
Improving the Light-Induced Spin Transition Efficiency in Ni(II)-Based Macrocyclic-Ligand Complexes


Abstract

1. Introduction
2. Results and Discussions
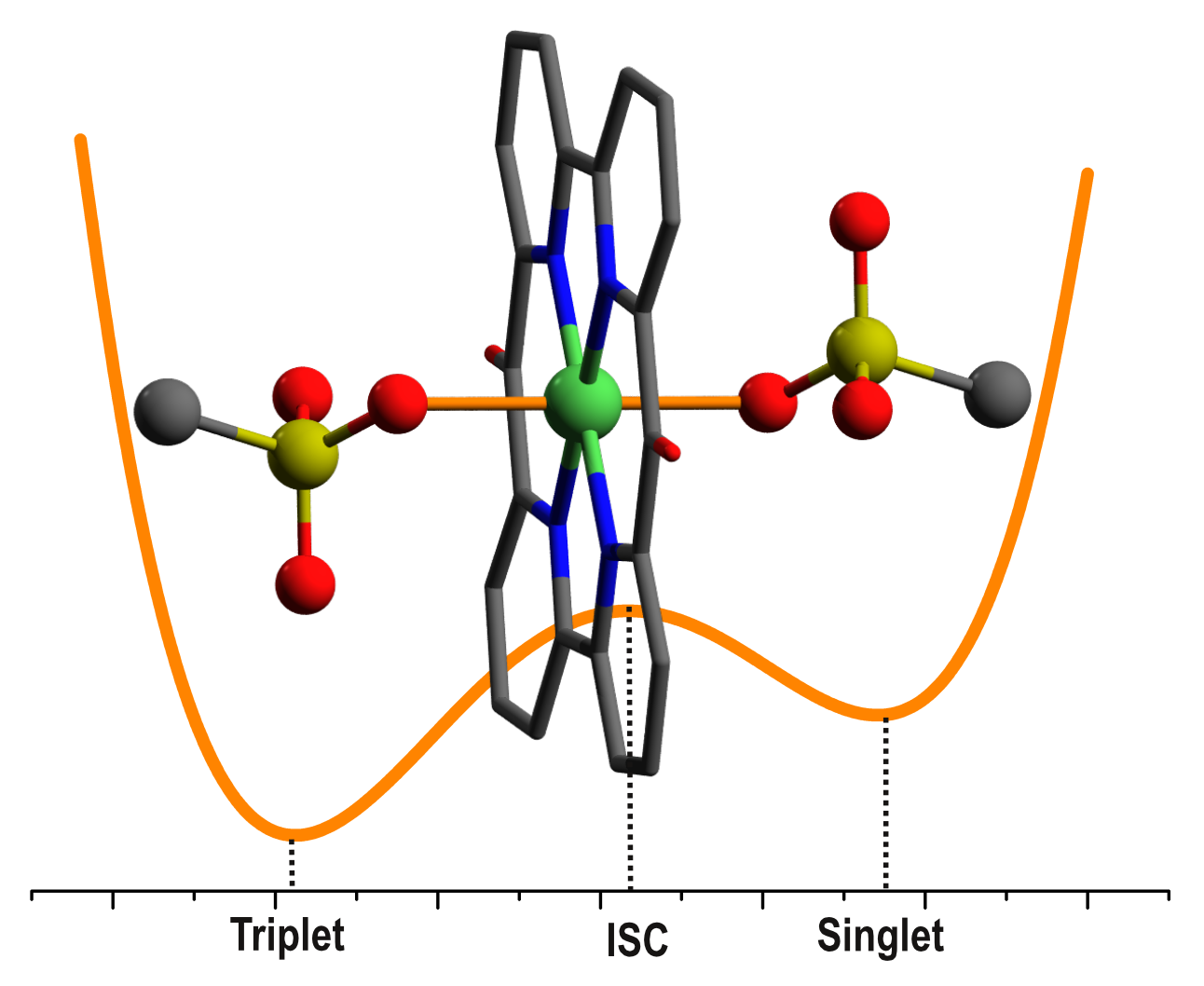
2.1. Structures and Energetics
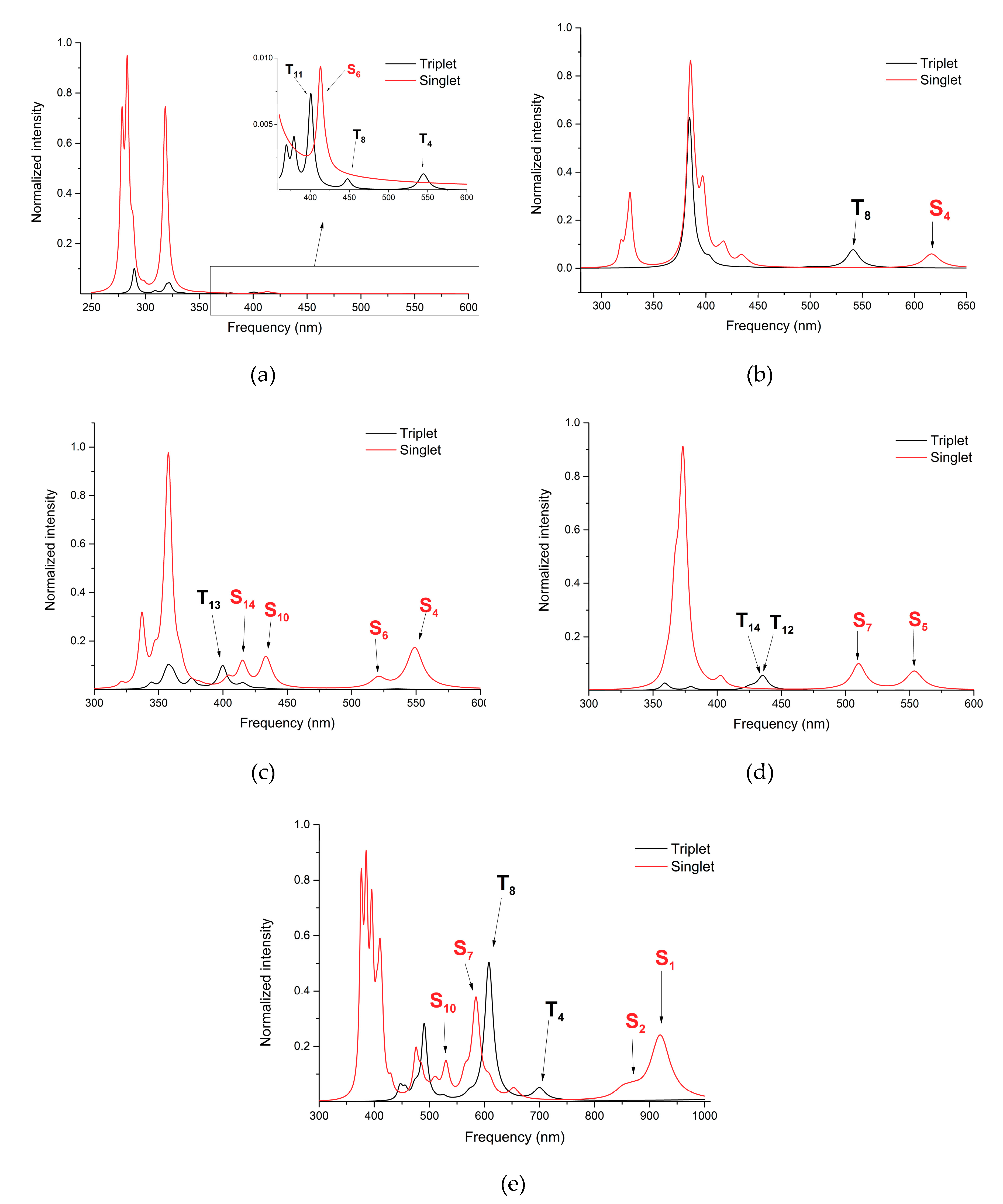
2.2. Singlet and Triplet Electronic Transitions
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kumar, K.; Ruben, M. Emerging trends in spin crossover (SCO) based functional materials and devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Quintero, C.; Felix, G.; Suleimanov, I.; Costa, J.; Molnár, G.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Hybrid spin-crossover nanostructures. Beilstein J. Nanotech. 2014, 5, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Yersin, H.; Strasser, J. Triplets in metal-organic compounds. Chemical tunability of relaxation dynamics. Coord. Chem. Rev. 2000, 208, 331–364. [Google Scholar] [CrossRef]
- Kjaer, K.; Zhang, W.; Alonso-Mori, R.; Bergmann, U.; Chollet, M.; Hadt, R.; Hartsock, R.; Harlang, T.; Kroll, T.; Kubicek, K.; et al. Ligand manipulation of charge transfer excited state relaxation and spin crossover in [Fe(2,2 ‘- bipyridine)(2)(CN)(2)]. Struct. Dynam. 2017, 4, 044030. [Google Scholar] [CrossRef]
- Zhang, W.; Kjaer, K.; Alonso-Mori, R.; Bergmann, U.; Chollet, M.; Fredin, L.; Hadt, R.; Hartsock, R.; Harlang, T.; Kroll, T.; et al. Manipulating charge transfer excited state relaxation and spin crossover in iron coordination complexes with ligand substitution. Chem. Sci. 2017, 8, 515–523. [Google Scholar] [CrossRef]
- Meng, Y.; Sato, O.; Liu, T. Manipulating metal-to-metal charge transfer for materials with switchable functionality. Angew. Chem. Int. Ed. 2018, 57, 12216–12226. [Google Scholar] [CrossRef]
- Farcaș, A.; Beu, T.; Bende, A. The influence of the metal-ligand charge transfer effects on the structural stability and the strength of the spin-orbit coupling in Ni(II)-based metal-ligand complexes. Int. J. Quant. Chem. 2019. under review. [Google Scholar]
- Penfold, T.; Gindensperger, E.; Daniel, C.; Marian, C. Spin-vibronic mechanism for intersystem crossing. Chem. Rev. 2018, 118, 6975–7025. [Google Scholar] [CrossRef]
- Marian, C. Spin-orbit coupling and intersystem crossing in molecules. Wires Comput. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Heitz, M.; Ribbing, C.; Daniel, C. Spin-orbit induced radiationless transitions in organometallics: Quantum simulation of the intersystem crossing processes in the photodissociation of HCo(CO)(4). J. Chem. Phys. 1997, 106, 1421–1428. [Google Scholar] [CrossRef]
- Vlassa, M.; Bende, A. Theoretical investigation of polymer chain stability in the metal coordinated azorubine and cyclam complex. Chem. Phys. 2015, 457, 152–159. [Google Scholar] [CrossRef]
- Lawthers, I.; Mcgarvey, J. Spin-state relaxation dynamics in iron(III) complexes—Photochemical perturbation of the 2t reversible 6a spin equilibrium by pulsed-laser irradiation in the ligand-to-metal charge-transfer absorption-band. J. Am. Chem. Soc. 1984, 106, 4280–4282. [Google Scholar] [CrossRef]
- Decurtins, S.; Gütlich, P.; Kohler, C.; Spiering, H.; Hauser, A. Light-induced excited spin state trapping in a transition-metal complex—The hexa-1-propyltetrazole-iron(II) tetrafluoroborate spin-crossover system. Chem. Phys. Lett. 1984, 105, 1–4. [Google Scholar] [CrossRef]
- Hauser, A. Light-induced spin crossover and the high-spin -> low-spin relaxation. In Spin Crossover in Transition Metal Compounds II; Gütlich, P., Goodwin, H.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 155–198. [Google Scholar] [CrossRef]
- Ohkoshi, S.; Imoto, K.; Tsunobuchi, Y.; Takano, S.; Tokoro, H. Light-induced spin-crossover magnet. Nat. Chem. 2011, 3, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ridier, K.; Molnár, G.; Salmon, L.; Nicolazzi, W.; Bousseksou, A. Hysteresis, nucleation and growth phenomena in spin-crossover solids. Solid State Sci. 2017, 74, A1–A22. [Google Scholar] [CrossRef]
- Venkataramani, S.; Jana, U.; Dommaschk, M.; Sonnichsen, F.; Tuczek, F.; Herges, R. Magnetic bistability of molecules in homogeneous solution at room temperature. Science 2011, 331, 445–448. [Google Scholar] [CrossRef]
- Dommaschk, M.; Peters, M.; Gutzeit, F.; Schütt, C.; Näther, C.; Sönnichsen, F.D.; Tiwari, S.; Riedel, C.; Boretius, S.; Herges, R. Photoswitchable magnetic resonance imaging contrast by improved light-driven coordination-induced spin state switch. J. Am. Chem. Soc. 2015, 137, 7552–7555. [Google Scholar] [CrossRef]
- Rosner, B.; Milek, M.; Witt, A.; Gobaut, B.; Torelli, P.; Fink, R.; Khusniyarov, M. Reversible photoswitching of a spin-crossover molecular complex in the solid state at room temperature. Angew. Chem. Int. Ed. 2015, 54, 12976–12980. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Meng, Y.; Zheng, H.; Zhu, H.; Shi, Q.; Liu, T. Synergic on/off photoswitching spin state and magnetic coupling between spin crossover centers. Inorg. Chem. 2017, 56, 10674–10680. [Google Scholar] [CrossRef]
- Chastanet, G.; Lorenc, M.; Bertoni, R.; Desplanches, C. Light-induced spin crossover-Solution and solid-state processes. CR Chim. 2018, 21, 1075–1094. [Google Scholar] [CrossRef]
- Kepp, K. Consistent descriptions of metal-ligand bonds and spin-crossover in inorganic chemistry. Coord. Chem. Rev. 2013, 257, 196–209. [Google Scholar] [CrossRef]
- Khusnutdinova, J.; Milstein, D. Metal-ligand cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef] [PubMed]
- Worner, H.; Arrell, C.; Banerji, N.; Cannizzo, A.; Chergui, M.; Das, A.; Hamm, P.; Keller, U.; Kraus, P.; Liberatore, E.; et al. Charge migration and charge transfer in molecular systems. Struct. Dynam. 2017, 4, 061508. [Google Scholar] [CrossRef] [PubMed]
- Bressler, C.; Milne, C.; Pham, V.; ElNahhas, A.; van der Veen, R.; Gawelda, W.; Johnson, S.; Beaud, P.; Grolimund, D.; Kaiser, M.; et al. Femtosecond XANES study of the light-induced spin crossover dynamics in an Iron(II) complex. Science 2009, 323, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, A.; Milne, C.; Consani, C.; Gawelda, W.; Bressler, C.; van Mourik, F.; Chergui, M. Light-induced spin crossover in Fe(II)-based complexes: The full photocycle unraveled by ultrafast optical and X-ray spectroscopies. Coord. Chem. Rev. 2010, 254, 2677–2686. [Google Scholar] [CrossRef]
- Vanko, G.; Glatzel, P.; Pham, V.; Abela, R.; Grolimund, D.; Borca, C.; Johnson, S.; Milne, C.; Bressler, C. Picosecond time-resolved X-ray emission spectroscopy: Ultrafast spin-state determination in an Iron complex. Angew. Chem. Int. Ed. 2010, 49, 5910–5912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Alonso-Mori, R.; Bergmann, U.; Bressler, C.; Chollet, M.; Galler, A.; Gawelda, W.; Hadt, R.; Hartsock, R.; Kroll, T.; et al. Tracking excited-state charge and spin dynamics in iron coordination complexes. Nature 2014, 509, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gaffney, K. Mechanistic studies of photoinduced spin crossover and electron transfer in inorganic complexes. Acc. Chem. Res. 2015, 48, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Chabera, P.; Kjaer, K.; Prakash, O.; Honarfar, A.; Liu, Y.; Fredin, L.; Harlang, T.; Lidin, S.; Uhlig, J.; Sundstrom, V.; et al. Fe-II hexa N-heterocyclic carbene complex with a 528 ps metal-to-ligand charge-transfer excited-state lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef]
- Chabera, P.; Liu, Y.; Prakash, O.; Thyrhaug, E.; El Nahhas, A.; Honarfar, A.; Essen, S.; Fredin, L.; Harlang, T.; Kjaer, K.; et al. A low-spin Fe(III) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. [Google Scholar] [CrossRef]
- Daku, L.; Aquilante, F.; Robinson, T.; Hauser, A. Accurate spin-state energetics of transition metal complexes. 1. CCSD(T), CASPT2, and DFT study of [M(NCH)(6)](2+) (M = Fe, Co). J. Chem. Theor. Comput. 2012, 8, 4216–4231. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, N.; Soupart, A.; Heitz, M. Methodological CASPT2 study of the valence excited states of an iron-porphyrin complex. J. Mol. Model. 2017, 23, 53. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Mussard, B.; Holmes, A.; Sharma, S. Cheap and near exact CASSCF with large active spaces. J. Chem. Theor. Comput. 2017, 13, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Garino, C.; Salassa, L. The photochemistry of transition metal complexes using density functional theory. Philos. T. R. Soc. A 2013, 371, 20120134. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Freitag, L.; Gonzalez, L. Using computational chemistry to design Ru photosensitizers with directional charge transfer. Coord. Chem. Rev. 2015, 304, 146–165. [Google Scholar] [CrossRef]
- Saureu, S.; de Graaf, C. TD-DFT study of the light-induced spin crossover of Fe(III) complexes. Phys. Chem. Chem. Phys. 2016, 18, 1233–1244. [Google Scholar] [CrossRef]
- Ronca, E.; De Angelis, F.; Fantacci, S. Time-dependent density functional theory modeling of spin-orbit coupling in ruthenium and osmium solar cell sensitizers. J. Phys. Chem. C 2014, 118, 17067–17078. [Google Scholar] [CrossRef]
- Sousa, C.; de Graaf, C.; Rudavskyi, A.; Broer, R.; Tatchen, J.; Etinski, M.; Marian, C. Ultrafast deactivation mechanism of the excited singlet in the light-induced spin crossover of [Fe(2,2 ‘-bipyridine)(3)](2+). Chem. Eur. J. 2013, 19, 17541–17551. [Google Scholar] [CrossRef]
- Atkins, A.; Talotta, F.; Freitag, L.; Boggio-Pasqua, M.; Gonzalez, L. Assessing excited state energy gaps with time-dependent density functional theory on Ru(II) complexes. J. Chem. Theor. Comp. 2017, 13, 4123–4145. [Google Scholar] [CrossRef]
- Sousa, C.; Llunell, M.; Domingo, A.; de Graaf, C. Theoretical evidence for the direct (MLCT)-M-3-HS deactivation in the light-induced spin crossover of Fe((II))-polypyridyl complexes. Phys. Chem. Chem. Phys. 2018, 20, 2351–2355. [Google Scholar] [CrossRef]
- Farcas, A.A.; Beu, T.A.; Bende, A. Light-induced spin transitions in Ni(II)-based macrocyclic-ligand complexes: A DFT study. J. Photoch. Photobio. A 2019, 376, 316–323. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Principles of Molecular Photochemistry: An Introduction; University Science Books: Sausalito, CA, USA, 2009. [Google Scholar]
- Thies, S.; Bornholdt, C.; Köhler, F.; Sönnichsen, F.D.; Näther, C.; Tuczek, F.; Herges, R. Coordination-induced spin crossover (CISCO) through axial bonding of substituted pyridines to nickel–porphyrins: σ-Donor versus π-acceptor effects. Chem. Eur. J. 2010, 16, 10074–10083. [Google Scholar] [CrossRef] [PubMed]
- Dommaschk, M.; Gutzeit, F.; Boretius, S.; Haag, R.; Herges, R. Coordination-induced spin-state-switch (CISSS) in water. Chem. Commun. 2014, 50, 12476–12478. [Google Scholar] [CrossRef] [PubMed]
- Dommaschk, M.; Thoms, V.; Schütt, C.; Näther, C.; Puttreddy, R.; Rissanen, K.; Herges, R. Coordination-induced spin-state switching with nickel chlorin and nickel isobacteriochlorin. Inorg. Chem. 2015, 54, 9390–9392. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, H.; Krahmer, J.; Fischer, K.; Schwager, B.; Flöser, B.; Näther, C.; Tuczek, F. Coordination-induced spin-state switching with Nickel(II) salpn complexes: Electronic versus steric effects and influence of intermolecular interactions. Eur. J. Inorg. Chem. 2018, 576–585. [Google Scholar] [CrossRef]
- Ioannidis, E.I.; Kulik, H.J. Towards quantifying the role of exact exchange in predictions of transition metal complex properties. J. Chem. Phys. 2015, 143, 034104. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, E.I.; Kulik, H.J. Ligand-field-dependent behavior of meta-GGA exchange in transition-metal complex spin-state ordering. J. Phys. Chem. A 2017, 121, 874–884. [Google Scholar] [CrossRef]
- Siig, O.S.; Kepp, K.P. Iron(II) and Iron(III) spin crossover: Toward an optimal density functional. J. Phys. Chem. A 2018, 122, 4208–4217. [Google Scholar] [CrossRef]
- Kepp, K.P. Theoretical study of spin crossover in 30 iron complexes. Inorg. Chem. 2016, 55, 2717–2727. [Google Scholar] [CrossRef]
- Phonsri, W.; Harding, P.; Liu, L.; Telfer, S.G.; Murray, K.S.; Moubaraki, B.; Ross, T.M.; Jameson, G.N.L.; Harding, D.J. Solvent modified spin crossover in an iron(III) complex: Phase changes and an exceptionally wide hysteresis. Chem. Sci. 2017, 8, 3949–3959. [Google Scholar] [CrossRef]
- Joliat-Wick, E.; Weder, N.; Klose, D.; Bachmann, C.; Spingler, B.; Probst, B.; Alberto, R. Light-induced H2 evolution with a macrocyclic cobalt diketo-pyrphyrin as a proton-reducing catalyst. Inorg. Chem. 2018, 57, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Kohler, L.; Niklas, J.; Johnson, R.C.; Zeller, M.; Poluektov, O.G.; Mulfort, K.L. Molecular cobalt catalysts for H2 generation with redox activity and proton relays in the second coordination sphere. Inorg. Chem. 2019, 58, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- De Souza, B.; Farias, G.; Neese, F.; Izsák, R. Predicting phosphorescence rates of light organic molecules using time-dependent density functional theory and the path integral approach to dynamics. J. Chem. Theor. Comput. 2019, 15, 1896–1904. [Google Scholar] [CrossRef] [PubMed]
- Gourlaouen, C.; Daniel, C. Spin-orbit effects in square-planar Pt(II) complexes with bidentate and terdentate ligands: Theoretical absorption/emission spectroscopy. Dalton Trans. 2014, 43, 17806–17819. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Marquetand, P.; Gonzalez, L. A general method to describe intersystem crossing dynamics in trajectory surface hopping. Int. J. Quant. Chem. 2015, 115, 1215–1231. [Google Scholar] [CrossRef]
- Atkins, A.; Gonzalez, L. Trajectory surface-hopping dynamics including intersystem crossing in [Ru(bpy)(3)](2+). J. Phys. Chem. Lett. 2017, 8, 3840–3845. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wires Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wires Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Delcey, M.; Pierloot, K.; Phung, Q.; Vancoillie, S.; Lindh, R.; Ryde, U. Accurate calculations of geometries and singlet-triplet energy differences for active-site models of [NiFe] hydrogenase. Phys. Chem. Chem. Phys. 2014, 16, 7927–7938. [Google Scholar] [CrossRef] [PubMed]
- Runge, E.; Gross, E. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Petersilka, M.; Gossmann, U.; Gross, E. Excitation energies from time-dependent density-functional theory. Phys. Rev. Lett. 1996, 76, 1212–1215. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Marom, N.; Kronik, L. Density functional theory of transition metal phthalocyanines, I: Electronic structure of NiPc and CoPc-self-interaction effects. Appl. Phys. A-Mater. 2009, 95, 159–163. [Google Scholar] [CrossRef]
- Marom, N.; Kronik, L. Density functional theory of transition metal phthalocyanines, II: Electronic structure of MnPc and FePc-symmetry and symmetry breaking. Appl. Phys. A-Mater. 2009, 95, 165–172. [Google Scholar] [CrossRef]
- Neese, F. Metal and ligand hyperfine couplings in transition metal complexes: The effect of spin-orbit coupling as studied by coupled perturbed Kohn-Sham theory. J. Chem. Phys. 2003, 118, 3939–3948. [Google Scholar] [CrossRef]
- Foster, J.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.; Weinhold, F. Natural localized molecular-orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO v3.1, Version 3.1. Available online: http://nbo6.chem.wisc.edu/ (accessed on 22 November 2019).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gaussview, Version 5.0.9; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conf. | Geom. | O⊥···Ni (Å) | N∥···Ni (Å) | Econf (eV) | Conf. | Geom. | O⊥···Ni (Å) | N∥···N (Å) | Econf (eV) |
|---|---|---|---|---|---|---|---|---|---|
| C1 | Sing. | 2.709 2.709 | 1.890, 1.890 1.889, 1.889 | 0.329 | C2 | Sing. | 2.654 2.654 | 1.902, 1.902 1.902, 1.902 | 0.587 |
| MECP | 2.461 2.460 | 1.923, 1.923 1.921, 1.921 | 0.432 | MECP | 2.461 2.461 | 1.921, 1.921 1.920, 1.920 | 0.616 | ||
| Trip. | 2.127 2.127 | 1.994, 1.995 1.995, 1.995 | 0.000 | Trip. | 2.106 2.106 | 2.006, 2.006 2.003, 2.003 | 0.000 | ||
| C3 | Sing. | - | 1.851, 1.850 1.850, 1.850 | - | C4 | Sing. | 2.573 2.566 | 1.921, 1.921 1.915, 1.915 | 0.448 |
| MECP | 2.459 2.459 | 1.918, 1.918 1.917, 1.917 | - | MECP | 2.439 2.438 | 1.937, 1.937 1.940, 1.940 | 0.642 | ||
| Trip. | 2.164 2.164 | 1.979, 1.979 1.979, 1.979 | - | Trip. | 2.130 2.130 | 1.997, 1.997 2.001, 2.001 | 0.000 | ||
| C5 | Sing. | 2.636 2.635 | 1.904, 1.904 1.902, 1.902 | 0.479 | C6 | Sing. | 2.466 2.912 | 1.802, 1.803 1.893, 1.894 | 0.000 |
| MECP | 2.403 2.403 | 1.929, 1.929 1.931, 1.931 | 0.565 | MECP | 2.236 2.240 | 1.817, 1.817 1.907, 1.907 | 0.474 | ||
| Trip. | 2.140 2.140 | 1.978, 1.978 1.983, 1.983 | 0.000 | Trip. | 2.131 2.133 | 1.809, 1.810 1.895, 1.896 | 0.193 |
| Conf. | S * | fosc | T ** | fosc | Conf. | S | fosc | T | fosc |
|---|---|---|---|---|---|---|---|---|---|
| C1 | S6 (413) | 0.0010 | T4 (545) | 0.0001 | C2 | S4 (617) | 0.0099 | T8 (541) | 0.0129 |
| S10 (354) | 0.0002 | T8 (448) | 0.0001 | S8 (440) | 0.0017 | T14 (502) | 0.0007 | ||
| S14 (319) | 0.1002 | T11 (401) | 0.0009 | S11 (417) | 0.0122 | T15 (495) | 0.0001 | ||
| C4 | S4 (549) | 0.0081 | T4 (535) | 0.0001 | C5 | S5 (554) | 0.0071 | T6 (542) | 0.0001 |
| S6 (520) | 0.0016 | T9 (431) | 0.0001 | S7 (510) | 0.0099 | T12 (436) | 0.0054 | ||
| S10 (436) | 0.0016 | T13 (400) | 0.0046 | S12 (403) | 0.0037 | T14 (429) | 0.0005 | ||
| S14 (415) | 0.0036 | ||||||||
| C6 | S1 (920) | 0.0159 | T4 (700) | 0.0029 | |||||
| S2 (852) | 0.0020 | T8 (608) | 0.0340 | ||||||
| S5 (654) | 0.0026 | T9 (573) | 0.0012 | ||||||
| S7 (586) | 0.0055 | T15 (490) | 0.0115 | ||||||
| S10 (530) | 0.0081 | T16 (490) | 0.0071 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farcaș, A.-A.; Bende, A. Improving the Light-Induced Spin Transition Efficiency in Ni(II)-Based Macrocyclic-Ligand Complexes. Molecules 2019, 24, 4249. https://doi.org/10.3390/molecules24234249
Farcaș A-A, Bende A. Improving the Light-Induced Spin Transition Efficiency in Ni(II)-Based Macrocyclic-Ligand Complexes. Molecules. 2019; 24(23):4249. https://doi.org/10.3390/molecules24234249
Chicago/Turabian StyleFarcaș, Alex-Adrian, and Attila Bende. 2019. "Improving the Light-Induced Spin Transition Efficiency in Ni(II)-Based Macrocyclic-Ligand Complexes" Molecules 24, no. 23: 4249. https://doi.org/10.3390/molecules24234249
APA StyleFarcaș, A.-A., & Bende, A. (2019). Improving the Light-Induced Spin Transition Efficiency in Ni(II)-Based Macrocyclic-Ligand Complexes. Molecules, 24(23), 4249. https://doi.org/10.3390/molecules24234249