
A Survey of Molecular Imaging of Opioid Receptors

 , , , and
, , , and
Abstract
1. Introduction
2. Radiotracers for the PET Imaging of ORs
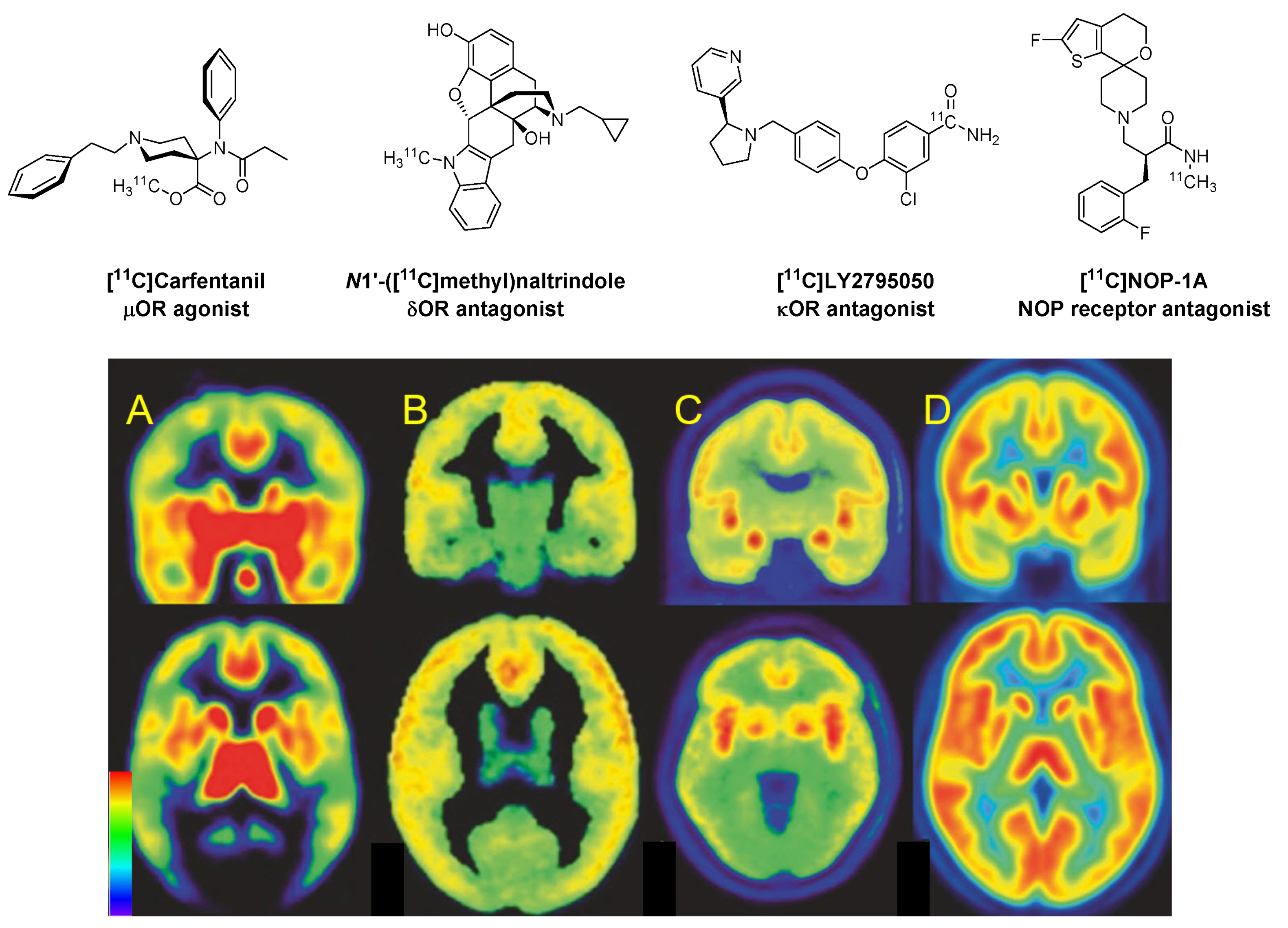
2.1. µOR Ligands and Non-Selective Ligands
2.2. Delta Ligands
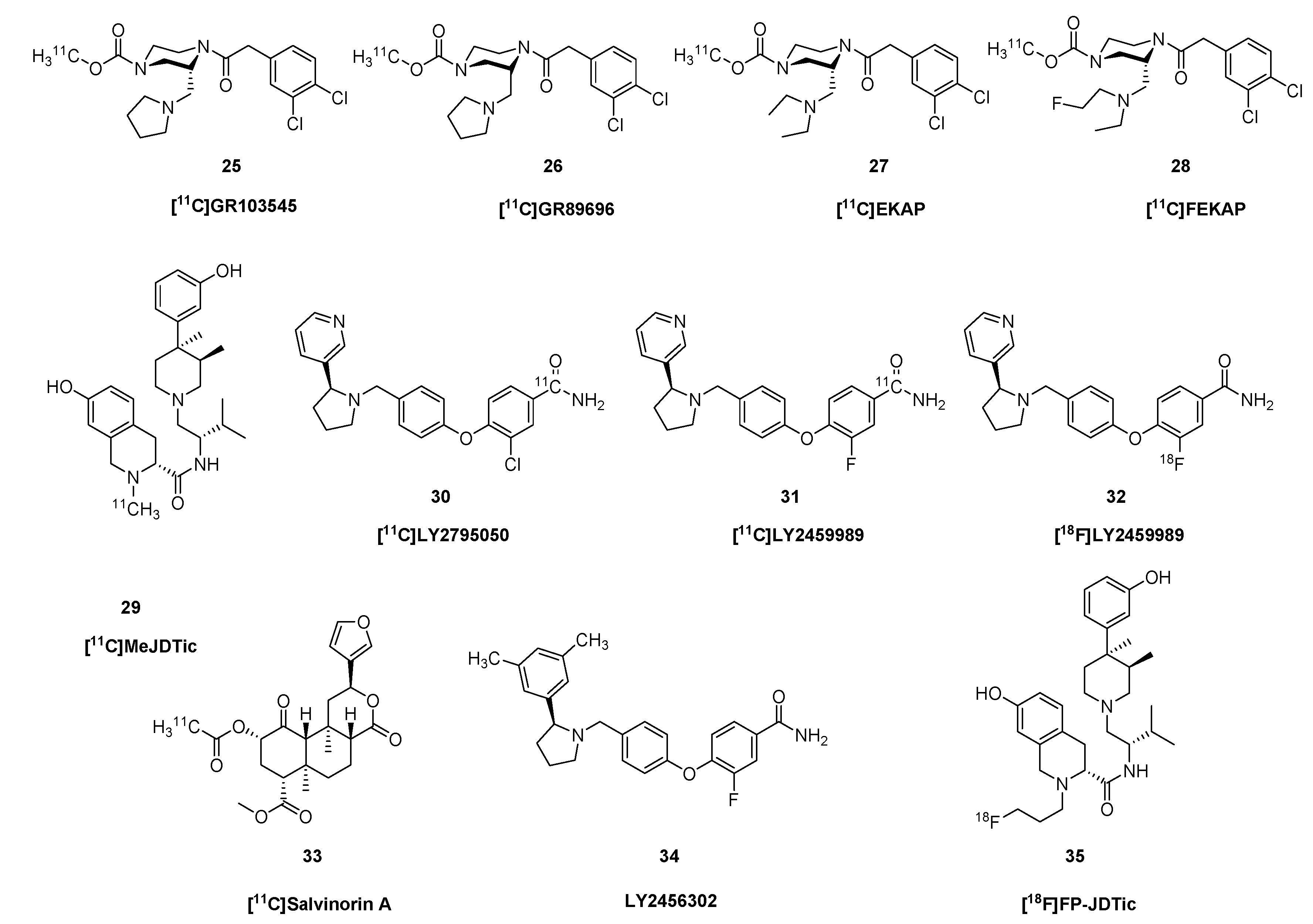
2.3. Kappa Ligands
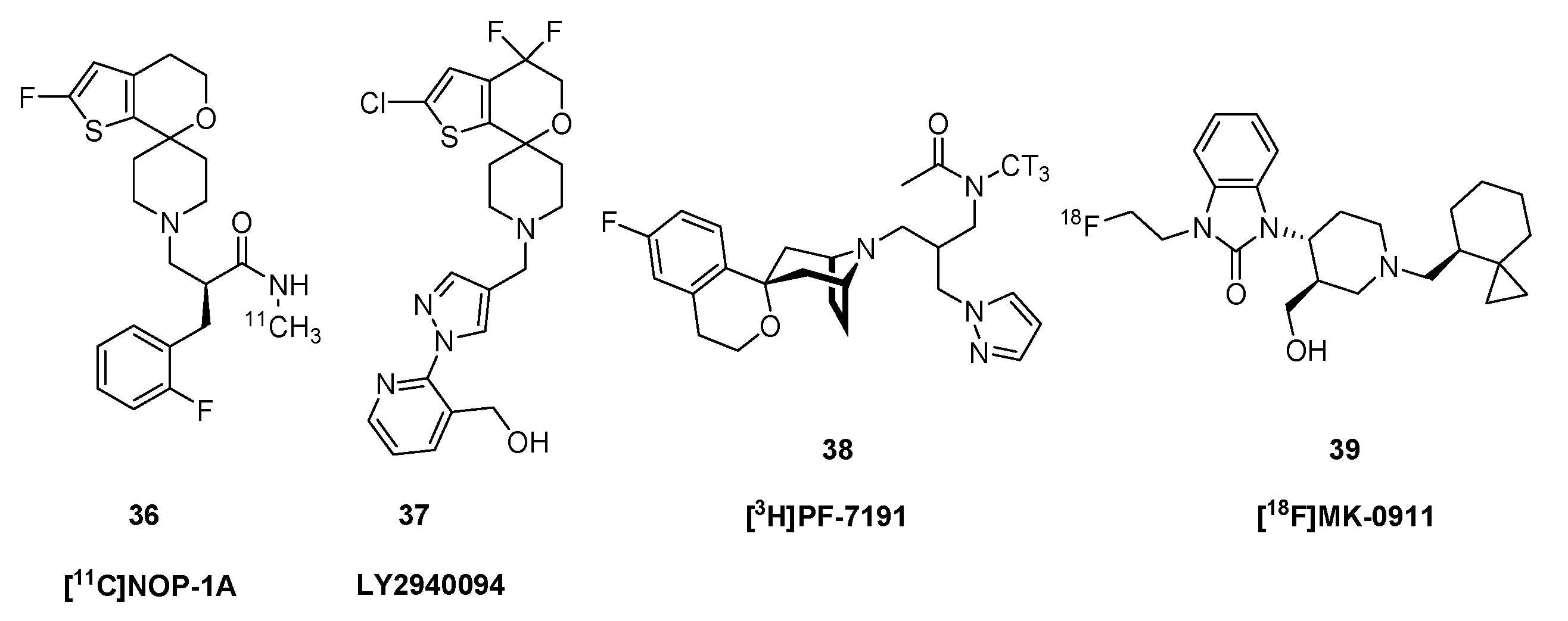
2.4. Nociceptin and Opioid-like 1 Receptors (ORL1)
3. Clinical Studies
3.1. Age and Gender
3.2. Epilepsy
3.3. Movement Disorders
3.4. Pain
3.5. Personality, Drug Dependence, and Psychiatric Disorders
4. Conclusions and Outlook
Funding
Conflicts of Interest
Abbreviations
| Compound | Name, Synonyms |
| 3-O-Ac-[18F]FcyF (10) | 3-O-[18F]acetylcyclofoxy, 3-O-Acetyl-6-deoxy-6-beta-[18F]fluoronaltrexone |
| β-Endorphin | Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val- Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-Tyr-Lys-Lys-Gly-Glu CAS RN: [61214-51-5] |
| BEMP | 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine CAS RN: [98015-45-3] |
| [11C]BPN (14) | [11C]buprenorphine, [6-O-methyl-11C]buprenorphine |
| Caf | carfentanil, 1-(2-phenylethyl)-4-[(1-oxopropyl)phenylamino]-4-piperidine carboxylic acid methyl ester, R-31,833, 4-carboxymethyl-fentanyl, CAS RN: [59708-52-09] |
| [11C]Caf (8) | [11C]carfentanil |
| [3H]DAMGO | [3,5-3H]Tyr-D-Ala-Gly-(NMe)Phe-Gly-ol |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene, CAS RN: [6674-22-2] |
| DIPEA | ethyldiisopropylamine, N,N-diisopropylamine, Hünig’s base |
| DMF | N,N-dimethylformamide |
| [11C]DPN (15) | [11C]diprenorphine, [6-O-methyl-11C]diprenorphine |
| dppf | 1,1’-ferrocenediyl-bis(diphenylphosphine), CAS RN: [121450-46-8] |
| Dynorphin A [1-17] | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-T rp-Asp-Asn-Gln CAS RN: [80448-90-4] |
| Dynorphin A [1-8] | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile, CAS RN: [75790-53-3] |
| Dynorpin B [1-13] | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr, CAS RN: [83335-41-5] |
| [11C]EKAP (27) | (R)-(methyl-11C) 4-(2-(3,4-dichlorophenyl)acetyl)-3-((diethylamino)methyl) piperazine-1-carboxylate |
| cyF | cyclofoxy, 6-deoxy-6β-[18F]fluoro-naltrexone, N-cyclopropylmethyl-6-deoxy-6β-fluoro-noroxymorphone, 17-cyclopropylmethyl-4,5α-epoxy-6β-fluoro-morphinan-3,14-diol CAS RN: [103223-57-0] |
| [18F]FcyF (11) | [18F]cyclofoxy, 6-Deoxy-6-β-[18F]fluoro-naltrexone, N-cyclopropylmethyl-6-deoxy-6β-[18F]fluoro-noroxymorphone, CAS RN: [103223-58-1] |
| FDOPA | 6-fluoro-L-dopa, 2-Fluoro-5-hydroxy-L-tyrosine, CAS RN: [144334-59-8] |
| [18F]FE-DPN (16) | 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine |
| [11C]FEKAP (28) | (R)-(methyl-11C)4-(2-(3,4-dichlorophenyl)acetyl)-3-((ethyl(2-fluoroethyl)amino) methyl)piperazine-1-carboxylate |
| [18F]FE-BPN (19) | 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-buprenorphine |
| [18F]FP-norBPN (21) | N17-(3-[18F]fluoropropyl)-nor-buprenorphine |
| [18F]FP-norDPN (20) | N17-(3-[18F]fluoropropyl)-nor-diprenorphine |
| [18F]FE-NTI (23) | [18F]fluoroethyl-naltrindole, N1’-(2-[18F]Fluoroethyl)-naltrindole, [18F]BU97001 |
| [18F]FE-PEO (17) | 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-phenethyl-orvinol |
| [18F]FE-Tos | [18F]fluoroethyl tosylate |
| Foxy | 6-deoxy-6β-fluoro-oxymorphone, «fluorooxymorphone », 4,5α-Epoxy -6β-fluoro-17-methyl-morphinan-3,14-diol, CAS RN: [92593-44-7] |
| [3H]Foxy | [1,2-3H]-4,5α-epoxy-6β-fluoro-17-methyl-morphinan-3,14-diol, CAS RN: [96917-45-2] |
| [11C]GR103545 (25) | [(3,4-dichlorophenyl)acetyl]-(3R)-(1-pyrrolidinylmethyl)-1-piperazine carboxylic acid methyl-11C ester |
| [11C]GR89696 (26) | 4-[(3,4-dichlorophenyl)acetyl]-3-(R,S)-(1-pyrrolidinylmethyl)-1-piperazine carboxylic acid methyl-11C ester |
| HD | Huntington’s disease |
| [11C]LAAM (13) | N-(methyl-11C)-L-α-acetoxymethadol, N-(methyl-11C)-levo-alpha-acetyl methadol, N-(methyl-11C)-levomethadyl acetate |
| Leu5-enkephalin | H-Tyr-Gly-Gly-Phe-Leu-OH, CAS RN: [58569-55-4] |
| LY2459989 | 3-fluoro-4-[4-[[(2S)-2-(3-pyridyl)pyrrolidin-1-yl]methyl]phenoxy] benzamide |
| LY2795050 | 3-chloro-4-[4-[[(2S)-2-(3-pyridyl)pyrrolidin-1-yl]methyl]phenoxy] benzamide, CAS RN: [1346133-08-1] |
| LY2456302 (34) | (S)-4-(4-((2-(3,5-dimethylphenyl)pyrrolidin-1-yl)methyl)phenoxy)- 3-fluorobenzamide, CERC-501, CAS RN: [1174130-61-0] |
| LY2940094 | ([2-[4-[(2-chloro-4,4-difluoro-spiro[5H-thieno[2,3-c]pyran-7,4′-piperidine]- 1′-yl)methyl]-3-methyl-pyrazol-1-yl]-3-pyridyl]methanol), BTRX-246040, CAS RN: [1307245-86-0] |
| Met5-enkephalin | H-Tyr-Gly-Gly-Phe-Met-OH |
| [11C]MeJDTic (29) | (3S)-7-hydroxy-N-((1S)-1-[[3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl- 1-piperidinyl]methyl]-2-methylpropyl)-2-[11C]methyl-1,2,3,4-tetrahydro- 3-isoquinolinecarboxamide |
| [11C]MeNTI (22) | [11C]methyl-naltrindole, N1’-[11C]methyl-naltrindole |
| [18F]MK-0911 (39) | 1-(2-[18F]fluoroethyl)-3-[(3R,4R)-3-(hydroxymethyl)-1-[[(8S)-spiro[2.5]octan-8-yl] methyl]piperidin-4-yl]benzimidazol-2-one |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, CAS RN: [28289-54-5] |
| Oxycodone | 14-hydroxy-dihydrocodeinone, CAS RN: [76-42-6] |
| Naloxone | N17-allyl-14-hydroxy-dihydromorphinone, N17-Allyl-noroxymorphone, CAS RN: [465-65-6] |
| Naltrexone | N17-cyclopropylmethyl-14-hydroxy-dihydromorphinone, N17-cyclopropylmethyl-noroxymorphone, NTX CAS RN: [16590-41-3] |
| Nociceptine | Phe-Gly-Gly-Phe-Thr-Gly-Ala-Arg-Lys-Ser-Ala-Arg-Lys-Leu-Ala-Asn-Gln |
| N-substituted- [11C]quinolinimide (24) | 6-(2-{2-[4-(4-fluorobutyl)-benzenesulfonyl]-1,2,3,4-tetrahydro-isoquinolin-1-yl} -ethyl)-2-[11C]methyl-pyrrolo[3,4-b]pyridine-5,7-dione |
| NTI | naltrexone-indole, naltrindole, CAS RN: [111555-53-4] |
| [11C]NOP-1A (36) | (2S)-2-[(2-fluorophenyl)methyl]-3-(2-fluorospiro[4,5-dihydro-thieno[2,3-c]pyran- 7,4'-piperidine]-1'-yl)-N-[11C]methyl-propanamide |
| OR | opioid receptor |
| [11C]PEO (18) | [11C]phenethyl-orvinol, [6-O-methyl-11C]phenethyl-orvinol |
| PET | positron emission tomography |
| Pd2dba3 | Pd2(dba)3, tris(dibenzylideneacetone)dipalladium, CAS RN: [51364-51-3] |
| [3H]PL017 | [3,5-3H]Tyr-Pro-(NMe)Phe-d-Pro-NH2, 3-N-Me-Phe-[3H]morphiceptin, [3H] [MePhe3,D-Pro4]morphiceptin |
| Salvinorin A | methyl (2S,4aR,6aR,7R,9S,10aS,10bR)-9-acetyloxy-2-(furan-3-yl)-6a,10b-dimethyl- 4,10-dioxo-2,4a,5,6,7,8,9,10a-octahydro-1H-benzo[f]isochromene-7-carboxylate, CAS RN: [83729-01-5] |
| TBDMS | tert-butyldimethylsilyl protecting group |
| TDBPN | 3-O-trityl-6-O-desmethyl-buprenorphine, CAS RN: [157891-93-5] |
| TDDPN | 3-O-trityl-6-O-desmethyl-diprenorphine, CAS RN: [157891-92-4], “Luthra-precursor”, TDDPN was the first product of the company ABX advanced biochemical compounds Biomedizinische Forschungsreagenzien GmbH, Radeberg in 1997 |
| TDPEO | 3-O-trityl-6-O-desmethyl-phenethyl-orvionol, CAS RN: [1187551-69-4] |
| U-50488 | 3,4-dichloro-N-methyl-N-[(1R,2R)-2-(1-pyrrolidinyl)cyclohexyl]- benzene acetamide, NIH10533, CAS RN: [67198-13-4] |
References
- Gulland, J.M.; Robinson, R. Constitution of codeine and thebaine. Mem. Proc. Manchester Lit. Phil. Soc. 1925, 69-86, 79–86. [Google Scholar]
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1952, 74, 1109–1110. [Google Scholar]
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1956, 78, 1380–1393. [Google Scholar] [CrossRef]
- Bentley, K.W.; Cardwell, H.M.E. The Morphine-Thebaine group of alkaloids. Part V. The absolute stereochemistry of the morphine, benzylisoquinoline, aporphine, and tetrahydroberberine alkaloids. J. Chem. Soc. 1955, 3252–3260. [Google Scholar] [CrossRef]
- Rice, K.C. Synthetic opium alkaloids and derivatives. A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners. J. Org. Chem. 1980, 45, 3135–3137. [Google Scholar] [CrossRef]
- Lever, J.R. PET and SPECT imaging of the opioid system: Receptors, radioligands and avenues for drug discovery and development. Curr. Pharm. Des. 2007, 13, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, G.; Willoch, F. Imaging of opioid receptors in the central nervous system. Brain 2008, 131, 1171–1196. [Google Scholar]
- Dannals, R.F. Positron emission tomography radioligands for the opioid system. J. Label. Compd. Radiopharm. 2013, 56, 187–195. [Google Scholar] [CrossRef]
- Pert, C.B.; Snyder, S.H. Properties of opiate-receptor binding in rat brain. Proc. Natl. Acad. Sci. USA 1973, 70, 2243–2247. [Google Scholar] [CrossRef]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–579. [Google Scholar] [CrossRef]
- Di Giulio, A.M.; Majane, E.M.; Yang, H.Y. On the distribution of [met5]- and [leu5]-enkephalins in the brain of the rat, guinea-pig and calf. Br. J. Pharmacol. 1979, 66, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Kosterlitz, H.W.; Smith, T.W. The distribution of methionine-enkephalin and leucine-enkephalin in the brain and peripheral tissues. Br. J. Pharmacol. 1977, 61, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Chung, D. Isolation and Structure of an Untriakontapeptide with Opiate Activity from Camel Pituitary Glands. Proc. Natl. Acad. Sci. USA 1976, 73, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Inoue, A.; Kita, T.; Numa, S.; Chang, A.C.; Cohen, S.N.; Nunberg, J.; Schimke, T.R. Construction of bacterial plasmids that contain the nucleotide sequence for bovine corticotropin-beta-lipotropin precursor. Proc. Natl. Acad. Sci. USA 1978, 75, 6021–6025. [Google Scholar] [CrossRef]
- Noda, M.; Furutani, Y.; Takahashi, H.; Toyosato, M.; Hirose, T.; Inayama, S.; Nakanishi, S.; Numa, S. Cloning and sequence analysis of cDNA for bovine adrenal preproenkephalin. Nature 1982, 295, 202–206. [Google Scholar] [CrossRef]
- Horikawa, S.; Takai, T.; Toyosato, M.; Takahashi, H.; Noda, M.; Kakidani, H.; Kubo, T.; Hirose, T.; Inayama, S.; Hayashida, H.; et al. Isolation and structural organization of the human preproenkephalin B gene. Nature 1983, 306, 611–614. [Google Scholar] [CrossRef]
- Kakidani, H.; Furutani, Y.; Takahashi, H.; Noda, M.; Morimoto, Y.; Hirose, T.; Asai, M.; Inayama, S.; Nakanishi, S.; Numa, S. Cloning and sequence analysis of cDNA for porcine β-neo-endorphin/dynorphin precursor. Nature 1982, 298, 245–249. [Google Scholar] [CrossRef]
- Peciña, M.; Karp, J.F.; Mathew, S.; Todtenkopf, M.S.; Ehrich, E.W.; Zubieta, J.-K. Endogenous opioid system dysregulation in depression: Implications for new therapeutic approaches. Mol. Psychiatry 2019, 24, 576–587. [Google Scholar] [CrossRef]
- Su, T.P. Evidence for sigma opioid receptor: Binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J. Pharmacol. Exp. Ther. 1982, 223, 284–290. [Google Scholar]
- Eriksson, O.; Antoni, G. [11C]Carfentanil binds preferentially to mu-opioid receptor subtype 1 compared to subtype 2. Mol. Imaging 2015, 14, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Ma, Y.; Bell, A.; Esch, T.; Guarna, M.; Bilfinger, T.V.; Bianchi, E.; Stefano, G.B. Presence of morphine in rat amygdala: Evidence for the mu3 opiate receptor subtype via nitric oxide release in limbic structures. Med. Sci. Monit. 2004, 10, 433–439. [Google Scholar]
- Witkin, J.M.; Rorick-Kehn, L.M.; Benvenga, M.J.; Adams, B.L.; Gleason, S.D.; Knitowski, K.M.; Li, X.; Chaney, S.; Falcone, J.F.; Smith, J.W.; et al. Preclinical findings predicting efficacy and side-effect profile of LY2940094, an antagonist of nociceptin receptors. Pharma Res. Per. 2016, 4, e00275. [Google Scholar] [CrossRef] [PubMed]
- Janson, W.; Stein, C. Peripheral opioid analgesia. Curr. Pharm. Biotechnol. 2003, 4, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Fox, C.A.; Akil, H.; Watson, S.J. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995, 18, 22–29. [Google Scholar] [CrossRef]
- Atweh, S.F.; Kuhar, M.J. Distribution and physiological significance of opioid receptors in the brain. Br. Med. Bull. 1983, 39, 47–52. [Google Scholar] [CrossRef][Green Version]
- Benyhe, S.; Zádor, F.; Ötvös, F. Biochemistry of opioid (morphine) receptors: Binding, structure and molecular modelling. Acta Biol. Szeged 2015, 59 (Suppl. 1), 17–37. [Google Scholar]
- Meng, F.; Xie, G.X.; Thompson, R.C.; Mansour, A.; Goldstein, A.; Watson, S.J.; Akil, H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 9954–9958. [Google Scholar] [CrossRef]
- Simonin, F.; Gavériaux-Ruff, C.; Befort, K.; Matthes, H.; Lannes, B.; Micheletti, G.; Mattéi, M.G.; Charron, G.; Bloch, B.; Kieffer, B. kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc. Natl. Acad. Sci. USA 1995, 92, 7006–7010. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, C.; Xue, J.-C.; Kunapuli, S.; DeRiel, J.K.; Liu-Chen, L.-Y. Cloning of a human kappa opioid receptor from the brain. Life Sci. 1995, 56, 201–207. [Google Scholar] [CrossRef]
- Witkin, J.M.; Statnick, M.A.; Rorick-Kehn, L.M.; Pintar, J.E.; Ansonoff, M.; Chen, Y.; Tucker, R.C.; Ciccocioppo, R. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol. Ther. 2014, 141, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Pert, C.B.; Kuhar, M.J.; Snyder, S.H. Autoradiograhic localization of the opiate receptor in rat brain. Life Sci. 1975, 16, 1849–1853. [Google Scholar] [CrossRef]
- Pert, C.B.; Snyder, S.H. Identification of opiate receptor binding in intact animals. Life Sci. 1975, 16, 1623–1634. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Y.; Hunkele, A.; Provasi, D.; Pasternak, G.W.; Filizola, M. Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput. Biol. 2019, 15, e1006689. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, S.G.; Lee, P.H.; Pugh, W.W.; Hong, J.S.; Chang, K.J. Characterization of the binding of a morphine (mu) receptor-specific ligand: Tyr-Pro-NMePhe-D-Pro-NH2, [3H]-PL17. Mol. Pharmacol. 1987, 31, 326–333. [Google Scholar]
- Pert, C.B.; Danks, J.A.; Channing, M.A.; Eckelman, W.C.; Larson, S.M.; Bennett, J.M.; Burke, T.R.J.; Rice, K.C. 3-[18F]Acetylcyclofoxy: A useful probe for the visualization of opiate receptors in living animals. FEBS Lett. 1984, 177, 281–286. [Google Scholar] [CrossRef]
- Larson, S.M.; Di Chiro, G. Comparative anatomo-functional imaging of two neuroreceptors and glucose metabolism: A PET study performed in the living baboon. J. Comput. Assist. Tomogr. 1985, 9, 676–681. [Google Scholar] [CrossRef]
- Rothman, R.B.; Bykov, V.; Reid, A.; De Costa, B.R.; Newman, A.H.; Jacobson, A.E.; Rice, K.C. A brief study of the selectivity of norbinaltorphimine, (−)-cyclofoxy, and (+)-cyclofoxy among opioid receptor subtypes in vitro. Neuropeptides 1988, 12, 181–187. [Google Scholar] [CrossRef]
- Ostrowski, N.L.; Burke, T.R.J.; Rice, K.C.; Pert, A.; Pert, C.B. The pattern of [3H]cyclofoxy retention in rat brain after in vivo injection corresponds to the in vitro opiate receptor distribution. Brain Res. 1987, 402, 275–286. [Google Scholar] [CrossRef]
- Rothman, R.; McLean, S.; Bykov, V.; Lessor, R.A.; Jacobson, A.E.; Rice, K.C.; Holaday, J.W. Chronic morphine upregulates a mu-opiate binding site labeled by [3H]cycloFOXY: A novel opiate antagonist suitable for positron emission tomography. Eur. J. Pharmacol. 1987, 142, 73–81. [Google Scholar] [CrossRef]
- Kawai, R.; Carson, R.E.; Dunn, B.; Newman, A.H.; Rice, K.C.; Blasberg, R.G. Regional brain measurement of Bmax and KD with the opiate antagonist cyclofoxy: Equilibrium studies in the conscious rat. J. Cereb. Blood Flow. Metab. 1991, 11, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Hartvig, P.; Bergström, K.; Lindberg, B.; Lundberg, P.O.; Lundqvist, H.; Långström, B.; Svärd, H.; Rane, A. Kinetics of 11C-labeled opiates in the brain of rhesus monkeys. J. Pharmacol. Exp. Ther. 1984, 230, 250–255. [Google Scholar] [PubMed]
- Hartvig, P.; Eckernäs, S.A.; Lindberg, B.S.; Lundqvist, H.; Antoni, G.; Rimland, A.; Långström, B. Regional distribution of the opioid receptor agonist N-(methyl-11C)pethidine in the brain of the rhesus monkey studied with positron emission tomography. Pharmacol. Toxicol. 1990, 66, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.K.; Fan, J.; Tu, Z.; Zerkel, P.; Mach, R.H.; Kharasch, E.D. Automated radiochemical synthesis and biodistribution of [¹¹C]l-α-acetylmethadol ([¹¹C]LAAM). Appl. Radiat. Isot. 2014, 91, 135–140. [Google Scholar] [CrossRef][Green Version]
- Weerts, E.M.; Kim, Y.K.; Wand, G.S.; Dannals, R.F.; Lee, J.S.; Frost, J.J.; McCaul, M.E. Differences in delta- and mu-opioid receptor blockade measured by positron emission tomography in naltrexone-treated recently abstinent alcohol-dependent subjects. Neuropsychopharmacology 2008, 33, 653–665. [Google Scholar] [CrossRef]
- Johansson, J.; Hirvonen, J.; Lovró, Z.; Ekblad, L.; Kaasinen, V.; Rajasilta, O.; Helin, S.; Tuisku, J.; Sirén, S.; Pennanen, M.; et al. Intranasal naloxone rapidly occupies brain mu-opioid receptors in human subjects. Neuropsychopharmacology 2019, 44, 1667–1673. [Google Scholar] [CrossRef]
- Lewis, J.W. Buprenorphine. Drug Alcohol Depen. 1985, 14, 363–372. [Google Scholar] [CrossRef]
- Lewis, J.W.; Husbands, S.M. The orvinols and related opioids—High affinity ligands with diverse efficacy profiles. Curr. Pharm. Des. 2004, 10, 717–732. [Google Scholar] [CrossRef]
- Husbands, S.M. Buprenorphine and related orvinols. In Research and Development of Opioid-Related Ligands; ACS Symposium Series; Ko, M.-C., Husbands, S.M., Eds.; American Chemical Society: Washington, DC, USA, 2013; Volume 1131, pp. 127–144. [Google Scholar]
- Cami-Kobeci, G.; Polgar, W.E.; Khroyan, T.V.; Toll, L.; Husbands, S.M. Structural determinants of opioid and NOP receptor activity in aerivatives of buprenorphine. J. Med. Chem. 2011, 54, 6531–6537. [Google Scholar] [CrossRef]
- Borsodi, A.; Bruchas, M.; Caló, G.; Chavkin, C.; Christie, M.J.; Civelli, O.; Connor, M.; Cox, B.M.; Devi, L.A.; Evans, C.; et al. Opioid receptors (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. In IUPHAR/BPS Guide to Pharmacology CITE, 2019(4); 2019. [Google Scholar] [CrossRef]
- Stefanucci, A.; Lei, W.; Pieretti, S.; Novellino, E.; Dimmito, M.P.; Marzoli, F.; Streicher, J.M.; Mollica, A. On resin click-chemistry-mediated synthesis of novel enkephalin analogues with potent anti-nociceptive activity. Sci. Rep. 2019, 9, 5771. [Google Scholar] [CrossRef]
- Corbett, A.D.; Paterson, S.J.; Kosterlitz, H.W. Selectivity of ligands for opioid receptors. In Handbook of Experimental Pharmacology Opioids I; Herz, A., Ed.; Springer-Verlag: New York, NY, USA, 1993; Volume 104/1, pp. 645–679. [Google Scholar]
- Zhang, S.; Tong, Y.; Tian, M.; Dehaven, R.N.; Cortesburgos, L.; Mansson, E.; Simonin, F.; Kieffer, B.; Yu, L. Dynorphin A as a potential endogenous ligand for four members of the opioid receptor gene family. J. Pharmacol. Exp. Ther. 1998, 286, 136–141. [Google Scholar] [PubMed]
- Valenzano, K.J.; Miller, W.; Chen, Z.; Shan, S.; Crumley, G.; Victory, S.F.; Davies, E.; Huang, J.-C.; Allie, N.; Nolan, S.J.; et al. DiPOA ([8-(3,3-Diphenyl-propyl)-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl]-acetic acid), a novel, systemically available, and peripherally restricted mu-opioid agonist with antihyperalgesic activity: I. In vitro pharmacological characterization and pharmacokinetic properties. J. Pharmacol. Exp. Ther. 2004, 310, 783–792. [Google Scholar] [PubMed]
- Miyazaki, T.; Choi, I.Y.; Rubas, W.; Anand, N.K.; Ali, C.; Evans, J.; Gursahani, H.; Hennessy, M.; Kim, G.; McWeeney, D.; et al. NKTR-181: A novel mu-opioid analgesic with inherently low abuse potential. J. Pharmacol. Exp. Ther. 2017, 363, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.Q.; Nabulsi, N.; Kim, S.J.; Tomasi, G.; Lin, S.F.; Mitch, C.; Quimby, S.; Barth, V.; Rash, K.; Masters, J.; et al. Synthesis and evaluation of 11C-LY2795050 as a kappa-opioid receptor antagonist radiotracer for PET imaging. J. Nucl. Med. 2013, 54, 455–463. [Google Scholar] [CrossRef]
- Henriksen, G.; Platzer, S.; Marton, J.; Hauser, A.; Berthele, A.; Schwaiger, M.; Marinelli, L.; Lavecchia, A.; Novellino, E.; Wester, H.-J. Syntheses, biological evaluation, and molecular modeling of 18F-labeled 4-anilidopiperidines as μ-opioid receptor imaging agents. J. Med. Chem. 2005, 48, 7720–7732. [Google Scholar] [CrossRef]
- Frost, J.J.; Wagner, H.N.J.; Dannals, R.F.; Ravert, H.T.; Links, J.M.; Wilson, A.A.; Burns, H.D.; Wong, D.F.; McPherson, R.W.; Rosenbaum, A.E.; et al. Imaging opiate receptors in the human brain by positron tomography. J. Comput. Assist. Tomogr. 1985, 9, 231–236. [Google Scholar] [CrossRef]
- Cometta-Morini, C.; Maguire, P.A.; Loew, G.H. Molecular determinants of mu receptor recognition for the fentanyl class of compounds. Mol. Pharmacol. 1992, 41, 185–196. [Google Scholar]
- Raynor, K.; Kong, H.; Chen, Y.; Yasuda, K.; Yu, L.; Bell, G.I.; Reisine, T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol. Pharmacol. 1994, 45, 330–334. [Google Scholar]
- Marton, J.; Schoultz, B.W.; Hjørnevik, T.; Drzezga, A.; Yousefi, B.H.; Wester, H.-J.; Willoch, F.; Henriksen, G. Synthesis and evaluation of a full-agonist orvinol for PET-Imaging of opioid receptors: [11C]PEO. J. Med. Chem. 2009, 52, 5586–5589. [Google Scholar] [CrossRef]
- Schoultz, B.W.; Hjørnevik, T.; Reed, B.J.; Marton, J.; Coello, C.S.; Willoch, F.; Henriksen, G. Synthesis and evaluation of three structurally related 18F-labeled orvinols of different intrinsic activities: 6-O-[18F]Fluoroethyl-diprenorphine ([18F]FDPN), 6-O-[18F]fluoroethyl-buprenorphine ([18F]FBPN), and 6-O-[18F]fluoroethyl-phenethyl-orvinol ([18F]FPEO). J. Med. Chem. 2014, 57, 5464–5469. [Google Scholar]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Design of peptidomimetic delta opioid receptor antagonists using the message-address concept. J. Med. Chem. 1990, 33, 1714–1720. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, B.W.; Hjornevik, T.; Willoch, F.; Marton, J.; Noda, A.; Murakami, Y.; Miyoshi, S.; Nishimura, S.; Arstad, E.; Drzezga, A.; et al. Evaluation of the kappa-opioid receptor-selective tracer [11C]GR103545 in awake rhesus macaques. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.-Q.; Kim, S.J.; Holden, D.; Lin, S.-F.; Need, A.; Rash, K.; Barth, V.; Mitch, C.; Navarro, A.; Kapinos, M.; et al. An improved antagonist radiotracer for the kappa-opioid receptor: Synthesis and characterization of 11C-LY2459989. J. Nucl. Med. 2014, 55, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, M.-Q.; Naganawa, M.; Gao, H.; Pracitto, R.; Shirali, A.; Lin, S.-F.; Teng, J.-K.; Ropchan, J.; Huang, Y. Novel kappa opioid receptor agonist as improved PET radiotracer: Development and in vivo evaluation. Mol. Pharm. 2019, 16, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, M.-Q.; Naganawa, M.; Kim, S.J.; Gao, H.; Kapinos, M.; Labaree, D.; Huang, Y. Development and in vivo evaluation of a kappa-opioid receptor agonist as a PET radiotracer with superior imaging characteristics. J. Nucl. Med. 2019, 60, 1023–1030. [Google Scholar] [CrossRef]
- Poisnel, G.; Oueslati, F.; Dhilly, M.; Delamare, J.; Perrio, C.; Debruyne, D.; Barré, L. [11C]-MeJDTic: A novel radioligand for kappa-opioid receptor positron emission tomography imaging. Nucl. Med. Biol. 2008, 35, 561–569. [Google Scholar] [CrossRef]
- Harding, W.W.; Tidgewell, K.; Byrd, N.; Cobb, H.; Dersch, C.M.; Butelman, E.R.; Rothman, R.B.; Prisinzano, T.E. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J. Med. Chem. 2005, 48, 4765–4771. [Google Scholar] [CrossRef]
- Pike, V.W.; Rash, K.S.; Chen, Z.; Pedregal, C.; Statnick, M.A.; Kimura, Y.; Hong, J.S.; Zoghbi, S.S.; Fujita, M.; Toledo, M.A.; et al. Synthesis and evaluation of radioligands for imaging brain nociceptin/orphanin FQ peptide (NOP) receptors with positron emission tomography. J. Med. Chem. 2011, 54, 2687–2700. [Google Scholar] [CrossRef]
- Hostetler, E.D.; Sanabria-Bohórquez, S.; Eng, W.; Joshi, A.D.; Patel, S.; Gibson, R.E.; O’Malley, S.; Krause, S.M.; Ryan, C.; Riffel, K.; et al. Evaluation of [18F]MK-0911, a positron emission tomography (PET) tracer for opioid receptor-like 1 (ORL1), in rhesus monkey and human. NeuroImage 2013, 68, 1–10. [Google Scholar] [CrossRef]
- Luthra, S.K.; Pike, V.W.; Brady, F.; Horlock, P.L.; Prenant, C.; Crouzel, C. Preparation of [11C]buprenorphine—A potential radioligand for the study of the opiate receptor system in vivo. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1987, 38, 65–66. [Google Scholar] [CrossRef]
- Luthra, S.K.; Pike, V.W.; Brady, F. The preparation of carbon-11 labelled diprenorphine: A new radioligand for the study of the opiate receptor system in vivo. J. Chem. Soc. Chem. Commun. 1985, 20, 1423–1425. [Google Scholar] [CrossRef]
- Lever, J.R.; Mazza, S.M.; Dannals, R.F.; Ravert, H.T.; Wilson, A.A.; Wagner, H.N. Facile synthesis of [11C]buprenorphine for positron emission tomographic studies of opioid receptors. Int. J. Radiat. Appl. Instrum. Part A Appl. Radiat. Isot. 1990, 41, 745–752. [Google Scholar] [CrossRef]
- Luthra, S.K.; Brady, F.; Turton, D.R.; Brown, D.J.; Dowsett, K.; Waters, S.L.; Jones, A.K.P.; Matthews, R.W.; Crowder, J.C. Automated radiosyntheses of [6-O-methyl-11C]diprenorphine and [6-O-methyl-11C]buprenorphine from 3-O-trityl protected precursors. Appl. Radiat. Isot. 1994, 45, 857–873. [Google Scholar] [CrossRef]
- Burns, H.D.; Lever, J.R.; Dannals, R.F.; Frost, J.J.; Wilson, A.A.; Ravert, H.T.; Subramanian, B.; Zeyman, S.E.; Langstrom, B.; Wagner, H.N., Jr. Synthesis of ligands for imaging opiate receptors by positron emission tomography: Carbon-11 labelled diprenorphine. J. Label. Compd. Radiopharm. 1984, 22, 1167–1169. [Google Scholar]
- Lever, J.R.; Dannals, R.F.; Wilson, A.A.; Ravert, H.T.; Wagner, H.N. Synthesis of carbon-11 labeled diprenorphine: A radioligand for positron emission tomographic studies of opiate receptors. Tetrahedron Lett. 1987, 28, 4015–4018. [Google Scholar] [CrossRef]
- Fairclough, M.; Prenant, C.; Brown, G.; McMahon, A.; Lowe, J.; Jones, A. The automated radiosynthesis and purification of the opioid receptor antagonist, [6-O-methyl-11C]diprenorphine on the GE TRACERlab FXFE radiochemistry module. J. Label. Compd. Radiopharm. 2014, 57, 388–396. [Google Scholar] [CrossRef]
- Galynker, I.; Schlyer, D.J.; Dewey, S.L.; Fowler, J.S.; Logan, J.; Gatley, S.J.; MacGregor, R.R.; Ferrieri, R.A.; Holland, M.J.; Brodie, J.; et al. Opioid receptor imaging and displacement studies with [6-O-[11C]methyl]buprenorphine in baboon brain. Nucl. Med. Biol. 1996, 23, 325–331. [Google Scholar] [CrossRef]
- Zubieta, J.; Greenwald, M.K.; Lombardi, U.; Woods, J.H.; Kilbourn, M.R.; Jewett, D.M.; Koeppe, R.A.; Schuster, C.R.; Johanson, C.E. Buprenorphine-induced changes in mu-opioid receptor availability in male heroin-dependent volunteers: A preliminary study. Neuropsychopharmacology 2000, 23, 326–334. [Google Scholar] [CrossRef]
- Melichar, J.K.; Hume, S.P.; Williams, T.M.; Daglish, M.R.; Taylor, L.G.; Ahmad, R.; Malizia, A.L.; Brooks, D.J.; Myles, J.S.; Lingford-Hughes, A.; et al. Using [11C]diprenorphine to image opioid receptor occupancy by methadone in opioid addiction: Clinical and preclinical studies. J. Pharmacol. Exp. Ther. 2005, 312, 309–315. [Google Scholar] [CrossRef]
- Frost, J.J.; Mayberg, H.S.; Sadzot, B.; Dannals, R.F.; Lever, J.R.; Ravert, H.T.; Wilson, A.A.; Wagner, H.N.J.; Links, J.M. Comparison of [11C]diprenorphine and [11C]carfentanil binding to opiate receptors in humans by positron emission tomography. J. Cereb. Blood Flow. Metab. 1990, 10, 484–492. [Google Scholar] [CrossRef]
- Schadrack, J.; Willoch, F.; Platzer, S.; Bartenstein, P.; Mahal, B.; Dworzak, D.; Wester, H.J.; Zieglgänsberger, W.; Tölle, T.R. Opioid receptors in the human cerebellum: Evidence from [11C]diprenorphine PET, mRNA expression and autoradiography. Neuroreport 1999, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Wester, H.-J.; Willoch, F.; Tölle, T.R.; Munz, F.; Herz, M.; Øye, I.; Schadrack, J.; Schwaiger, M.; Bartenstein, P. 6-O-(2-[18F]Fluoroethyl-6-O-desmethyldiprenorphine ([18F]DPN): Synthesis, biologic evaluation, and comparison with [11C]DPN in humans. J. Nucl. Med. 2000, 41, 1279–1286. [Google Scholar] [PubMed]
- Schoultz, B.W.; Reed, B.J.; Marton, J.; Willoch, F.; Henriksen, G. A fully automated radiosynthesis of [18F]fluoroethyl-diprenorphine on a single module by use of SPE cartridges for preparation of high quality 2-[18F]fluoroethyl tosylate. Molecules 2013, 18, 7271–7278. [Google Scholar] [CrossRef] [PubMed]
- Baumgärtner, U.; Buchholz, H.G.; Bellosevich, A.; Magerl, W.; Siessmeier, T.; Rolke, R.; Höhnemann, S.; Piel, M.; Rösch, F.; Wester, H.J.; et al. High opiate receptor binding potential in the human lateral pain system. NeuroImage 2006, 30, 692–699. [Google Scholar] [CrossRef]
- Henriksen, G.; Spilker, M.E.; Sprenger, T.; Hauser, A.; Platzer, S.; Boecker, H.; Toelle, T.R.; Schwaiger, M.; Wester, H.J. Gender dependent rate of metabolism of the opioid receptor-PET ligand [18F]fluoroethyldiprenorphine. Nuklearmedizin 2006, 45, 197–200. [Google Scholar]
- Vučković, S.; Prostran, M.; Ivanović, M.; Došen-Mićović, L.; Todorović, Z.; Nešić, Z.; Stojanović, R.; Divac, N.; Miković, Ž. Fentanyl analogs: Stuctrure-activity-relationship study. Curr. Med. Chem. 2009, 16, 2468–2474. [Google Scholar] [CrossRef]
- Vardanyan, R.S.; Hruby, V.J. Fentanyl-related compounds and derivatives: Current status and future prospects for pharmaceutical applications. Future Med. Chem. 2014, 6, 385–412. [Google Scholar] [CrossRef]
- Van Daele, P.G.; De Bruyn, M.F.; Boey, J.M.; Sanczuk, S.; Agten, J.T.; Janssen, P.A. Synthetic analgesics: N-(1-[2-arylethyl]-4-substituted 4-piperidinyl) N-arylalkanamides. Arzneim. Forsch. Drug Res. 1976, 26, 1521–1531. [Google Scholar]
- Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wilson, A.A.; Burns, H.D.; Wagner, H.N.J. Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef]
- Jewett, D.M.; Kilbourn, M.R. In vivo evaluation of new carfentanil-based radioligands for the mu opiate receptor. Nucl. Med. Biol. 2004, 31, 321–325. [Google Scholar] [CrossRef]
- Shafer, S.L. Carfentanil: A weapon of mass destruction. Can. J. Anesth. 2019, 66, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.J.; Douglass, K.H.; Mayberg, H.S.; Dannals, R.F.; Links, J.M.; Wilson, A.A.; Ravert, H.T.; Crozier, W.C.; Wagner, H.N.J. Multicompartmental analysis of [11C]-carfentanil binding to opiate receptors in humans measured by positron emission tomography. J. Cereb. Blood Flow. Metab. 1989, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Hirvonen, J.; Aalto, S.; Hagelberg, N.; Maksimow, A.; Ingman, K.; Oikonen, V.; Virkkala, J.; Någren, K.; Scheinin, H. Measurement of central mu-opioid receptor binding in vivo with PET and [11C]carfentanil: A test-retest study in healthy subjects. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Hardy, D.G. Novel analgesics and molecular rearrangements in the morphine-thebaine group. III. Alcohols of the 6,14-endo-ethenotetrahydrooripavine series and derived analogs of N-allylnormorphine and -norcodeine. J. Am. Chem. Soc. 1967, 89, 3281–3292. [Google Scholar] [CrossRef]
- Marton, J.; Henriksen, G. Design and synthesis of an 18F-labeled version of phenylethyl orvinol ([18F]FE-PEO) for PET-imaging of opioid receptors. Molecules 2012, 17, 11554–11569. [Google Scholar] [CrossRef]
- Riss, P.J.; Hong, Y.T.; Marton, J.; Caprioli, D.; Williamson, D.J.; Ferrari, V.; Saigal, N.; Roth, B.L.; Henriksen, G.; Fryer, T.D.; et al. Synthesis and evaluation of 18F-FE-PEO in rodents: An 18F-labeled full agonist for opioid receptor imaging. J. Nucl. Med. 2013, 54, 299–305. [Google Scholar] [CrossRef]
- Lever, J.R.; Kinter, C.M.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; Dannals, R.F. Synthesis of N1′-([11C]methyl)naltrindole ([11C]MeNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1995, 36, 137–145. [Google Scholar] [CrossRef]
- Madar, I.; Lever, J.R.; Kinter, C.M.; Scheffel, U.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; Dannals, R.F.; Frost, J.J. Imaging of delta opioid receptors in human brain by N1′-([11C]methyl)naltrindole and PET. Synapse 1996, 24, 19–28. [Google Scholar] [CrossRef]
- Smith, J.S.; Zubieta, J.K.; Price, J.C.; Flesher, J.E.; Madar, I.; Lever, J.R.; Kinter, C.M.; Dannals, R.F.; Frost, J.J. Quantification of delta-opioid receptors in human brain with N1′-([11C]methyl) naltrindole and positron emission tomography. J. Cereb. Blood Flow. Metab. 1999, 19, 956–966. [Google Scholar] [CrossRef]
- Mathews, W.B.; Kinter, C.M.; Palma, J.; Daniels, R.V.; Ravert, H.T.; Dannals, R.F.; Lever, J.R. Synthesis of N1′-([18F]fluoroethyl)naltrindole ([18F]FEtNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1999, 42, 43–54. [Google Scholar] [CrossRef]
- Tyacke, R.J.; Robinson, E.S.; Schnabel, R.; Lewis, J.W.; Husbands, S.M.; Nutt, D.J.; Hudson, A.L. N1′-fluoroethyl-naltrindole (BU97001) and N1′-fluoroethyl-(14-formylamino)-naltrindole (BU97018) potential delta-opioid receptor PET ligands. Nucl. Med. Biol. 2002, 29, 455–462. [Google Scholar] [CrossRef]
- Bourdier, T.; Poisnel, G.; Dhilly, M.; Delamare, J.; Henry, J.; Debruyne, D.; Barré, L. Synthesis and biological evaluation of N-substituted quinolinimides, as potential ligands for in vivo imaging studies of delta-opioid receptors. Bioconj. Chem. 2007, 18, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.G.; Birch, P.J.; Hayward, N.J.; Sheehan, M.J.; Rogers, H.; Tyers, M.B.; Judd, D.B.; Scopes, D.I.C.; Naylor, A. A series of novel, highly potent and selective agonists for the kappa-opioid receptor. Br. J. Pharmacol. 1990, 101, 944–948. [Google Scholar] [CrossRef]
- Birch, P.J.; Rogers, H.; Hayes, A.G.; Hayward, N.J.; Tyers, M.B.; Scopes, D.I.C.; Naylor, A.; Judd, D.B. Neuroprotective actions of GR89696, a highly potent and selective κ-opioid receptor agonist. Br. J. Pharmacol. 1991, 103, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.; Judd, D.B.; Lloyd, J.E.; Scopes, D.I.C.; Hayes, A.G.; Birch, P.J. A potent new class of kappa-receptor agonist: 4-substituted 1-(arylacetyl)-2-[(dialkylamino)methyl]piperazines. J. Med. Chem. 1993, 36, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Ravert, H.T.; Mathews, W.B.; Musachio, J.L.; Scheffel, U.; Finley, P.; Dannals, R.F. [11C]-methyl 4-[(3,4-dichlorophenyl)acetyl]-3-[(1-pyrrolidinyl)methyl]-1-piperazinecarboxylate ([11C]GR89696): Synthesis and in vivo binding to kappa opiate receptors. Nucl. Med. Biol. 1999, 26, 737–741. [Google Scholar] [CrossRef]
- Ravert, H.T.; Scheffel, U.; Mathews, W.B.; Musachio, J.L.; Dannals, R.F. [11C]-GR89696, a potent kappa opiate receptor radioligand; in vivo binding of the R and S enantiomers. Nucl. Med. Biol. 2002, 29, 47–53. [Google Scholar] [CrossRef]
- Talbot, P.S.; Narendran, R.; Butelman, E.R.; Huang, Y.; Ngo, K.; Slifstein, M.; Martinez, D.; Laruelle, M.; Hwang, D.R. 11C-GR103545, a radiotracer for imaging kappa-opioid receptors in vivo with PET: Synthesis and evaluation in baboons. J. Nucl. Med. 2005, 46, 484–494. [Google Scholar]
- Schoultz, B.W.; Arstad, E.; Marton, J.; Willoch, F.; Drzezga, A.; Wester, H.J.; Henriksen, G. A new method for radiosynthesis of 11C-labeled carbamate groups and its application for a highly efficient synthesis of the kappa-opioid receptor tracer [11C]GR103545. Open Med. Chem. J. 2008, 2, 72–74. [Google Scholar] [CrossRef]
- Wilson, A.A.; Gracia, A.; Houle, S.; Vasdev, N. Direct fixation of [11C]-CO2 by amines: Formation of [11C-carbonyl]-methylcarbamates. Org. Biomol. Chem. 2010, 8, 428–432. [Google Scholar] [CrossRef]
- Nabulsi, N.B.; Zheng, M.-Q.; Ropchan, J.; Labaree, D.; Ding, Y.-S.; Blumberg, L.; Huang, Y. [11C]GR103545: Novel one-pot radiosynthesis with high specific activity. Nucl. Med. Biol. 2011, 38, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, G.; Nabulsi, N.; Zheng, M.Q.; Weinzimmer, D.; Ropchan, J.; Blumberg, L.; Brown-Proctor, C.; Ding, Y.S.; Carson, R.E.; Huang, Y. Determination of in vivo Bmax and Kd for 11C-GR103545, an agonist PET tracer for kappa-opioid receptors: A study in nonhuman primates. J. Nucl. Med. 2013, 54, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Naganawa, M.; Jacobsen, L.K.; Zheng, M.-Q.; Lin, S.-F.; Banerjee, A.; Byon, W.; Weinzimmer, D.; Tomasi, G.; Nabulsi, N.; Grimwood, S.; et al. Evaluation of the agonist PET radioligand [11C]GR103545 to image kappa opioid receptor in humans: Kinetic model selection, test–retest reproducibility and receptor occupancy by the antagonist PF-04455242. NeuroImage 2014, 99, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cai, Z.; Zheng, M.-Q.; Holden, D.; Naganawa, M.; Lin, S.-F.; Ropchan, J.; Labaree, D.; Kapinos, M.; Lara-Jaime, T.; et al. Novel 18F-labeled kappa-opioid receptor antagonist as PET radiotracer: Synthesis and in vivo evaluation of 18F-LY2459989 in nonhuman primates. J. Nucl. Med. 2018, 59, 140–146. [Google Scholar] [CrossRef]
- Fichna, J.; Schicho, R.; Janecka, A.; Zjawiony, J.K.; Storr, M. Selective natural kappa opioid and cannabinoid receptor agonists with a potential role in the treatment of gastrointestinal dysfunction. Drug News Perspect. 2009, 22, 383–392. [Google Scholar] [CrossRef][Green Version]
- Butelman, E.R.; Kreek, M.J. Salvinorin A, a kappa-opioid receptor agonist hallucinogen: Pharmacology and potential template for novel pharmacotherapeutic agents in neuropsychiatric disorders. Front. Pharmacol. 2015, 6, 190. [Google Scholar]
- Zjawiony, J.K.; Machado, A.S.; Menegatti, R.; Ghedini, P.C.; Costa, E.A.; Pedrino, G.R.; Lukas, S.E.; Franco, O.L.; Silva, O.N.; Fajemiroye, J.O. Cutting-edge search for safer opioid pain relief: Retrospective review of Salvinorin A and its analogs. Front. Psychiatry 2019, 10(157), 1–11. [Google Scholar] [CrossRef]
- Hooker, J.M.; Xu, Y.; Schiffer, W.; Shea, C.; Carter, P.; Fowler, J.S. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. NeuroImage 2008, 41, 1044–1050. [Google Scholar] [CrossRef]
- Placzek, M.S.; Van de Bittner, G.C.; Wey, H.Y.; Lukas, S.E.; Hooker, J.M. Immediate and persistent effects of Salvinorin A on the kappa opioid receptor in rodents, monitored in vivo with PET. Neuropsychopharmacology 2015, 40, 2865–2872. [Google Scholar] [CrossRef]
- Thomas, J.B.; Atkinson, R.N.; Rothman, R.B.; Fix, S.E.; Mascarella, S.W.; Vinson, N.A.; Xu, H.; Dersch, C.M.; Lu, Y.F.; Cantrell, B.E.; et al. Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J. Med. Chem. 2001, 44, 2687–2690. [Google Scholar] [CrossRef]
- Schmitt, S.; Delamare, J.; Tirel, O.; Fillesoye, F.; Dhilly, M.; Perrio, C. N-[18F]-FluoropropylJDTic for kappa-opioid receptor PET imaging: Radiosynthesis, pre-clinical evaluation, and metabolic investigation in comparison with parent JDTic. Nucl. Med. Biol. 2017, 44, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Zheng, M.Q.; Nabulsi, N.; Labaree, D.; Ropchan, J.; Najafzadeh, S.; Carson, R.E.; Huang, Y.; Morris, E.D. Determination of the in vivo selectivity of a new kappa-opioid receptor antagonist PET tracer 11C-LY2795050 in the rhesus monkey. J. Nucl. Med. 2013, 54, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Naganawa, M.; Zheng, M.Q.; Nabulsi, N.; Tomasi, G.; Henry, S.; Lin, S.F.; Ropchan, J.; Labaree, D.; Tauscher, J.; Neumeister, A.; et al. Kinetic modeling of 11C-LY2795050, a novel antagonist radiotracer for PET imaging of the kappa opioid receptor in humans. J. Cereb. Blood Flow. Metab. 2014, 34, 1818–1825. [Google Scholar] [CrossRef]
- Naganawa, M.; Zheng, M.-Q.; Henry, S.; Nabulsi, N.; Lin, S.-F.; Ropchan, J.; Labaree, D.; Najafzadeh, S.; Kapinos, M.; Tauscher, J.; et al. Test–Retest Reproducibility of Binding Parameters in Humans with 11C-LY2795050, an Antagonist PET Radiotracer for the kappa Opioid Receptor. J. Nucl. Med. 2015, 56, 243–248. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Naganawa, M.; Dickinson, G.L.; Zheng, M.-Q.; Henry, S.; Vandenhende, F.; Witcher, J.; Bell, R.; Nabulsi, N.; Lin, S.-F.; Ropchan, J.; et al. Receptor occupancy of the kappa-opioid antagonist LY2456302 measured with positron emission tomography and the novel radiotracer 11C-LY2795050. J. Pharmacol. Exp. Ther. 2016, 356, 260–266. [Google Scholar] [CrossRef]
- Placzek, M.S.; Schroeder, F.A.; Che, T.; Wey, H.-Y.; Neelamegam, R.; Wang, C.; Roth, B.L.; Hooker, J.M. Discrepancies in kappa opioid agonist binding revealed through PET Imaging. ACS Chem. Neurosci. 2019, 10, 384–395. [Google Scholar] [CrossRef]
- Chesis, P.L.; Welch, M.J. Synthesis and in vitro characterization of fluorinated U-50488 analogs for PET studies of kappa opioid receptors. Int. J. Radiat. Applicat. Instrum. Part A Appl. Radiat. Isot. 1990, 41, 267–273. [Google Scholar] [CrossRef]
- Zaveri, N.T. Nociceptin opioid receptor (NOP) as a therapeutic target: Progress in translation from preclinical research to clinical utility. J. Med. Chem. 2016, 59, 7011–7028. [Google Scholar] [CrossRef]
- Kimura, Y.; Fujita, M.; Hong, J.S.; Lohith, T.G.; Gladding, R.L.; Zoghbi, S.S.; Tauscher, J.A.; Goebl, N.; Rash, K.S.; Chen, Z.; et al. Brain and whole-body imaging in rhesus monkeys of 11C-NOP-1A, a promising PET radioligand for nociceptin/orphanin FQ peptide receptors. J. Nucl. Med. 2011, 52, 1638–1645. [Google Scholar] [CrossRef]
- Lohith, T.G.; Zoghbi, S.S.; Morse, C.L.; Araneta, M.F.; Barth, V.N.; Goebl, N.A.; Tauscher, J.T.; Pike, V.W.; Innis, R.B.; Fujita, M. Brain and whole-body imaging of nociceptin/orphanin FQ peptide receptor in humans using the PET Ligand 11C-NOP-1A. J. Nucl. Med. 2012, 53, 385–392. [Google Scholar] [CrossRef]
- Lohith, T.G.; Zoghbi, S.S.; Morse, C.L.; Araneta, M.D.F.; Barth, V.N.; Goebl, N.A.; Tauscher, J.T.; Pike, V.W.; Innis, R.B.; Fujita, M. Retest imaging of [11C]NOP-1A binding to nociceptin/orphanin FQ peptide (NOP) receptors in the brain of healthy humans. NeuroImage 2014, 87, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Raddad, E.; Chappell, A.; Meyer, J.; Wilson, A.A.; Ruegg, C.E.; Tauscher, J.; Statnick, M.A.; Barth, V.; Zhang, X.; Verfaille, S.J. Occupancy of nociceptin/orphanin FQ peptide receptors by the antagonist LY2940094 in rats and healthy human subjects. Drug Metab. Dispos. 2016, 44, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Drummond, E.; Brodney, M.A.; Cianfrogna, J.; Drozda, S.E.; Grimwood, S.; Vanase-Frawley, M.A.; Villalobos, A. Design, synthesis and evaluation of [3H]PF-7191, a highly specific nociceptin opioid peptide (NOP) receptor radiotracer for in vivo receptor occupancy (RO) studies. Bioorg. Med. Chem. Lett. 2014, 24, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Zubieta, J.K.; Dannals, R.F.; Frost, J.J. Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am. J. Psychiarty 1999, 156, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Vijay, A.; Cavallo, D.; Goldberg, A.; de Laat, B.; Nabulsi, N.; Huang, Y.; Krishnan-Sarin, S.; Morris, E.D. PET imaging reveals lower kappa opioid receptor availability in alcoholics but no effect of age. Neuropsychopharmacology 2018, 43, 2539–2547. [Google Scholar] [CrossRef]
- Vijay, A.; Wang, S.; Worhunsky, P.; Zheng, M.-Q.; Nabulsi, N.; Ropchan, J.; Krishnan-Sarin, S.; Huang, Y.; Morris, E.D. PET imaging reveals sex differences in kappa opioid receptor availability in humans, in vivo. Am. J. Nucl. Med. Mol. Imaging 2016, 6, 205–214. [Google Scholar] [PubMed]
- Mayberg, H.S.; Sadzot, B.; Meltzer, C.C.; Fisher, R.S.; Lesser, R.P.; Dannals, R.F.; Lever, J.R.; Wilson, A.A.; Ravert, H.T.; Wagner, H.N.J.; et al. Quantification of mu and non–mu opiate receptors in temporal lobe epilepsy using positron emission tomography. Ann. Neurol. 1991, 30, 3–11. [Google Scholar] [CrossRef]
- Madar, I.; Lesser, R.P.; Krauss, G.; Zubieta, J.K.; Lever, J.R.; Kinter, C.M.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; Dannals, R.F.; et al. Imaging of δ- and μ-opioid receptors in temporal lobe epilepsy by positron emission tomography. Ann. Neurol. 1997, 41, 358–367. [Google Scholar] [CrossRef]
- Hammers, A.; Asselin, M.-C.; Hinz, R.; Kitchen, I.; Brooks, D.J.; Duncan, J.S.; Koepp, M.J. Upregulation of opioid receptor binding following spontaneous epileptic seizures. Brain 2007, 130, 1009–1016. [Google Scholar] [CrossRef]
- McGinnity, C.J.; Shidahara, M.; Feldmann, M.; Keihaninejad, S.; Riaño Barros, D.A.; Gousias, I.S.; Duncan, J.S.; Brooks, D.J.; Heckemann, R.A.; Turkheimer, F.E.; et al. Quantification of opioid receptor availability following spontaneous epileptic seizures: Correction of [11C]diprenorphine PET data for the partial-volume effect. NeuroImage 2013, 79, 72–80. [Google Scholar] [CrossRef]
- Koepp, M.J.; Richardson, M.P.; Brooks, D.J.; Duncan, J.S. Focal cortical release of endogenous opioids during reading induced seizures. Lancet 1998, 352, 952–955. [Google Scholar] [CrossRef]
- Cohen, R.M.; Carson, R.E.; Aigner, T.G.; Doudet, D.J. Opiate receptor avidity is reduced in non-motor impaired MPTP-lesioned rhesus monkeys. Brain Res. 1998, 806, 292–296. [Google Scholar] [CrossRef]
- Cohen, R.M.; Carson, R.E.; Wyatt, R.J.; Doudet, D.J. Opiate receptor avidity is reduced bilaterally in rhesus monkeys unilaterally lesioned with MPTP. Synapse 1999, 33, 282–288. [Google Scholar] [CrossRef]
- Piccini, P.; Weeks, R.A.; Brooks, D.J. Alterations in opioid receptor binding in Parkinson’s disease patients with levodopa-induced dyskinesias. Ann. Neurol. 1997, 42, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.L.; Von Spiczak, S.; Edwards, M.; Valente, E.-M.; Hammers, A.; Bhatia, K.P.; Brooks, D.J. Opioid binding in DYT1 primary torsion dystonia: An 11C-diprenorphine PET study. Mov. Dis. 2004, 19, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Von Spiczak, S.; Whone, A.L.; Hammers, A.; Asselin, M.-C.; Turkheimer, F.; Tings, T.; Happe, S.; Paulus, W.; Trenkwalder, C.; Brooks, D.J. The role of opioids in restless legs syndrome: An [11C]diprenorphine PET study. Brain 2005, 128, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Weeks, R.A.; Cunningham, V.J.; Piccini, P.; Waters, S.; Harding, A.E.; Brooks, D.J. 11C-Diprenorphine binding in Huntington’s disease: A comparison of region of interest analysis with statistical parametric mapping. J. Cereb. Blood Flow. Metab. 1997, 17, 943–949. [Google Scholar] [CrossRef]
- Willoch, F.; Tölle, T.R.; Wester, H.-J.; Munz, F.; Petzold, A.; Schwaiger, M.; Conrad, B.; Bartenstein, P. Central pain after pontine infarction is associated with changes in opioid receptor binding: A PET study with 11C-Diprenorphine. AJNR Am. J. Neuroradiol. 1999, 20, 686–690. [Google Scholar]
- Willoch, F.; Schindler, F.; Wester, H.-J.; Empl, M.; Straube, A.; Schwaiger, M.; Conrad, B.; Tölle, T.R. Central poststroke pain and reduced opioid receptor binding within pain processing circuitries: A [11C]diprenorphine PET study. Pain 2004, 108, 213–220. [Google Scholar] [CrossRef]
- Maarrawi, J.; Peyron, R.; Mertens, P.; Costes, N.; Magnin, M.; Sindou, M.; Laurent, B.; Garcia-Larrea, L. Differential brain opioid receptor availability in central and peripheral neuropathic pain. Pain 2007, 127, 183–194. [Google Scholar] [CrossRef]
- Campbell, C.M.; Bounds, S.C.; Kuwabara, H.; Edwards, R.R.; Campbell, J.N.; Haythornthwaite, J.A.; Smith, M.T. Individual variation in sleep quality and duration is related to cerebral mu opioid receptor binding potential during tonic laboratory pain in healthy subjects. Pain Med. 2013, 14, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, B.; Fuchs, P.N.; Sheth, R.; Dannals, R.F.; Campbell, J.N.; Frost, J.J. Pain activation of human supraspinal opioid pathways as demonstrated by [11C]-carfentanil and positron emission tomography (PET). Pain 2002, 99, 589–598. [Google Scholar] [CrossRef]
- Sprenger, T.; Valet, M.; Boecker, H.; Henriksen, G.; Spilker, M.E.; Willoch, F.; Wagner, K.J.; Wester, H.J.; Tölle, T.R. Opioidergic activation in the medial pain system after heat pain. Pain 2006, 122, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Zubieta, J.K.; Smith, Y.R.; Bueller, J.A.; Xu, Y.; Kilbourn, M.R.; Jewett, D.M.; Meyer, C.R.; Koeppe, R.A.; Stohler, C.S. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 2001, 293, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Zubieta, J.K.; Smith, Y.R.; Bueller, J.A.; Xu, Y.; Kilbourn, M.R.; Jewett, D.M.; Meyer, C.R.; Koeppe, R.A.; Stohler, C.S. Mu-opioid receptor-mediated antinociceptive responses differ in men and women. J. Neurosci. 2002, 22, 5100–5107. [Google Scholar] [CrossRef] [PubMed]
- DosSantos, M.F.; Martikainen, I.K.; Nascimento, T.D.; Love, T.M.; Deboer, M.D.; Maslowski, E.C.; Monteiro, A.A.; Vincent, M.B.; Zubieta, J.K.; DaSilva, A.F. Reduced basal ganglia mu-opioid receptor availability in trigeminal neuropathic pain: A pilot study. Mol. Pain 2012, 8, 74. [Google Scholar] [CrossRef]
- Scott, D.J.; Stohler, C.S.; Koeppe, R.A.; Zubieta, J.K. Time-course of change in [11C]carfentanil and [11C]raclopride binding potential after a nonpharmacological challenge. Synapse 2007, 61, 707–714. [Google Scholar] [CrossRef]
- Hjornevik, T.; Schoultz, B.W.; Marton, J.; Gjerstad, J.; Drzezga, A.; Henriksen, G.; Willoch, F. Spinal long-term potentiation is associated with reduced opioid neurotransmission in the rat brain. Clin. Physiol. Funct. Imaging 2010, 30, 285–293. [Google Scholar] [CrossRef]
- Boecker, H.; Sprenger, T.; Spilker, M.E.; Henriksen, G.; Koppenhoefer, M.; Wagner, K.J.; Valet, M.; Berthele, A.; Tolle, T.R. The runner’s high: Opioidergic mechanisms in the human brain. Cereb. Cortex 2008, 18, 2523–2531. [Google Scholar] [CrossRef]
- Saanijoki, T.; Tuominen, L.; Tuulari, J.J.; Nummenmaa, L.; Arponen, E.; Kalliokoski, K.; Hirvonen, J. Opioid release after high-intensity interval training in healthy human subjects. Neuropsychopharmacol 2018, 43, 246–254. [Google Scholar] [CrossRef]
- Maarrawi, J.; Peyron, R.; Mertens, P.; Costes, N.; Magnin, M.; Sindou, M.; Laurent, B.; Garcia-Larrea, L. Motor cortex stimulation for pain control induces changes in the endogenous opioid system. Neurology 2007, 69, 827–834. [Google Scholar] [CrossRef] [PubMed]
- DosSantos, M.F.; Love, T.M.; Martikainen, I.K.; Nascimento, T.D.; Fregni, F.; Cummiford, C.; Deboer, M.D.; Zubieta, J.K.; DaSilva, A.F. Immediate Effects of tDCS on the mu-opioid system of a chronic pain patient. Front. Psychiatry 2012, 3, 93. [Google Scholar] [PubMed]
- Maarrawi, J.; Peyron, R.; Mertens, P.; Costes, N.; Magnin, M.; Sindou, M.; Laurent, B.; Garcia-Larrea, L. Brain opioid receptor density predicts motor cortex stimulation efficacy for chronic pain. Pain 2013, 154, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Sims-Williams, H.; Matthews, J.C.; Talbot, P.S.; Love-Jones, S.; Brooks, J.C.; Patel, N.K.; Pickering, A.E. Deep brain stimulation of the periaqueductal gray releases endogenous opioids in humans. NeuroImage 2017, 146, 833–842. [Google Scholar] [CrossRef]
- Ly, H.G.; Dupont, P.; Geeraerts, B.; Bormans, G.; Van Laere, K.; Tack, J.; Van Oudenhove, L. Lack of endogenous opioid release during sustained visceral pain: A [11C]carfentanil PET study. Pain 2013, 154, 2072–2077. [Google Scholar] [CrossRef]
- Baier, B.; Bense, S.; Birklein, F.; Buchholz, H.-G.; Mischke, A.; Schreckenberger, M.; Dieterich, M. Evidence for modulation of opioidergic activity in central vestibular processing: A [18F] diprenorphine PET study. Hum. Brain Mapp. 2010, 31, 550–555. [Google Scholar] [CrossRef]
- Wager, T.D.; Scott, D.J.; Zubieta, J.K. Placebo effects on human mu-opioid activity during pain. Proc. Natl. Acad. Sci. USA 2007, 104, 11056–11061. [Google Scholar] [CrossRef]
- DosSantos, M.F.; Martikainen, I.K.; Nascimento, T.D.; Love, T.M.; DeBoer, M.D.; Schambra, H.M.; Bikson, M.; Zubieta, J.K.; DaSilva, A.F. Building up analgesia in humans via the endogenous mu-opioid system by combining placebo and active tDCS: A preliminary report. PLoS ONE 2014, 9, e102350. [Google Scholar] [CrossRef]
- Dougherty, D.D.; Kong, J.; Webb, M.; Bonab, A.A.; Fischman, A.J.; Gollub, R.L. A combined [11C]diprenorphine PET study and fMRI study of acupuncture analgesia. Behav. Brain Res. 2008, 193, 63–68. [Google Scholar] [CrossRef]
- Harris, R.E.; Zubieta, J.K.; Scott, D.J.; Napadow, V.; Gracely, R.H.; Clauw, D.J. Traditional Chinese acupuncture and placebo (sham) acupuncture are differentiated by their effects on mu-opioid receptors (MORs). NeuroImage 2009, 47, 1077–1085. [Google Scholar] [CrossRef]
- Xiang, X.-H.; Chen, Y.-M.; Zhang, J.-M.; Tian, J.-H.; Han, J.-S.; Cui, C.-L. Low- and high-frequency transcutaneous electrical acupoint stimulation induces different effects on cerebral mu-opioid receptor availability in rhesus monkeys. J. Neurosci. Res. 2014, 92, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Klega, A.; Buchholz, H.-G.; Rolke, R.; Magerl, W.; Schirrmacher, R.; Schirrmacher, E.; Birklein, F.; Treede, R.-D.; Schreckenberger, M. Basal opioid receptor binding is associated with differences in sensory perception in healthy human subjects: A [18F]diprenorphine PET study. NeuroImage 2010, 49, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, N.; Aalto, S.; Tuominen, L.; Pesonen, U.; Någren, K.; Hietala, J.; Scheinin, H.; Pertovaara, A.; Martikainen, I.K. Striatal mu-opioid receptor availability predicts cold pressor pain threshold in healthy human subjects. Neurosci. Lett. 2012, 521, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.J.; Pitcher, M.H.; Stone, L.S.; Tarum, F.; Niu, G.; Chen, X.; Kiesewetter, D.O.; Schweinhardt, P.; Bushnell, M.C. Chronic neuropathic pain reduces opioid receptor availability with associated anhedonia in rat. Pain 2018, 159, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, T.; Willoch, F.; Miederer, M.; Schindler, F.; Valet, M.; Berthele, A.; Spilker, M.E.; Förderreuther, S.; Straube, A.; Stangier, I.; et al. Opioidergic changes in the pineal gland and hypothalamus in cluster headache: A ligand PET study. Neurology 2006, 66, 1108–1110. [Google Scholar] [CrossRef]
- DaSilva, A.F.; Nascimento, T.D.; DosSantos, M.F.; Lucas, S.; van Holsbeeck, H.; DeBoer, M.; Maslowski, E.; Love, T.; Martikainen, I.K.; Koeppe, R.A.; et al. Mu-Opioid activation in the prefrontal cortex in migraine attacks—Brief report I. Ann. Clin. Transl. Neurol. 2014, 1, 439–444. [Google Scholar] [CrossRef]
- Linnman, C.; Catana, C.; Petkov, M.P.; Chonde, D.B.; Becerra, L.; Hooker, J.; Borsook, D. Molecular and functional PET-fMRI measures of placebo analgesia in episodic migraine: Preliminary findings. NeuroImage Clin. 2018, 17, 680–690. [Google Scholar] [CrossRef]
- Schreckenberger, M.; Klega, A.; Gründer, G.; Buchholz, H.-G.; Scheurich, A.; Schirrmacher, R.; Schirrmacher, E.; Müller, C.; Henriksen, G.; Bartenstein, P. Opioid receptor PET reveals the psychobiologic correlates of reward processing. J. Nucl. Med. 2008, 49, 1257–1261. [Google Scholar] [CrossRef]
- Tuominen, L.; Salo, J.; Hirvonen, J.; Någren, K.; Laine, P.; Melartin, T.; Isometsä, E.; Viikari, J.; Raitakari, O.; Keltikangas-Järvinen, L.; et al. Temperament trait harm avoidance associates with mu-opioid receptor availability in frontal cortex: A PET study using [11C]carfentanil. NeuroImage 2012, 61, 670–676. [Google Scholar] [CrossRef]
- Karjalainen, T.; Tuominen, L.; Manninen, S.; Kalliokoski, K.K.; Nuutila, P.; Jääskeläinen, I.P.; Hari, R.; Sams, M.; Nummenmaa, L. Behavioural activation system sensitivity is associated with cerebral mu-opioid receptor availability. Soc. Cogn. Affect. Neurosci. 2016, 11, 1310–1316. [Google Scholar] [CrossRef]
- Nummenmaa, L.; Manninen, S.; Tuominen, L.; Hirvonen, J.; Kalliokoski, K.K.; Nuutila, P.; Jääskeläinen, I.P.; Hari, R.; Dunbar, R.I.; Sams, M. Adult attachment style is associated with cerebral mu-opioid receptor availability in humans. Hum. Brain Mapp. 2015, 36, 3621–3628. [Google Scholar] [CrossRef]
- Karjalainen, T.; Seppälä, K.; Glerean, E.; Karlsson, H.K.; Lahnakoski, J.M.; Nuutila, P.; Jääskeläinen, I.P.; Hari, R.; Sams, M.; Nummenmaa, L. Opioidergic Regulation of Emotional Arousal: A Combined PET–fMRI Study. Cereb. Cortex 2018, 29, 4006–4016. [Google Scholar] [CrossRef]
- Nummenmaa, L.; Tuominen, L.; Dunbar, R.; Hirvonen, J.; Manninen, S.; Arponen, E.; Machin, A.; Hari, R.; Jääskeläinen, I.P.; Sams, M. Social touch modulates endogenous mu-opioid system activity in humans. NeuroImage 2016, 138, 242–247. [Google Scholar] [CrossRef]
- Manninen, S.; Tuominen, L.; Dunbar, R.I.; Karjalainen, T.; Hirvonen, J.; Arponen, E.; Hari, R.; Jääskeläinen, I.P.; Sams, M.; Nummenmaa, L. Social laughter triggers endogenous opioid release in humans. J. Neurosci. 2017, 37, 6125–6131. [Google Scholar] [CrossRef]
- Prossin, A.R.; Koch, A.E.; Campbell, P.L.; Barichello, T.; Zalcman, S.S.; Zubieta, J.-K. Acute experimental changes in mood state regulate immune function in relation to central opioid neurotransmission: A model of human CNS-peripheral inflammatory interaction. Mol. Psychiatry 2016, 21, 243–251. [Google Scholar] [CrossRef]
- Kennedy, S.E.; Koeppe, R.A.; Young, E.A.; Zubieta, J.K. Dysregulation of Endogenous Opioid Emotion Regulation Circuitry in Major Depression in Women. Arch. Gen. Psychiatry. 2006, 63, 1199–1208. [Google Scholar] [CrossRef]
- Miller, J.M.; Zanderigo, F.; Purushothaman, P.D.; DeLorenzo, C.; Rubin-Falcone, H.; Ogden, R.T.; Keilp, J.; Oquendo, M.A.; Nabulsi, N.; Huang, Y.H.; et al. Kappa opioid receptor binding in major depression: A pilot study. Synapse 2018, 72, e22042. [Google Scholar] [CrossRef]
- Matuskey, D.; Dias, M.; Naganawa, M.; Pittman, B.; Henry, S.; Li, S.; Gao, H.; Ropchan, J.; Nabulsi, N.; Carson, R.E.; et al. Social status and demographic effects of the kappa opioid receptor: A PET imaging study with a novel agonist radiotracer in healthy volunteers. Neuropsychopharmacology 2019, 44, 1714–1719. [Google Scholar] [CrossRef]
- Ashok, A.H.; Myers, J.; Marques, T.R.; Rabiner, E.A.; Howes, O.D. Reduced mu opioid receptor availability in schizophrenia revealed with [11C]carfentanil positron emission tomographic Imaging. Nat. Commun. 2019, 10, 4493–4502. [Google Scholar] [CrossRef]
- Prossin, A.R.; Zalcman, S.S.; Heitzeg, M.M.; Koch, A.E.; Campbell, P.L.; Phan, K.L.; Stohler, C.S.; Zubieta, J.K. Dynamic interactions between plasma IL-1 family cytokines and central endogenous opioid neurotransmitter function in humans. Neuropsychopharmacology 2015, 40, 554–565. [Google Scholar] [CrossRef]
- Schrepf, A.; Harper, D.E.; Harte, S.E.; Wang, H.; Ichesco, E.; Hampson, J.P.; Zubieta, J.K.; Clauw, D.J.; Harris, R.E. Endogenous opioidergic dysregulation of pain in fibromyalgia: A PET and fMRI study. Pain 2016, 157, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Kim, Y.K.; Bencherif, B.; Boyd, S.J.; Nelson, R.; Copersino, M.; Endres, C.J.; Dannals, R.F.; Frost, J.J. Imaging brain mu-opioid receptors in abstinent cocaine users: Time course and relation to cocaine craving. Biol. Psychiatry 2005, 57, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Kim, Y.K.; Bencherif, B.; Boyd, S.J.; Nelson, R.; Copersino, M.L.; Dannals, R.F.; Frost, J.J. Brain mu-opioid receptor binding: Relationship to relapse to cocaine use after monitored abstinence. Psychopharmacology 2008, 200, 475–486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martinez, D.; Slifstein, M.; Matuskey, D.; Nabulsi, N.; Zheng, M.-Q.; Lin, S.-F.; Ropchan, J.; Urban, N.; Grassetti, A.; Chang, D.; et al. Kappa-opioid receptors, dynorphin, and cocaine addiction: A positron emission tomography study. Neuropsychopharmacology 2019, 44, 1720–1727. [Google Scholar] [CrossRef]
- Narendran, R.; Tollefson, S.; Himes, M.L.; Paris, J.; Lopresti, B.; Ciccocioppo, R.; Scott Mason, N. Nociceptin receptors upregulated in cocaine use disorder: A positron emission tomography imaging study using [11C]NOP-1A. Am. J. Psychiarty 2019, 176, 468–476. [Google Scholar] [CrossRef]
- Heinz, A.; Reimold, M.; Wrase, J.; Hermann, D.; Croissant, B.; Mundle, G.; Dohmen, B.M.; Braus, D.F.; Schumann, G.; Machulla, H.J.; et al. Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: A positron emission tomography study using carbon 11C-labeled carfentanil. Arch. Gen. Psychiatry 2005, 62, 57–64. [Google Scholar] [CrossRef]
- Turton, S.; Myers, J.F.M.; Mick, I.; Colasanti, A.; Venkataraman, A.; Durant, C.; Waldman, A.; Brailsford, A.; Parkin, M.C.; Dawe, G.; et al. Blunted endogenous opioid release following an oral dexamphetamine challenge in abstinent alcohol-dependent individuals. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Williams, T.M.; Davies, S.J.C.; Taylor, L.G.; Daglish, M.R.C.; Hammers, A.; Brooks, D.J.; Nutt, D.J.; Lingford-Hughes, A. Brain opioid receptor binding in early abstinence from alcohol dependence and relationship to craving: An [11C]diprenorphine PET study. Eur. Neuropsychopharmacol. 2009, 19, 740–748. [Google Scholar] [CrossRef]
- Hermann, D.; Hirth, N.; Reimold, M.; Batra, A.; Smolka, M.N.; Hoffmann, S.; Kiefer, F.; Noori, H.R.; Sommer, W.H.; Reischl, G.; et al. Low mu-opioid receptor status in alcohol dependence identified by combined positron emission tomography and post-mortem brain analysis. Neuropsychopharmacology 2016, 42, 606–614. [Google Scholar] [CrossRef]
- Weerts, E.M.; Wand, G.S.; Kuwabara, H.; Munro, C.A.; Dannals, R.F.; Hilton, J.; Frost, J.J.; McCaul, M.E. Positron emission tomography imaging of Mu- and Delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcohol. Clin. Exp. Res. 2011, 35, 2162–2173. [Google Scholar] [CrossRef]
- Wand, G.S.; Weerts, E.M.; Kuwabara, H.; Frost, J.J.; Xu, X.; McCaul, M.E. Naloxone-induced cortisol predicts mu opioid receptor binding potential in specific brain regions of healthy subjects. Psychoneuroendocrinology 2011, 36, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Wand, G.S.; Weerts, E.M.; Kuwabara, H.; Wong, D.F.; Xu, X.; McCaul, M.E. The relationship between naloxone-induced cortisol and mu opioid receptor availability in mesolimbic structures is disrupted in alcohol dependent subjects. Alcohol 2012, 46, 511–517. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ray, R.; Ruparel, K.; Newberg, A.; Wileyto, E.P.; Loughead, J.W.; Divgi, C.; Blendy, J.A.; Logan, J.; Zubieta, J.-K.; Lerman, C. Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proc. Natl. Acad. Sci. USA 2011, 108, 9268–9273. [Google Scholar] [CrossRef] [PubMed]
- Domino, E.F.; Hirasawa-Fujita, M.; Ni, L.; Guthrie, S.K.; Zubieta, J.K. Regional brain [11C]carfentanil binding following tobacco smoking. Progr. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 59, 100–104. [Google Scholar] [CrossRef]
- Nuechterlein, E.B.; Ni, L.; Domino, E.F.; Zubieta, J.K. Nicotine-specific and non-specific effects of cigarette smoking on endogenous opioid mechanisms. Progr. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 69, 69–77. [Google Scholar] [CrossRef]
- Weerts, E.M.; Wand, G.S.; Kuwabara, H.; Xu, X.; Frost, J.J.; Wong, D.F.; McCaul, M.E. Association of smoking with mu-opioid receptor availability before and during naltrexone blockade in alcohol-dependent subjects. Addict. Biol. 2014, 19, 733–742. [Google Scholar] [CrossRef]
- Kuwabara, H.; Heishman, S.J.; Brasic, J.R.; Contoreggi, C.; Cascella, N.; Mackowick, K.M.; Taylor, R.; Rousset, O.; Willis, W.; Huestis, M.A.; et al. Mu opioid receptor binding correlates with nicotine dependence and reward in smokers. PLoS ONE 2014, 9, e113694. [Google Scholar] [CrossRef]
- Guterstam, J.; Jayaram-Lindström, N.; Cervenka, S.; Frost, J.J.; Farde, L.; Halldin, C.; Franck, J. Effects of amphetamine on the human brain opioid system—A positron emission tomography study. Int. J. Neuropsychopharmacol. 2013, 16, 763–769. [Google Scholar] [CrossRef]
- Colasanti, A.; Searle, G.E.; Long, C.J.; Hill, S.P.; Reiley, R.R.; Quelch, D.; Erritzoe, D.; Tziortzi, A.C.; Reed, L.J.; Lingford-Hughes, A.; et al. Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol. Psychiatry 2012, 72, 371–377. [Google Scholar] [CrossRef]
- Mick, I.; Myers, J.; Stokes, P.R.A.; Erritzoe, D.; Colasanti, A.; Bowden-Jones, H.; Clark, L.; Gunn, R.N.; Rabiner, E.A.; Searle, G.E.; et al. Amphetamine induced endogenous opioid release in the human brain detected with [11C]carfentanil PET: Replication in an independent cohort. Int. J. Neuropsychopharmacol. 2014, 17, 2069–2074. [Google Scholar] [CrossRef]
- Quelch, D.R.; Katsouri, L.; Nutt, D.J.; Parker, C.A.; Tyacke, R.J. Imaging endogenous opioid peptide release with [11C]carfentanil and [3H]diprenorphine: Influence of agonist-induced internalization. J. Cereb. Blood Flow. Metab. 2014, 34, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Mick, I.; Myers, J.; Ramos, A.C.; Stokes, P.R.A.; Erritzoe, D.; Colasanti, A.; Gunn, R.N.; Rabiner, E.A.; Searle, G.E.; Waldman, A.D.; et al. Blunted endogenous opioid release following an oral amphetamine challenge in pathological gamblers. Neuropsychopharmacology 2016, 41, 1742–1750. [Google Scholar] [CrossRef] [PubMed]
- Majuri, J.; Joutsa, J.; Arponen, E.; Forsback, S.; Kaasinen, V. Dopamine synthesis capacity correlates with mu-opioid receptor availability in the human basal ganglia: A triple-tracer PET study. NeuroImage 2018, 183, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, B.; Guarda, A.S.; Colantuoni, C.; Ravert, H.T.; Dannals, R.F.; Frost, J.J. Regional mu-opioid receptor binding in insular cortex is decreased in bulimia nervosa and correlates inversely with fasting behavior. J. Nucl. Med. 2005, 46, 1349–1351. [Google Scholar] [PubMed]
- Karlsson, H.K.; Tuominen, L.; Tuulari, J.J.; Hirvonen, J.; Parkkola, R.; Helin, S.; Salminen, P.; Nuutila, P.; Nummenmaa, L. Obesity is associated with decreased mu-opioid but unaltered dopamine D2 receptor availability in the brain. J. Neurosci. 2015, 35, 3959–3965. [Google Scholar] [CrossRef] [PubMed]
- Joutsa, J.; Karlsson, H.K.; Majuri, J.; Nuutila, P.; Helin, S.; Kaasinen, V.; Nummenmaa, L. Binge eating disorder and morbid obesity are associated with lowered mu-opioid receptor availability in the brain. Psychiatry Res. Neuroimaging 2018, 276, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, H.K.; Tuulari, J.J.; Tuominen, L.; Hirvonen, J.; Honka, H.; Parkkola, R.; Helin, S.; Salminen, P.; Nuutila, P.; Nummenmaa, L. Weight loss after bariatric surgery normalizes brain opioid receptors in morbid obesity. Mol. Psychiatry 2016, 21, 1057–1062. [Google Scholar] [CrossRef]
- Tuominen, L.; Tuulari, J.; Karlsson, H.; Hirvonen, J.; Helin, S.; Salminen, P.; Parkkola, R.; Hietala, J.; Nuutila, P.; Nummenmaa, L. Aberrant mesolimbic dopamine–opiate interaction in obesity. NeuroImage 2015, 122, 80–86. [Google Scholar] [CrossRef]
- Tuulari, J.J.; Tuominen, L.; de Boer, F.E.; Hirvonen, J.; Helin, S.; Nuutila, P.; Nummenmaa, L. Feeding releases endogenous opioids in humans. J. Neurosci. 2017, 37, 8284–8291. [Google Scholar] [CrossRef]
- Nummenmaa, L.; Saanijoki, T.; Tuominen, L.; Hirvonen, J.; Tuulari, J.J.; Nuutila, P.; Kalliokoski, K. Mu-opioid receptor system mediates reward processing in humans. Nat. Commun. 2018, 9, 1500. [Google Scholar] [CrossRef]
- Saanijoki, T.; Nummenmaa, L.; Tuulari, J.J.; Tuominen, L.; Arponen, E.; Kalliokoski, K.K.; Hirvonen, J. Aerobic exercise modulates anticipatory reward processing via the mu-opioid receptor system. Hum. Brain Mapp. 2018, 39, 3972–3983. [Google Scholar] [CrossRef] [PubMed]
- Hiura, M.; Sakata, M.; Ishii, K.; Toyohara, J.; Oda, K.; Nariai, T.; Ishiwata, K. Central mu-opioidergic system activation evoked by heavy and severe-intensity cycling exercise in humans: A pilot study using positron emission tomography with 11C-carfentanil. Int. J. Sports Med. 2017, 38, 19–26. [Google Scholar] [PubMed]
- Pert, C.B.; Snyder, S.H. Opiate receptor binding—Enhancement by opiate administration in vivo. Biochem. Pharmacol. 1976, 25, 847–853. [Google Scholar] [CrossRef]
- Burns, J.A.; Kroll, D.S.; Feldman, D.E.; Liu, C.K.; Manza, P.; Wiers, C.E.; Volkow, N.D.; Wang, G.-J. Molecular imaging of opioid and dopamine systems: Insights into the pharmacogenetics of opioid use disorders. Front. Psychiatry 2019, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.-H.; Wang, B.; Kou, Z.-Z.; Bai, Y.; Chen, T.; Dong, Y.-L.; Li, H.; Li, Y.-Q. Endomorphins: Promising endogenous opioid peptides for the development of novel analgesics. Neurosignals 2017, 25, 98–116. [Google Scholar] [CrossRef] [PubMed]
- Redila, V.A.; Chavkin, C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology 2008, 200, 59–70. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Ki [nM] | Action | Compound Class | Ref. | |||
|---|---|---|---|---|---|---|---|
| μ-OR | δ-OR | κ-OR | NOP | ||||
| Met5-enkephalin | 261 | 9.9 | - | - | agonist δOR | EOP | Stefanucci [52] |
| Leu5-enkephalin | 513 | 10.7 | - | - | agonist δOR | EOP | Stefanucci [52] |
| β-Endorphin | 2.1 | 2.4 | 96 | - | agonist μOR, κOR | EOP | Corbett [53] |
| Dinorphin A | 1.6 | 1.25 | 0.05 | 386 | agonist κOR | EOP | Zhang [54] |
| Nociceptin | 437 | 2846 | 147 | 0.08 | agonist | EOP | Cami-Cobeci [50] |
| Morphine | 2.06 | >10,000 | 134 | >10,000 | agonist μOR | EM | Valenzano [55] |
| Oxycodone | 16 | 7680 | 43,000 | - | agonist μOR | EM | Miyazaki [56] |
| Naltrexone | 0.62 | 12.3 | 1.88 | - | antagonist | EM | Zheng [57] |
| Carfentanil | 0.07 | - | - | - | agonist μOR | 4-AP | Henriksen [58] |
| Carfentanila | 0.051 | 4.7 | 13 | - | agonist μOR | 4-AP | Frost [59] |
| Carfentanilb | 0.024 | 3.28 | 43.1 | - | agonist μOR | 4-AP | Cometta [60] |
| Cyclofoxya | 2.62 | 89 | 9.3 | - | antagonist μOR, κOR | EM | Rothman [38] |
| DPN | 0.07 | 0.23 | 0.02 | - | antagonist | orvinol | Raynor [61] |
| BPN | 1.5 | 6.1 | 2.5 | 77.4 | partial μOR agonist, κOR antagonist | orvinol | Cami-Cobeci [50] |
| PEO | 0.18 | 5.1 | 0.12 | - | full agonist | orvinol | Marton [62] |
| FE-DPN | 0.24 | 8.00 | 0.20 | - | antagonist | orvinol | Schoultz [63] |
| FE-BPN | 0.24 | 2.10 | 0.12 | - | mixed agonist/antagonist | orvinol | Schoultz [63] |
| FE-PEO | 0.10 | 0.49 | 0.08 | - | full agonist | orvinol | Schoultz [63] |
| NTIb | 3.8 | 0.03 | 332 | - | antagonist δOR | EM | Portoghese [64] |
| MeNTIb | 14 | 0.02 | 65 | - | antagonist δOR | EM | Portoghese [64] |
| GR103545 | 16.2 | 536 | 0.02 | - | agonist κOR | ArAP | Schoultz [65] |
| LY2459989a | 7.68 | 91.3 | 0.18 | - | antagonist κOR | APPB | Zheng [66] |
| LY2795050 | 25.8 | 153 | 0.72 | - | antagonist κOR | APPB | Zheng [57] |
| FEKAP | 7.4 | 139 | 0.43 | - | agonist κOR | ArAP | Li [67] |
| EKAP | 8.6 | 386 | 0.28 | - | agonist | ArAP | Li [68] |
| MeJDTic | 8.88 | 118 | 1.01 | - | antagonist κOR | JDTic | Poisnel [69] |
| Salvinorin A | >1000 | 5790 | 1.9 | - | agonist κOR | NND | Harding [70] |
| NOP-1A | - | - | - | 0.15 | antagonist NOP | FDPTP | Pike [71] |
| MK-0911 | 94 | - | - | 0.6 | antagonist NOP | SPB | Hostetler [72] |
| Condition | Ligand | Main Finding | Ref. |
|---|---|---|---|
| Healthy aging | [11C]Caf (8) (µOR) | 20% decrease in frontal cortex (females) | [137] |
| Epilepsy | [11C]Caf (8) (µOR) | Increased in ipsilateral temporal lobe, decreased in amygdala | [140] |
| Epilepsy | [11C]DPN (15) (mixed ligand) | No change | [140] |
| Parkinson’s disease | [11C]DPN (15) (mixed ligand) | 20–30% decrease in striatum and thalamus only in those patients with iatrogenic DOPA-dyskinesia | [147] |
| Huntington’s disease | [11C]DPN (15) (mixed ligand) | 30% reduced in caudate/putamen | [150] |
| Pontine infarct central pain | [11C]DPN (15) (mixed ligand) | Reduced throughout pain network | [151] |
| Capsaicin-induced acute pain | [11C]Caf (8) (µOR) | Up to 50% decrease contralateral thalamus, in proportion to subjective severity | [155] |
| Sustained painful stimulus of the jaw muscle with saline injection | [11C]Caf (8) (µOR) | Blateral decrease in binding in the ipsilateral amygdala (5%) and contralateral ventro-lateral thalamus (7%) | [157] |
| painful heat | [11C]Caf (8) (µOR) | Placebo effect on binding changes | [170] |
| Acupuncture therapy with sham acupuncture control | [11C]Caf (8) (µOR) | Persistent 10–30% increases in µ-OR binding in pain-related brain regions | [173] |
| Harm avoidance trait in healthy males | [18F]FE-DPN (16) (mixed ligand) | Trait correlated positively with binding in vental striatum, suggesting link with substance abuse | [181] |
| Correlation with BOLD signal responses A large group of healthy women, binding. to viewing emotionally arousing scenes | [11C]Caf (8) (µOR) | Negative correlation in amygdala, hippocampus, thalamus, and hypothalamus | [185] |
| Major depressive disorder | [11C]GR103545 (25) (κOR) | No difference from controls | [190] |
| Detoxified cocaine addicts | [11C]Caf (8) (µOR) | Increased in frontal and cingulate cortex, which correlated with the extent of craving | [195] |
| Detoxified alcohol-dependent subjects | [11C]MeNTI (22) (δOR) | Globally 10–20% increased binding inverse relationship in some regions with intensity of craving | [203] |
| Obesity (BMI > 40) | [11C]Caf (8) (µOR) | Globally 20% lower compared to lean volunteers | [218] |
| Feeding, regardless of the hedonic experience in non-obese subjects | [11C]Caf (8) (µOR) | Widespread decreases in binding | [222] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cumming, P.; Marton, J.; Lilius, T.O.; Olberg, D.E.; Rominger, A. A Survey of Molecular Imaging of Opioid Receptors. Molecules 2019, 24, 4190. https://doi.org/10.3390/molecules24224190
Cumming P, Marton J, Lilius TO, Olberg DE, Rominger A. A Survey of Molecular Imaging of Opioid Receptors. Molecules. 2019; 24(22):4190. https://doi.org/10.3390/molecules24224190
Chicago/Turabian StyleCumming, Paul, János Marton, Tuomas O. Lilius, Dag Erlend Olberg, and Axel Rominger. 2019. "A Survey of Molecular Imaging of Opioid Receptors" Molecules 24, no. 22: 4190. https://doi.org/10.3390/molecules24224190
APA StyleCumming, P., Marton, J., Lilius, T. O., Olberg, D. E., & Rominger, A. (2019). A Survey of Molecular Imaging of Opioid Receptors. Molecules, 24(22), 4190. https://doi.org/10.3390/molecules24224190