; Pereira, L.; Nascimento, D.O.; Conde, L.; Morrot, A. Human Kinetoplastid Protozoan infections: Where Are we Going Next? Front. Immunol. 2018, 9, 1493. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gürtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Invest. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.S.; Barrett, M.P.; Dranoff, G.; Faraday, C.J.; Gimpelewicz, C.R.; Hailu, A.; Jones, C.L.; Kelly, J.M.; Lazdins-Helds, J.K.; Mäser, P.; et al. Drug Discovery for Kinetoplastid Diseases: Future Directions. ACS Infect. Dis. 2019, 5, 152–157. [Google Scholar] [CrossRef]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human african trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef]
- De Rycker, M.; Baragaña, B.; Duce, S.L.; Gilbert, I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature 2018, 559, 498–506. [Google Scholar] [CrossRef]
- Alcântara, L.M.; Ferreira, T.C.S.; Gadelha, F.R.; Miguel, D.C. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 430–439. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Njogu, P.M.; Guantai, E.M.; Pavadai, E.; Chibale, K. Computer-Aided Drug Discovery Approaches against the Tropical Infectious Diseases Malaria, Tuberculosis, Trypanosomiasis, and Leishmaniasis. ACS Infect. Dis. 2016, 2, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Amisigo, C.M.; Antwi, C.A.; Adjimani, J.P.; Gwira, T.M. In vitro anti-trypanosomal effects of selected phenolic acids on Trypanosoma brucei. PLoS One 2019, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Osman, S.M.; Almeida, I.A.; Cardoso, V.; Alasmary, F.A.S.; AlOthman, Z.; Vermelho, A.B.; Gratteri, P.; Supuran, C.T. Appraisal of anti-protozoan activity of nitroaromatic benzenesulfonamides inhibiting carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani. J. Enzyme Inhib. Med. Chem. 2019, 34, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tang, Y.; Li, N.G.; Zhu, Y.; Duan, J.A. Bioactivity and chemical synthesis of caffeic acid phenethyl ester and its derivatives. Molecules 2014, 19, 16458–16476. [Google Scholar] [CrossRef]
- Otero, E.; Robledo, S.M.; Díaz, S.; Carda, M.; Muñoz, D.; Paños, J.; Vélez, I.D.; Cardona, W. Synthesis and leishmanicidal activity of cinnamic acid esters: Structure-activity relationship. Med. Chem. Res. 2014, 23, 1378–1386. [Google Scholar] [CrossRef]
- Rodrigues, M.P.; Tomaz, D.C.; Ângelo de Souza, L.; Onofre, T.S.; Aquiles de Menezes, W.; Almeida-Silva, J.; Suarez-Fontes, A.M.; Rogéria de Almeida, M.; Manoel da Silva, A.; Bressan, G.C.; et al. Synthesis of cinnamic acid derivatives and leishmanicidal activity against Leishmania braziliensis. Eur. J. Med. Chem. 2019, 183, 111688. [Google Scholar] [CrossRef]
- Da Silva, E.R.; Brogi, S.; Grillo, A.; Campiani, G.; Gemma, S.; Vieira, P.C.; Maquiaveli, C. do C. Cinnamic acids derived compounds with antileishmanial activity target Leishmania amazonensis arginase. Chem. Biol. Drug Des. 2019, 93, 139–146. [Google Scholar] [CrossRef]
- Dos Santos, A.P. de A.; Fialho, S.N.; de Medeiros, D.S.S.; Garay, A.F.G.; Diaz, J.A.R.; Gómez, M.C.V.; Teles, C.B.G.; Calderon, L. de A. Antiprotozoal action of synthetic cinnamic acid analogs. Rev. Soc. Bras. Med. Trop. 2018, 51, 849–853. [Google Scholar]
- Otero, E.; García, E.; Palacios, G.; Yepes, L.M.; Carda, M.; Agut, R.; Vélez, I.D.; Cardona, W.I.; Robledo, S.M. Triclosan-caffeic acid hybrids: Synthesis, leishmanicidal, trypanocidal and cytotoxic activities. Eur. J. Med. Chem. 2017, 141, 73–83. [Google Scholar] [CrossRef]
- Lima, T.C.; Souza, R.J.; Santos, A.D.C.; Moraes, M.H.; Biondo, N.E.; Barison, A.; Steindel, M.; Biavatti, M.W. Evaluation of leishmanicidal and trypanocidal activities of phenolic compounds from Calea uniflora Less. Nat. Prod. Res. 2016, 30, 551–557. [Google Scholar] [CrossRef]
- Steverding, D.; da Nóbrega, F.R.; Rushworth, S. a.; de Sousa, D.P. Trypanocidal and cysteine protease inhibitory activity of isopentyl caffeate is not linked in Trypanosoma brucei. Parasitol. Res. 2016, 115, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Glaser, J.; Schultheis, M.; Hazra, S.; Hazra, B.; Moll, H.; Schurigt, U.; Holzgrabe, U. Antileishmanial lead structures from nature: Analysis of structure-activity relationships of a compound library derived from caffeic acid bornyl ester. Molecules 2014, 19, 1394–1410. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fu, J.; Shan, L.; Sun, Q.; Zhang, W. Synthesis, preliminary bioevaluation and computational analysis of caffeic acid analogues. Int. J. Mol. Sci. 2014, 15, 8808–8820. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.A.; Kaiser, M.; Wünsch, B.; Schmidt, T.J. Structure–Activity Relationships of Cinnamate Ester Analogs as Potent Antiprotozoal Agents. ChemMedChem 2019, 14. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; Van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Umetrics SIMCA 14.1 2015. Umeå, Sweden.
- Chemical Computing Group ULC Molecular Operating Environment (MOE), 2018.01. 1010 Sherbooke St.West, Suite #910, Montr. QC, Canada, H3A 2R7. 2019.
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef]
- Andrada, M.F.; Vega-Hissi, E.G.; Estrada, M.R.; Garro Martinez, J.C. Impact assessment of the rational selection of training and test sets on the predictive ability of QSAR models. SAR QSAR Environ. Res. 2017, 28, 1011–1023. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Snarey, M.; Terrett, N.K.; Willett, P.; Wilton, D.J. Comparison of algorithms for dissimilarity-based compound selection. J. Mol. Graph. Model. 1997, 15, 372–385. [Google Scholar] [CrossRef]
- Hopfinger, A.J.; Patel, H.C. Application of Genetic Algorithms to the General QSAR Problem and to Guiding Molecular Diversity Experiments. In Principles of QSAR and Drug Design, Genetic Algorithms in Molecular Modeling; Devillers, J., Ed.; Academic Press: London, UK, 1996; pp. 131–157. ISBN 9780122138102. [Google Scholar]
- Sukumar, N.; Prabhu, G.; Saha, P. Applications of genetic algorithms in QSAR/QSPR modeling. In Applications of Metaheuristics in Process Engineering; Valadi, J., Siarry, P., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 315–324. ISBN 9783319065083. [Google Scholar]
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment; OECD Publishing: Paris, France, 2007; ISBN 9789264085442. [Google Scholar]
- Topliss, J.G.; Costello, R.J. Chance Correlations in Structure-Activity Studies Using Multiple Regression Analysis. J. Med. Chem. 1972, 15, 1066–1068. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Beware of q^2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. On some aspects of variable selection for partial least squares regression models. QSAR Comb. Sci. 2008, 27, 302–313. [Google Scholar] [CrossRef]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “rm2” metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Shi, L.M.; Fang, H.; Tong, W.; Wu, J.; Perkins, R.; Blair, R.M.; Branham, W.S.; Dial, S.L.; Moland, C.L.; Sheehan, D.M. QSAR Models Using a Large Diverse Set of Estrogens. J. Chem. Inf. Comput. Sci. 2001, 41, 186–195. [Google Scholar] [CrossRef]
- Schüuürmann, G.; Ebert, R.-U.; Chen, J.; Wang, B.; Kühne, R. External Validation and Prediction Employing the Predictive Squared Correlation Coefficient-Test Set Activity Mean vs Training Set Activity Mean. J. Chem. Inf. Model. 2008, 48, 2140–2145. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Evaluation of model predictive ability by external validation techniques. J. Chemom. 2010, 24, 194–201. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate It? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef]
- Polishchuk, P. Interpretation of Quantitative Structure-Activity Relationship Models: Past, Present, and Future. J. Chem. Inf. Model. 2017, 57, 2618–2639. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “two QSARs. ” J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Sun, G.; Fan, T.; Sun, X.; Liu, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. Prediction on the mutagenicity of nitroaromatic compounds using quantum chemistry descriptors based QSAR and machine learning derived classification methods. Ecotoxicol. Environ. Saf. 2019, 186, 109822. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Wold, S.; Eriksson, L.; Clementi, S. Statistical Validation of QSAR Results. In Chemometric Methods in Molecular Design; Dr. van de Waterbeemd, H., Ed.; VCH Verlagsgesellschaft mbH: Weinheim, Germany, 1995; pp. 309–338. ISBN 9783527615452. [Google Scholar]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. On the development and validation of QSAR models. In Computational Toxicology. Methods in Molecular Biology; Reisfeld, B., Mayeno, A., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 930, pp. 499–526. ISBN 9781627030588. [Google Scholar]
- Cruciani, G.; Crivori, P.; Carrupt, P.A.; Testa, B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; WileyVCH: Weinheim, Germany, 2000; ISBN 9783527299133. [Google Scholar]
- Hall, L.H.; Kier, L.B. The Molecular Connectivity Chi Indexes and Kappa Shape Indexes in Structure-Property Modeling. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH, Inc.: New York, NY, USA, 1991; pp. 367–422. [Google Scholar]
- MathWorks MATLAB R2018b 2018.
- Chemical Computing Group (CCG) Support and Training. Available online: https://www.chemcomp.com/Support.htm, (accessed on 05 October 2019).
- Gramatica, P.; Sangion, A. A Historical Excursus on the Statistical Validation Parameters for QSAR Models: A Clarification Concerning Metrics and Terminology. J. Chem. Inf. Model. 2016, 56, 1127–1131. [Google Scholar] [CrossRef]
- Eriksson, L.; Byrne, T.; Johansson, E.; Trygg, J.; Vikström, C. Multi-and Megavariate Data Analysis Basic Principles and Applications; Umetrics Academy: Malmö, Sweden, 2013; ISBN 9197373052. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor * | Frequency Per Total of Descriptors (%) | Frequency Per Number of Equations (%) |
|---|---|---|
| ASA– | 12.9 | 54 |
| PEOE_VSA_FPOL | 11.4 | 48 |
| chi1v | 7.1 | 30 |
| PEOE_VSA_FHYD | 7.1 | 30 |
| vsurf_D1 | 5.7 | 24 |
| Function | Definition | Observations a | ||
|---|---|---|---|---|
| P1 | P2 | P3 b | ||
| F1 | 1) > 0.8 2) > 0.5 3) > 0.6 4) > 0.5 5) > 0.5 7) | 1) 2) 3) | 1) 2) | |
| F2 | 1) 2) 3) 4) 5) 6) 7) 8) | 1) 2) 3) | 1) and 2) | |
| F3 c | -- | -- | -- | |
| Model | TR a | Ndb | F1 | F2 c | F3 c | Model | TR a | Ndb | F1 | F2 c | F3 c |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M1-1 | 1 | 3 | 8 | 1.49 | - | M6-1 | 2 | 5 | 10 | 2.47 | - |
| M1-2 | 1 | 3 | 6 | −0.07 | - | M6-2 | 2 | 5 | 12 | 2.99 | 0.598 |
| M1-3 | 1 | 3 | 9 | 2.26 | - | M6-3 | 2 | 5 | 10 | 2.48 | - |
| M1-4 | 1 | 3 | 8 | 1.23 | - | M6-4 | 2 | 5 | 12 | 2.62 | 0.525 |
| M1-5 | 1 | 3 | 10 | 2.31 | - | M6-5 | 2 | 5 | 12 | 2.81 | 0.562 |
| M2-1 | 1 | 4 | 12 | 2.53 | 0.632 | M7-1 | 3 | 3 | 9 | 1.64 | - |
| M2-2 | 1 | 4 | 4 | −1.00 | - | M7-2 | 3 | 3 | 8 | 0.61 | - |
| M2-3 | 1 | 4 | 11 | 2.08 | - | M7-3 | 3 | 3 | 9 | 0.72 | - |
| M2-4 | 1 | 4 | 12 | 2.18 | 0.546 | M7-4 | 3 | 3 | 9 | 0.66 | - |
| M2-5 | 1 | 4 | 9 | 1.82 | - | M7-5 | 3 | 3 | 8 | 0.65 | - |
| M3-1 | 1 | 5 | 6 | −0.04 | - | M8-1 | 3 | 4 | 9 | 1.45 | - |
| M3-2 | 1 | 5 | 12 | 2.04 | 0.409 | M8-2 | 3 | 4 | 7 | −0.20 | - |
| M3-3 | 1 | 5 | 10 | 2.20 | - | M8-3 | 3 | 4 | 8 | 0.98 | - |
| M3-4 | 1 | 5 | 7 | 1.54 | - | M8-4 | 3 | 4 | 8 | 0.91 | - |
| M3-5 | 1 | 5 | 3 | −3.18 | - | M8-5 | 3 | 4 | 7 | 0.63 | - |
| M4-1 | 2 | 3 | 2 | −6.56 | - | M9-1 | 3 | 5 | 10 | 1.85 | - |
| M4-2 | 2 | 3 | 2 | −6.56 | - | M9-2 | 3 | 5 | 10 | 2.34 | - |
| M4-3 | 2 | 3 | 9 | 1.56 | - | M9-3 | 3 | 5 | 9 | 1.64 | - |
| M4-4 | 2 | 3 | 9 | 1.56 | - | M9-4 | 3 | 5 | 9 | 1.67 | - |
| M4-5 | 2 | 3 | 10 | 2.19 | - | M9-5 | 3 | 5 | 9 | 1.67 | - |
| M5-1 | 2 | 4 | 12 | 2.77 | 0.692 | M10-1 | 3 | 6 | 11 | 2.62 | - |
| M5-2 | 2 | 4 | 11 | 2.55 | - | M10-2 | 3 | 6 | 11 | 2.60 | - |
| M5-3 | 2 | 4 | 11 | 2.55 | - | M10-3 | 3 | 6 | 9 | 1.87 | - |
| M5-4 | 2 | 4 | 10 | 2.28 | - | M10-4 | 3 | 6 | 9 | 1.88 | - |
| M5-5 | 2 | 4 | 11 | 2.47 | - | M10-5 | 3 | 6 | 10 | 2.02 | - |
| Model | Equation |
|---|---|
| M5-1 | 0.59011 − 0.0263602× ASA– + 16.9485 × PEOE_VSA_FPOL + 0.162194 × a_IC − 0.00750344× vsurf_W3 |
| M6-2 | 18.0466 − 0.025593× ASA– − 17.8311× PEOE_VSA_FHYD − 0.00420797× Q_VSA_POL + 0.177629× a_IC − 0.00880367× vsurf_W3 |
| Descriptor * | Frequency Per Total of Descriptors (%) | Frequency Per Number of Equations (%) |
|---|---|---|
| vsurf_EWmin3 | 13.3 | 53.3 |
| std_dim2 | 8.9 | 35.6 |
| vsurf_EWmin2 | 4.4 | 17.8 |
| ASA_P | 2.8 | 11.1 |
| chi1v_C | 2.8 | 11.1 |
| FCASA+ | 2.8 | 11.1 |
| h_pKb | 2.8 | 11.1 |
| Q_VSA_FPPOS | 2.8 | 11.1 |
| Model | TR a | Ndb | F1 | F2 c | F3 c | Model | TR a | ndb | F1 | F2 c | F3 c |
|---|---|---|---|---|---|---|---|---|---|---|---|
| M11-1 | 1 | 3 | 2 | −32.2 | - | M15-4 | 2 | 4 | 11 | 1.91 | - |
| M11-2 | 1 | 3 | 2 | −37.1 | - | M15-5 | 2 | 4 | 11 | 1.99 | - |
| M11-3 | 1 | 3 | 2 | −31.6 | - | M16-1 | 2 | 5 | 12 | 2.62 | 0.525 |
| M11-4 | 1 | 3 | 2 | −31.0 | - | M16-2 | 2 | 5 | 12 | 2.66 | 0.531 |
| M11-5 | 1 | 3 | 2 | −33.2 | - | M16-3 | 2 | 5 | 12 | 2.57 | 0.514 |
| M12-1 | 1 | 4 | 12 | 2.33 | 0.584 | M16-4 | 2 | 5 | 11 | 2.11 | - |
| M12-2 | 1 | 4 | 12 | 2.17 | 0.543 | M16-5 | 2 | 5 | 12 | 2.25 | 0.450 |
| M12-3 | 1 | 4 | 12 | 2.42 | 0.606 | M17-1 | 3 | 3 | 11 | 1.52 | - |
| M12-4 | 1 | 4 | 12 | 2.60 | 0.651 | M17-2 | 3 | 3 | 12 | 1.85 | 0.616 |
| M12-5 | 1 | 4 | 12 | 2.60 | 0.651 | M17-3 | 3 | 3 | 10 | 1.36 | - |
| M13-1 | 1 | 5 | 12 | 2.39 | 0.479 | M17-4 | 3 | 3 | 11 | 1.46 | - |
| M13-2 | 1 | 5 | 12 | 2.59 | 0.518 | M17-5 | 3 | 3 | 11 | 1.62 | - |
| M13-3 | 1 | 5 | 12 | 2.79 | 0.558 | M18-1 | 3 | 4 | 11 | 2.59 | - |
| M13-4 | 1 | 5 | 12 | 2.43 | 0.486 | M18-2 | 3 | 4 | 12 | 2.34 | 0.586 |
| M13-5 | 1 | 5 | 12 | 2.60 | 0.519 | M18-3 | 3 | 4 | 12 | 2.14 | 0.534 |
| M14-1 | 2 | 3 | 11 | 2.56 | - | M18-4 | 3 | 4 | 12 | 2.40 | 0.599 |
| M14-2 | 2 | 3 | 12 | 2.32 | 0.774 | M18-5 | 3 | 4 | 12 | 2.32 | 0.579 |
| M14-3 | 2 | 3 | 11 | 1.48 | - | M19-1 | 3 | 5 | 12 | 2.47 | 0.495 |
| M14-4 | 2 | 3 | 11 | 2.18 | - | M19-2 | 3 | 5 | 11 | 1.65 | - |
| M14-5 | 2 | 3 | 11 | 1.93 | - | M19-3 | 3 | 5 | 11 | 1.76 | - |
| M15-1 | 2 | 4 | 11 | 2.31 | - | M19-4 | 3 | 5 | 12 | 1.97 | 0.395 |
| M15-2 | 2 | 4 | 11 | 1.46 | - | M19-5 | 3 | 5 | 11 | 1.45 | - |
| M15-3 | 2 | 4 | 7 | 0.65 | - |
| Model | Equation |
|---|---|
| M13-3 | 6.76902 − 0.008234 × ASA– − 15.0033 × Q_RPC– + 3.09437× Q_VSA_FPPOS − 1.78347× std_dim2 − 0.801448× vsurf_EWmin3 |
| M14-2 | –1.45307 + 0.39037× chi1_C + 0.149959× lip_don − 0.771908× vsurf_EWmin3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernal, F.A.; Schmidt, T.J. A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues. Molecules 2019, 24, 4358. https://doi.org/10.3390/molecules24234358
Bernal FA, Schmidt TJ. A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues. Molecules. 2019; 24(23):4358. https://doi.org/10.3390/molecules24234358
Chicago/Turabian StyleBernal, Freddy A., and Thomas J. Schmidt. 2019. "A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues" Molecules 24, no. 23: 4358. https://doi.org/10.3390/molecules24234358
APA StyleBernal, F. A., & Schmidt, T. J. (2019). A Comprehensive QSAR Study on Antileishmanial and Antitrypanosomal Cinnamate Ester Analogues. Molecules, 24(23), 4358. https://doi.org/10.3390/molecules24234358