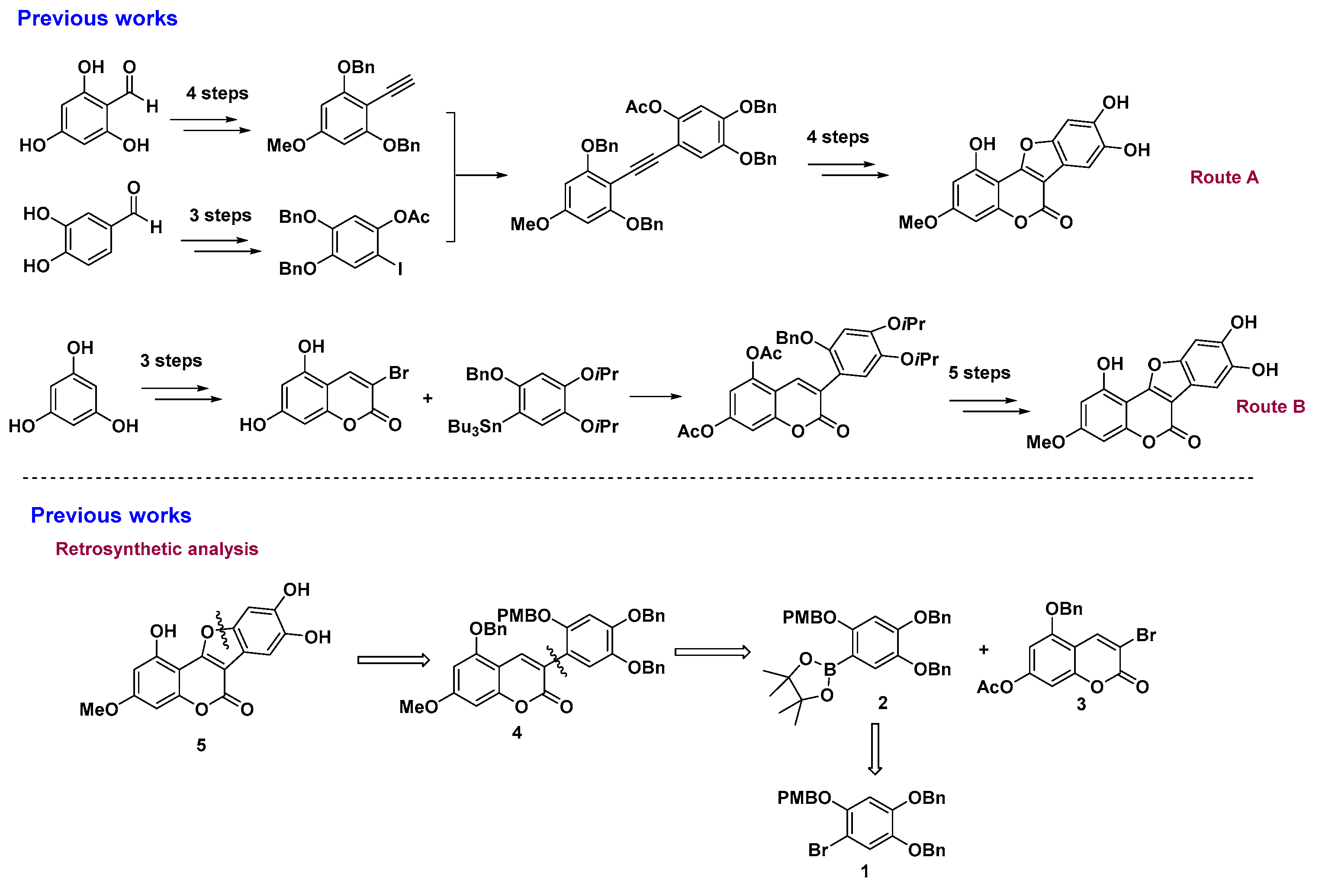
3.2. Chemical Synthesis
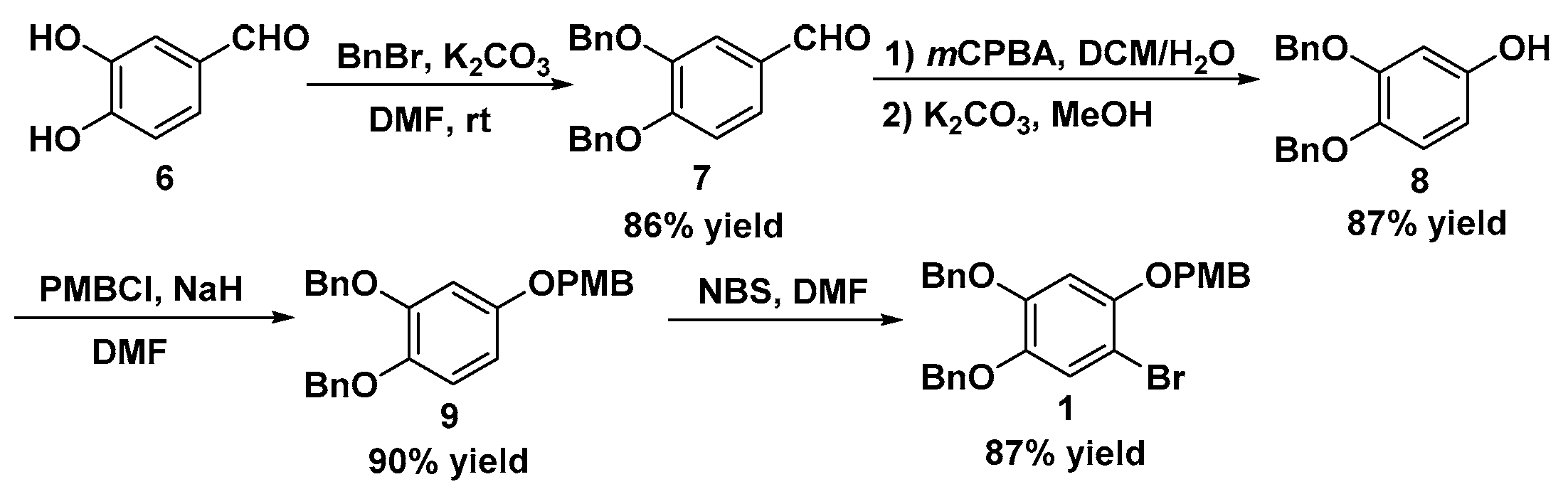
Preparation of 3,4-bis(benzyloxy)benzaldehyde 7: A 250-mL reaction vessel with a magnetic stirring bar was equipped with 3,4-dihydroxybenzaldehyde (5.13 g, 37.2 mmol), K2CO3 (25.70 g, 186.0 mmol), BnBr (19.0 g, 111.6 mmol), and DMF (100 mL). The mixture was stirred at room temperature for 4 h. After the reaction was completed, water (500 mL) was added, and the mixture was extracted with ethyl acetate (500 mL). The organic layer was washed with water (250 mL × 2) and brine (250 mL). After drying over MgSO4, the extracts were filtered and concentrated. The residue was firstly stirred in hexane (200 mL) overnight and then filtered to afford the intermediate 7 without any purification for the next step. White solid; 10.17 g, 86% yield; melting point (m.p.) 89–90 °C; Rf: 0.43 (EA:hexane = 1:4); IR (cm−1): 3026, 2819, 2726, 1676, 1596, 1580, 1512, 1435, 1396, 1386, 1349, 1269, 1245, 1231, 1211, 1135, 1021; 1H-NMR (CDCl3, 300 MHz): 5.24 (s, 2H), 5.28 (s, 2H), 7.05 (d, J = 8.1 Hz, 1H), 7.34–7.52 (m, J = 12.0 Hz, 12 Hz), 9.84 (s, 1H); 13C-NMR (CDCl3, 75 MHz): 70.4, 70.5, 112.0, 112.7, 126.2, 126.6, 126.8, 127.5, 127.6, 128.1, 128.2, 129.9, 135.8, 136.1, 148.8, 153.9, 190.3 ppm; HR-MS (ESI) calculated for C21H19O3 [M + H] 319.1334, found 319.1330.
Preparation of 3,4-bis(benzyloxy)phenol 8: To a solution of compound 7 (8.60 g, 27.0 mmol) in DCM (135 mL) was added m-chloroperbenzoic acid (7.00 g, 40.5 mmol), and then the mixture was stirred at room temperature for 15 h. The reaction was quenched with saturated aqueous Na2S2O3 solution (25 mL) and saturated aqueous Na2CO3 solution (125 mL). The mixture was firstly extracted with DCM (125 mL) and then successively washed with saturated aqueous Na2CO3 solution (125 mL × 2) and brine (75 mL). After drying over Na2SO4, the solvent was removed under vacuum. Next, the residue was firstly dissolved in MeOH (135 mL) and then treated with K2CO3 (4.10 g, 29.7 mmol). The mixture was stirred at room temperature for 30 min. After the reaction was completed, the solvent was removed under vacuum, and water (100 mL) was added. HCl solution (3 M) was added to adjust the pH to 3. The mixture was extracted with ethyl acetate (250 mL × 2). The combined organic layer was washed with brine (200 mL) and dried over Na2SO4. Finally, the extracts were filtered and concentrated to afford the intermediate 8 without any purification for the next step. White solid; 7.19 g, 87% yield; m.p. 105–106 °C; Rf: 0.25 (EA:hexane = 1:4); IR (cm−1): 3031, 2926, 1608, 1508, 1453, 1435, 1281, 1269, 1245, 1165, 1003; 1H-NMR (CDCl3, 300 MHz): 5.09 (s, 2H), 5.11 (s, 2H), 6.30–6.33 (m, 1H), 6.51 (s, 1H), 6.79–6.83 (m, 1H), 7.28–7.44 (m, 10H); 13C-NMR (CDCl3, 75 MHz): 70.6, 72.4, 102.9, 106.5, 117.2, 126.8, 127.2, 127.3, 127.4, 127.9, 128.0, 136.5, 137.1, 142.3, 149.8, 150.4 ppm; HR-MS (ESI) calculated for C20H19O3 [M + H] 307.1334, found 307.1336.
Preparation of 1,2-dibenzyloxy-4-(p-methoxybenzyl)oxybenzene 9: To a solution of compound 8 (6.64 g, 21.7 mmol) in DMF (20 mL) was added NaH (60%, dispersion in paraffin liquid) (1.05 g, 26.1 mmol) at 0 °C. The mixture was stirred for 30 min, and PMBCl (3.22 mL, 23.9 mmol) was added. The mixture was continually stirred at room temperature. After the reaction was finished according to TLC monitoring, the saturated NH4Cl solution (100 mL) was added, and the mixture was extracted with ethyl acetate (100 mL × 2). The organic layer was then washed with water (100 mL) and brine (50 mL) and dried over Na2SO4. The filtrate was concentrated, and the residue was stirred in hexane (75 mL) overnight. The mixture was filtered to provide the compound 9 without any purification for the next step. White solid; 8.32 g, 90% yield; m.p. 80–81 °C; Rf: 0.62 (EA:hexane = 1:4); IR (cm−1): 3062, 3035, 2914, 2867, 1610, 1589, 1515, 1468, 1418, 1392, 1380, 1272, 1228, 1171, 1116, 1004; 1H-NMR (CDCl3, 300 MHz): 3.84 (s, 3H), 4.92 (s, 2H), 5.12 (s, 2H), 5.15 (s, 2H), 6.47–6.51 (m, 1H), 6.68 (d, J = 8.7 Hz, 1H), 6.88 (d, J = 8.7 Hz, 1H), 6.93 (s, 1H), 6.95 (s, 1H), 7.33–7.46 (m, 12H); 13C-NMR (CDCl3, 75 MHz): 54.8, 69.8, 70.6, 72.2, 103.4, 105.2, 113.5, 116.6, 126.9, 127.1, 127.2, 127.3, 127.9, 128.0, 128.6, 128.8, 136.6, 137.2, 142.7, 149.7, 153.7, 159.0 ppm; HR-MS (ESI) calculated for C28H27O4 [M + H] 427.1909, found 427.1909.
Preparation of 1,2-dibenzyloxy-4-bromo-5-(p-methoxybenzyl)oxybenzene 1: To a solution of compound 8 (7.32 g, 17.2 mmol) in DMF (185 mL) was added the solution of NBS (3.21 g, 18.0 mmol) in DMF (35 mL) dropwise at 0 °C. After stirring for 30 min, the mixture was warmed to room temperature and stirred for additional 4 h. When the reaction was completed, ice water (500 mL) was added and a white solid appeared. After the suspension was filtered, the residue was washed with ice water, and dried under reduced pressure to give the intermediate 1. White solid; 7.54 g, 87% yield; m.p. 103–104 °C; Rf: 0.62 (EA:hexane = 1:4); IR (cm−1): 3058, 3012, 2910, 1589, 1520, 1420, 1395, 1375, 1225, 1180, 1123, 1001; 1H-NMR (CDCl3, 300 MHz): 3.84 (s, 3H), 4.96 (s, 2H), 5.09 (s, 2H), 5.10 (s, 2H), 6.63 (s, 1H), 6.92 (d, J = 8.7 Hz, 2H), 7.17 (s, 1H), 7.33–7.46 (m, 12H); 13C-NMR (CDCl3, 75 MHz): 54.8, 71.4, 71.5, 72.1, 103.0, 104.4, 113.5, 120.1, 126.9, 127.1, 127.4, 127.5, 128.0, 128.1, 128.2, 128.5, 136.3, 136.5, 143.7, 148.5, 149.5, 158.9 ppm; HR-MS (ESI) calculated for C28H25BrKO4 [M + K] 543.0573, found 543.0559.
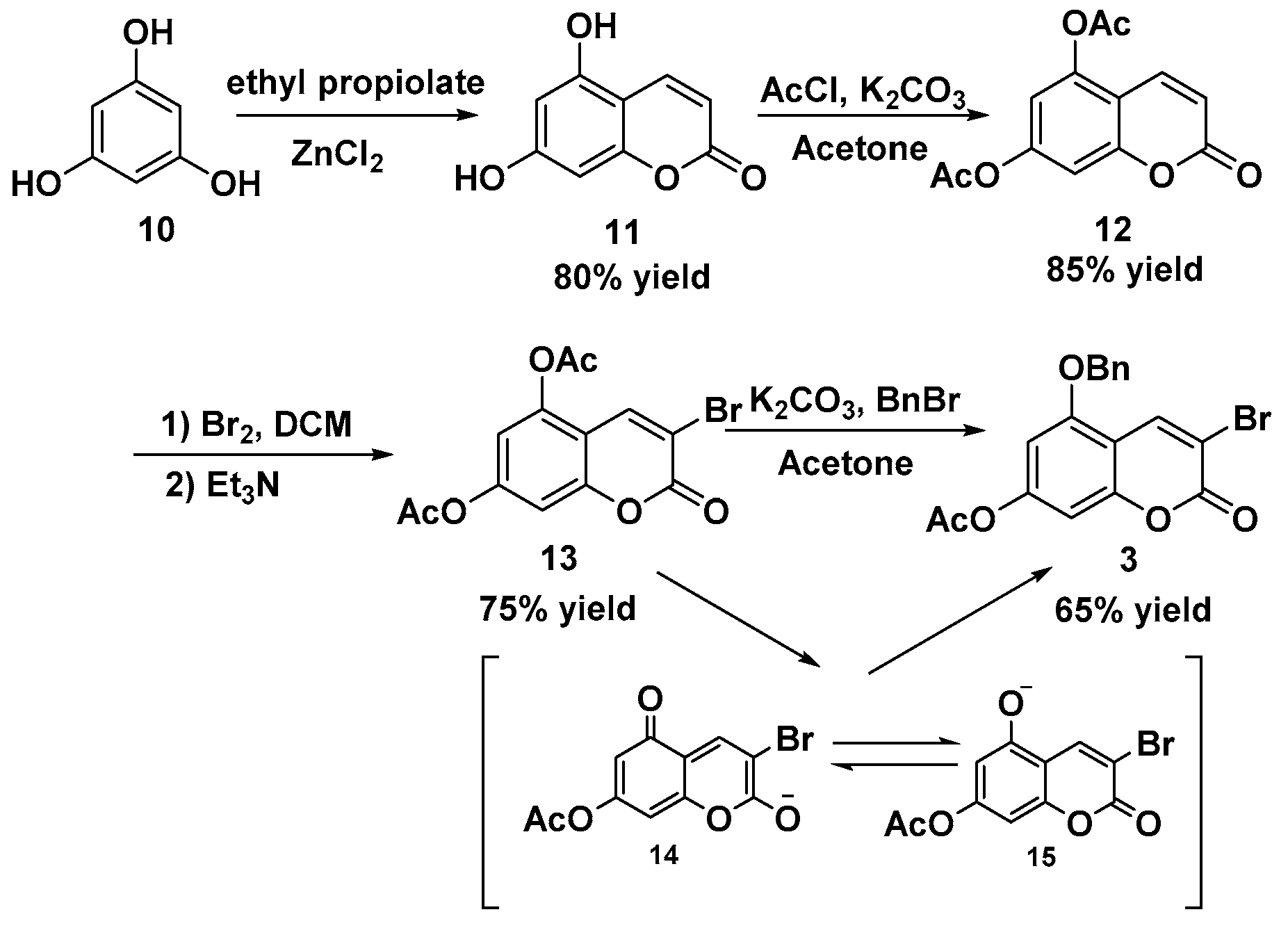
Preparation of 5,7-dihydroxy-2-chromenone 11: A stirred mixture of phloroglucinol dihydrate (18.90 g, 150.0 mmol), ethyl propiolate (17.7 mL, 180.0 mmol), and ZnCl2 (1.00 g, 7.5 mmol) was heated at 100 °C for 2 h. A flocculent precipitate formed during the reaction. After the mixture was filtered, the solid was recrystallized from water to afford the intermediate 11. Yellow solid; 21.36 g, 80% yield; m.p. 263–264 °C; Rf: 0.50 (EA:hexane = 3:1); IR (cm−1): 3421, 3054, 1686, 1615, 1574, 1474, 1365, 1302, 1244, 1156, 1071; 1H-NMR (DMSO-d6, 300 MHz): 5.94 (d, J = 9.6 Hz, 1H), 6.09 (m, 1H), 6.17 (d, J = 2.1 Hz, 1H), 7.86 (dd, J = 5.7, 9.6 Hz, 1H), 10.28 (s, 1H), 10.56 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 94.0, 98.2, 101.6, 108.6, 139.5, 155.9, 156.4, 160.7, 162.0; HR-MS (ESI) calculated for C9H7O4 [M+H] 179.0344, found 179.0330.
Preparation of 5,7-diacetoxyl-2-chromenone 12: A mixture of 11 (5.00 g, 28.0 mmol) and K2CO3 (19.40 g, 140.0 mmol) in dry acetone was heated to reflux (oil bath, 90 °C) for 1 h. Acetyl chloride (8.0 mL, 112.3 mmol) was added dropwise with syringe. The reaction suspension was stirred for another 1.5 h at 90 °C. The resulting solution was cooled down and filtrated out of the excess K2CO3. The solvent was concentrated in vacuo, and the residue was recrystallized by MeOH to give compound 12. White solid; 6.24 g, 85% yield; m.p. 129–130 °C; Rf: 0.28 (EA:hexane = 1:2); IR (cm−1): 3075, 1767, 1745, 1630, 1575, 1433, 1365, 1309, 1242, 1188, 1131, 1066, 1027; 1H-NMR (DMSO-d6, 300 MHz): 2.31 (s, 3H), 2.40 (s, 3H), 6.51 (d, J = 9.6 Hz, 1H), 7.11 (d, J = 2.1 Hz, 1H), 7.24 (dd, J = 0.6, 2.1 Hz, 1H), 8.07 (dd, J = 0.6, 9.6 Hz, 1H); 13C-NMR (DMSO-d6, 75 MHz): 20.6, 20.8, 108.0, 110.6, 112.9, 115.9, 137.9, 147.4, 152.6, 154.3, 159.2, 168.5, 168.8 ppm; HR-MS (ESI) calculated for C13H11O6 [M + H] 263.0556, found 263.0538.
Preparation of 3-bromo-5,7-diacetoxyl-2-chromenone 13: To a solution of 12 (2.00 g, 7.6 mmol) in DCM (20 mL) was added Br2 (1.95 mL, 38.0 mmol) at 0 °C. The mixture was stirred at 10 °C for 8 h. The reaction was quenched with a 2 M NaOH aqueous solution (25 mL). The brown solution was extracted with DCM (50 mL). The organic layer was washed with water (25 mL × 2) and brine (25 mL) and then dried over Na2SO4. The filtrate was concentrated in vacuo to afford the dibromochromanone. To the above dibromide in DCM (25 mL) was added Et3N (1.33 g, 13.2 mmol) at room temperature, which was then stirred for 1 h. The resulting solution was removed from the solvent in vacuo, and the residue was recrystallized by MeOH to give the desired product 13. White solid; 1.94 g, 75% yield; m.p. 130–131 °C; Rf: 0.51(EA:hexane = 1:2); IR (cm−1): 3065, 1765, 1743, 1619, 1430, 1291, 1239, 1192, 1132, 1067, 1022; 1H-NMR (CDCl3, 300 MHz): 2.34 (s, 3H), 2.44 (s, 3H), 7.04–7.06 (m, 2H), 8.11 (s, 1H); 13C-NMR (CDCl3, 75 MHz): 20.4, 20.6, 107.3, 110.4, 111.1, 112.2, 137.6, 145.9, 152.4, 153.4, 167.5, 167.6 ppm; HR-MS (ESI) calculated for C13H9BrNaO6 [M + Na] 362.9480, found 362.9476.
Preparation of 3-bromo-5-benzyloxy-7-acetoxyl-2-chromenone 3: To a solution of 13 (1.53 g, 4.5 mmol) and K2CO3 (1.88 g, 13.5 mmol) in acetone (30 mL) was added BnBr (0.81 mL, 6.8 mmol), and then the mixture was stirred at 56 °C overnight. The suspension was concentrated in vacuo, and water (75 mL) was added. The resulting solution was extracted with ethyl acetate (100 mL × 2). The combined organic layer was washed with brine (100 mL) and dried over Na2SO4. After the mixture was filtered, the filtrate was removed from the solvent and the residue was recrystallized by EtOH to give the desired product 3. White solid; 1.13 g, 65% yield; m.p. 176–177 °C; Rf: 0.60 (EA:hexane = 1:2); IR (cm−1): 3091, 2924, 2853, 1737, 1615, 1499, 1459, 1430, 1351, 1296, 1234, 1208, 1131, 1028; 1H-NMR (DMSO-d6, 300 MHz): 2.31 (s, 3H), 5.25 (s, 2H), 6.93 (d, J = 1.3 Hz, 1H), 6.98 (d, J = 1.6 Hz, 1H), 7.36–7.45 (m, 3H), 7.51–7.54 (m, 2H), 8.39 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 20.8, 70.6, 102.8, 102.9, 107.7, 108.7, 127.8, 128.2, 128.5, 135.8, 138.9, 153.9, 154.0, 154.6, 156.3, 168.5 ppm; HR-MS (ESI) calculated for C18H14BrO5 [M + H] 389.0025, found 389.0022.
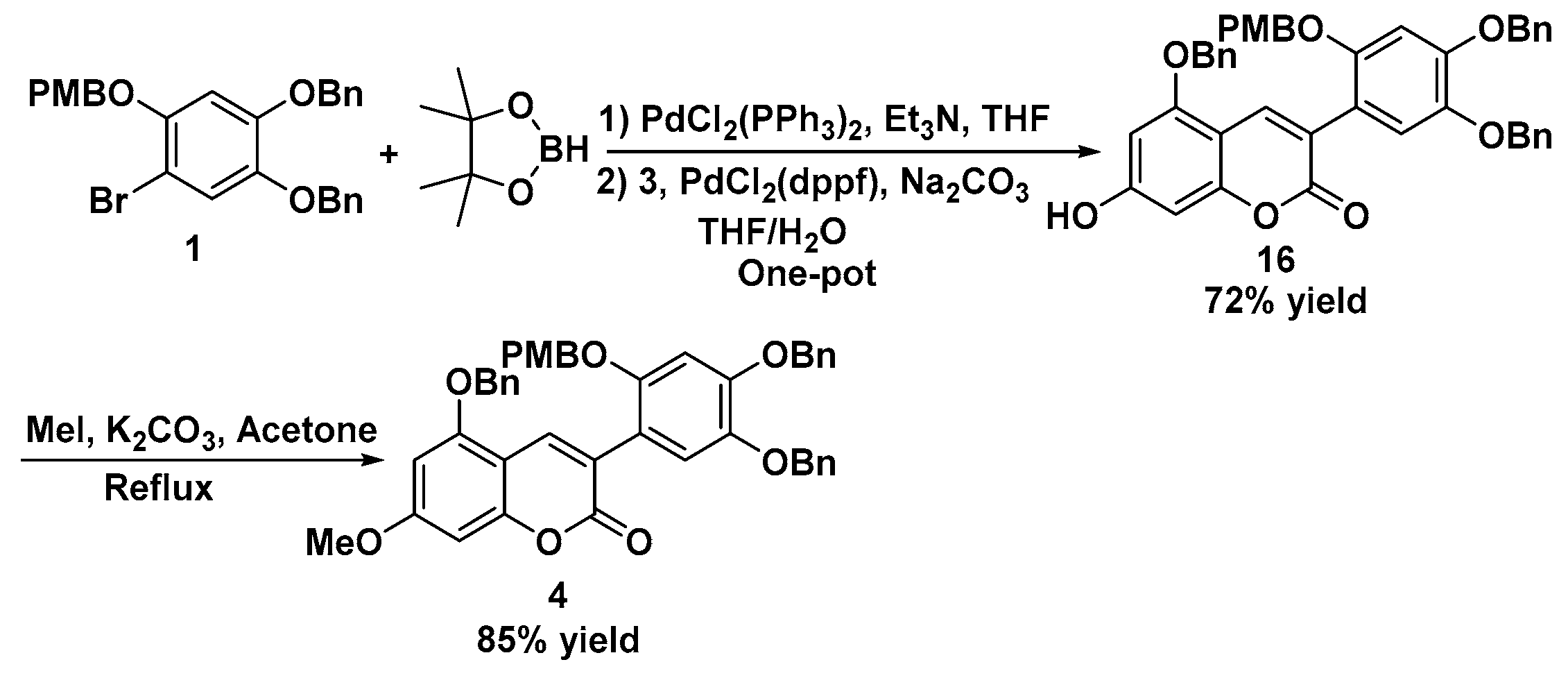
Preparation of 5-(benzyloxy)-3-(4,5-bis(benzyloxy)-2-((4-methoxybenzyl)oxy)phenyl)-7-hydroxy-2-chromenone 16: To a solution of 1 (6.06 g, 12.0 mmol) and Pd(PPh3)2Cl2 (420 mg, 0.6 mmol, 5 mol%) in THF (120 mL) was added TEA (14.4 mL, 103.0 mmol, 8.6 equivalents) and pinacoborane (13.0 mL, 90.0 mmol, 7.5 equivalents) under Ar atmosphere. The mixture was heated overnight at 80 °C. After being cooled, 3 (3.18 g, 8.0 mmol), Pd(dppf)Cl2 (293 mg, 0.4 mmol, 5 mol.%), and a solution of Na2CO3 (8.90 g, 84.0 mmol) in H2O (30 mL) was added under Ar atmosphere. The mixture was stirred at 90 °C for another 12 h, and a 3 M HCl solution was added to adjust the pH to 3. The mixture was filtered with diatomaceous earth, and the filtrate was extracted with ethyl acetate (200 mL × 3). The combined organic layer was washed with brine (200 mL) and then dried over Na2SO4. After drying and concentrating, the residue was purified by column chromatography with ethyl acetate/petroleum ether (1:3–1:1) to afford the desired product 16. Yellow solid; 3.98 g, 72% yield; m.p. 166–167 °C; Rf: 0.32 (EA:hexane = 1:2); IR (cm−1): 3425, 3012, 2988, 1725, 1613, 1520, 1443, 1400, 1387, 1312, 1214, 1189, 1102, 1043, 1004; 1H-NMR (DMSO-d6, 300 MHz): 3.69 (s, 3H), 4.95 (s, 2H), 5.03 (s, 2H), 5.19 (s, 2H), 5.20 (s, 2H), 6.37 (s, 1H), 6.46 (s, 1H), 6.77 (s, 1H), 6.80 (s, 1H), 6.98 (s, 1H), 7.09 (s, 1H), 7.20–7.46 (m, 17H), 7.88 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 55.0, 70.0, 70.4, 71.4, 93.7, 94.8, 96.6, 98.0, 102.2, 102.5, 113.6, 116.9, 118.5, 127.5, 127.6, 127.7, 127.8, 128.0, 128.2, 128.4, 128.5, 128.9, 129.1, 136.3, 136.6, 137.1, 137.4, 141.8, 149.2, 150.8, 155.5, 155.9, 158.8, 159.8, 161.8 ppm; HR-MS (ESI) calculated for C44H37O8 [M + H] 693.2488, found 693.2484.
Preparation of 5-(benzyloxy)-3-(4,5-bis(benzyloxy)-2-((4-methoxybenzyl)oxy)phenyl)-7-methoxy-2-chromenone 4: To a solution of 16 (1.38 g, 2.0 mmol) and K2CO3 (0.42 g, 3.0 mmol) in DMF (10 mL) was added MeI (250 uL, 4.0 mmol) slowly, and then the mixture was stirred at the room temperature. After TLC showed the reaction was completed, the mixture was diluted with ethyl acetate (50 mL), and the organic layer was washed with H2O (40 mL × 2) and brine (25 mL). After drying over Na2SO4, the filtrate was concentrated, and the residue was purified by column chromatography with ethyl acetate/petroleum ether (1:3–1:2) to afford the desired product 4. Yellow solid; 1.20 g, 85% yield; m.p. 130–132 °C; Rf: 0.46 (EA:hexane = 1:2); IR (cm−1): 3031, 3005, 2927, 2835, 1719, 1610, 1513, 1445, 1413, 1385, 1300, 1250, 1197, 1155, 1109, 1082, 1015; 1H-NMR (DMSO-d6, 300 MHz): 3.69 (s, 3H), 3.85 (s, 3H), 4.95 (s, 2H), 5.04 (s, 2H), 5.20 (s, 2H), 5.25 (s, 2H), 6.63 (d, J = 0.9 Hz, 2H), 6.76 (s, 1H), 6.79 (s, 1H), 7.00 (s, 1H), 7.11 (s, 1H), 7.20 (s, 1H), 7.23 (s, 1H), 7.30–7.44 (m, 11H), 7.46–7.49 (m, 4H), 7.90 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 55.0, 56.0, 70.2, 70.3, 70.4, 71.4, 93.0, 96.3, 102.2, 103.6, 113.6, 116.7, 118.4, 119.6, 127.6, 127.7, 127.8, 128.0, 128.2, 128.4, 128.5, 128.8, 129.1, 136.2, 136.4, 137.1, 137.4, 141.8, 149.4, 150.9, 155.5, 155.6, 158.8, 159.6, 163.0; HR-MS (ESI) calculated for C45H39O8 [M + H] 707.2645, found 707.2648.
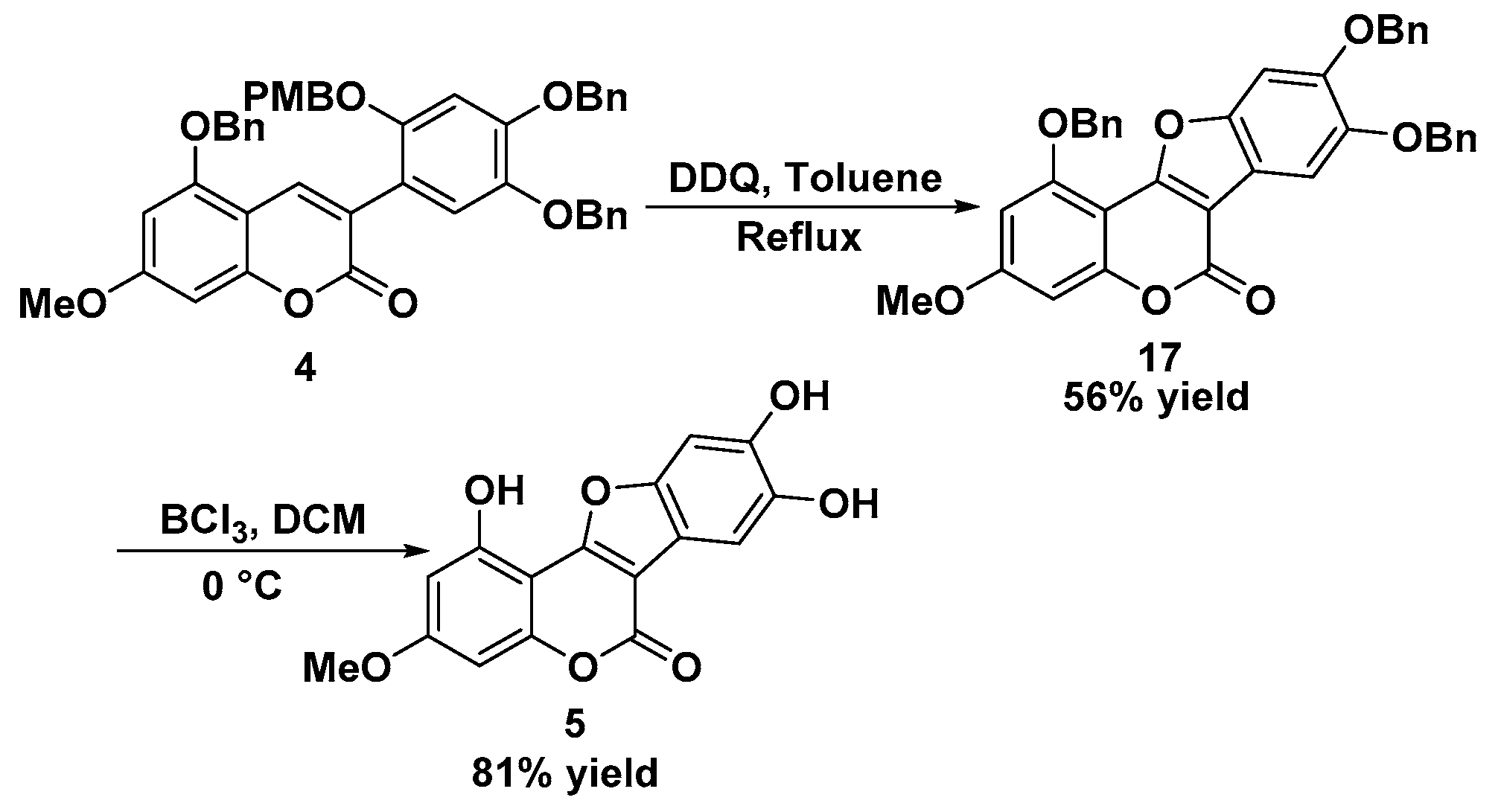
Preparation of 1,8,9-tris(benzyloxy)-3-methoxy-6H-benzofuro[3,2-c]chromen-6-one 17: To a solution of 4 (0.71 g, 1.0 mmol) in toluene (20 mL) was added DDQ (0.45 g, 2.0 mmol), and then the mixture was stirred under reflux for 24 h. After the reaction was finished, the mixture was concentrated and then purified by column chromatography (DCM) to give the product 17. White solid; 327 mg, 56% yield; m.p. 212–213 °C; Rf: 0.40 (EA:hexane = 1:2); IR (cm−1): 3031, 2930, 2850, 1736, 1604, 1450, 1401, 1352, 1301, 1273, 1197, 1161, 1137, 1081, 1049; 1H-NMR (CDCl3, 300 MHz): 3.88 (s, 3H), 5.25 (s, 2H), 5.27 (s, 2H), 5.31 (s, 2H), 6.48 (d, J = 2.1 Hz, 1H), 6.63 (d, J = 2.1 Hz, 1H), 7.20 (s, 1H), 7.37–7.63 (m, 15H), 7.68 (s, 1H); 13C-NMR (CDCl3, 75 MHz): 55.8, 70.8, 71.8, 72.0, 94.1, 97.1, 99.4, 105.5, 116.2, 126.8, 127.3, 127.5, 127.9, 128.0, 128.1, 128.5, 128.6, 128.7, 136.2, 136.8, 137.0, 148.0, 148.5, 150.1, 155.4, 155.9, 158.5, 159.7, 162.8; HR-MS (ESI) calculated for C37H28NaO7 [M + Na] 607.1733, found 607.1718.
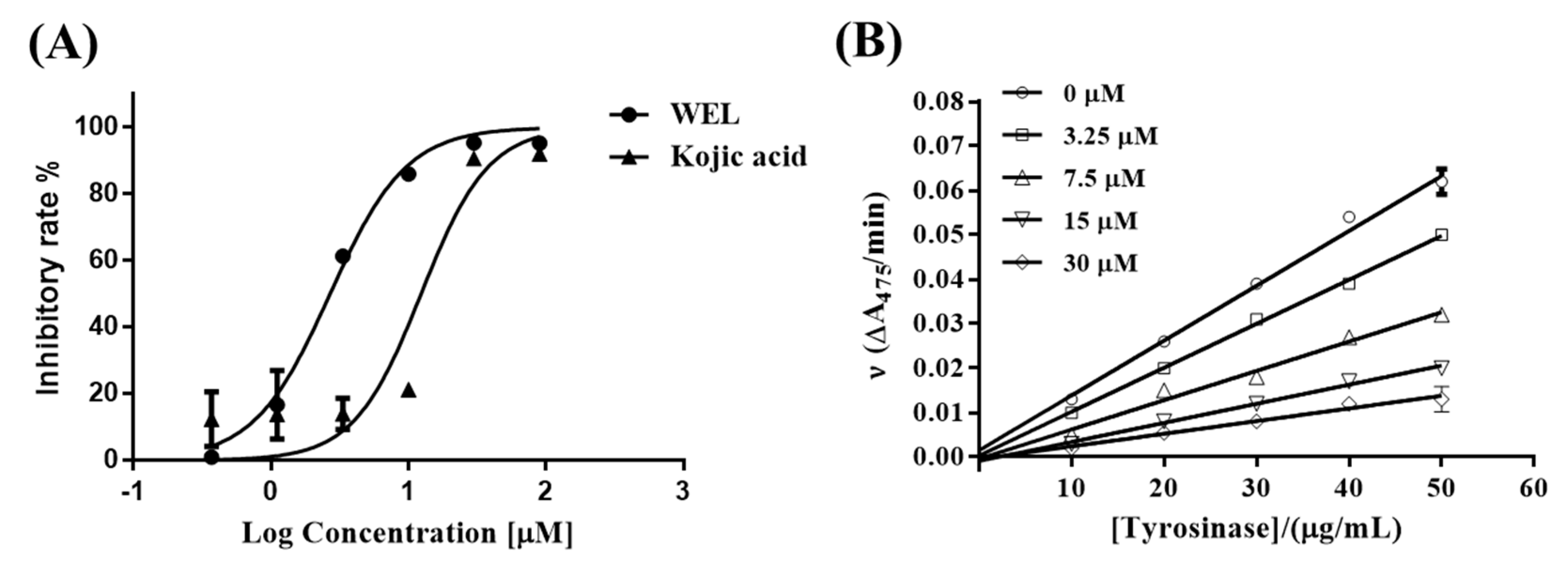
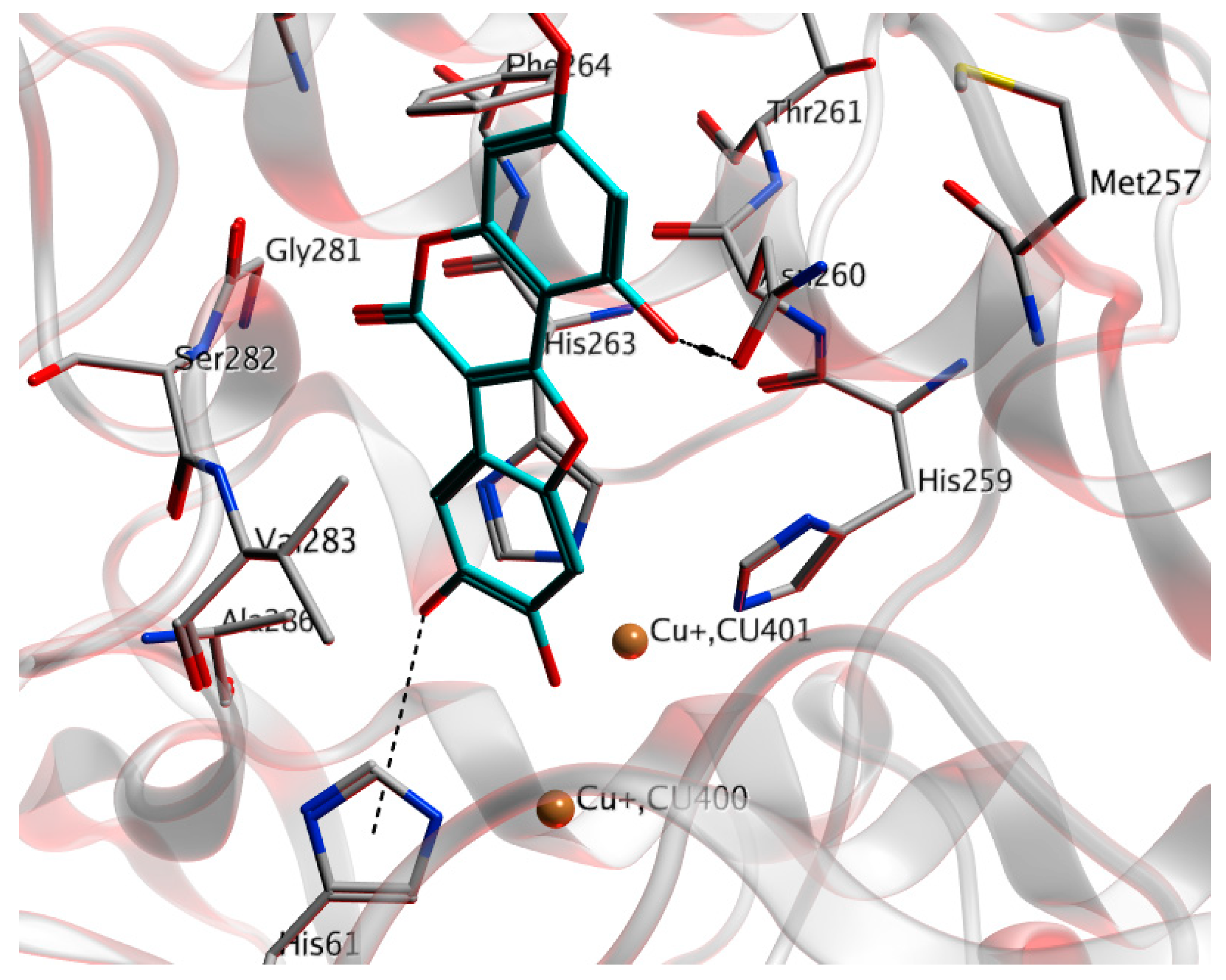
Preparation of WEL 5: To a solution of 17 (0.58 g, 1.0 mmol) in DCM (15 mL) was added BCl3 (3 mL, 1 M) slowly at 0 °C under Ar atmosphere. After the reaction was finished, the mixture was concentrated and then purified by column chromatography (MeOH:DCM = 1:20) to give the product 5. Gray solid; 254 mg, 81% yield; m.p. >300 °C; Rf: 0.48 (MeOH:DCM = 1:10); IR (cm−1): 3397, 2955, 2923, 2853, 1710, 1670, 1612, 1447, 1324, 1284, 1206, 1151, 1070, 1047; 1H-NMR (DMSO-d6, 300 MHz): 3.82 (s, 3H), 6.45 (d, J = 1.8 Hz, 1H), 6.62 (d, J = 1.9 Hz, 1H), 7.16 (s, 1H), 7.24 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 55.7, 93.2, 96.7, 98.1, 98.8, 101.7, 104.5, 113.7, 144.3, 145.4, 148.8, 154.8, 155.2, 157.7, 158.9, 162.2; HR-MS (ESI) calculated for C16H11O7 [M + H] 315.0505, found 315.0504.
Characterization data of WEL reported by Yang’s group were as follows: 1H-NMR (DMSO-d6, 300 MHz): 3.77 (s, 3H), 6.40 (d, J = 2.1 Hz, 1H), 6.55 (d, J = 1.8 Hz, 1H), 7.15 (s, 1H), 7.23 (s, 1H); 13C-NMR (DMSO-d6, 75 MHz): 55.7, 93.2, 96.7, 98.1, 98.9, 101.7, 104.6, 113.8, 144.3, 145.4, 148.9, 154.8, 155.3, 157.8, 158.9, 162.2.
All above chemicals were commercially available and used without further purification. Analytical thin-layer chromatography was performed on glass plates precoated with silica gel impregnated with a fluorescent indicator (254 nm). The plates were visualized by exposure to ultraviolet light. 1H-NMR spectra were recorded on a Bruker DRX (300 MHz), and 13C-NMR spectra were recorded on a Bruker DRX (75 MHz). HR-MS spectra were taken on an Agilent Technologies Accurate-Mass Q-TQF LC/MS 6520.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






