
Development and Validation of a Sensitive UHPLC-MS/MS Method for the Measurement of Gardneramine in Rat Plasma and Tissues and Its Application to Pharmacokinetics and Tissue Distribution Study

 and
and
Abstract
1. Introduction
2. Results and Discussion
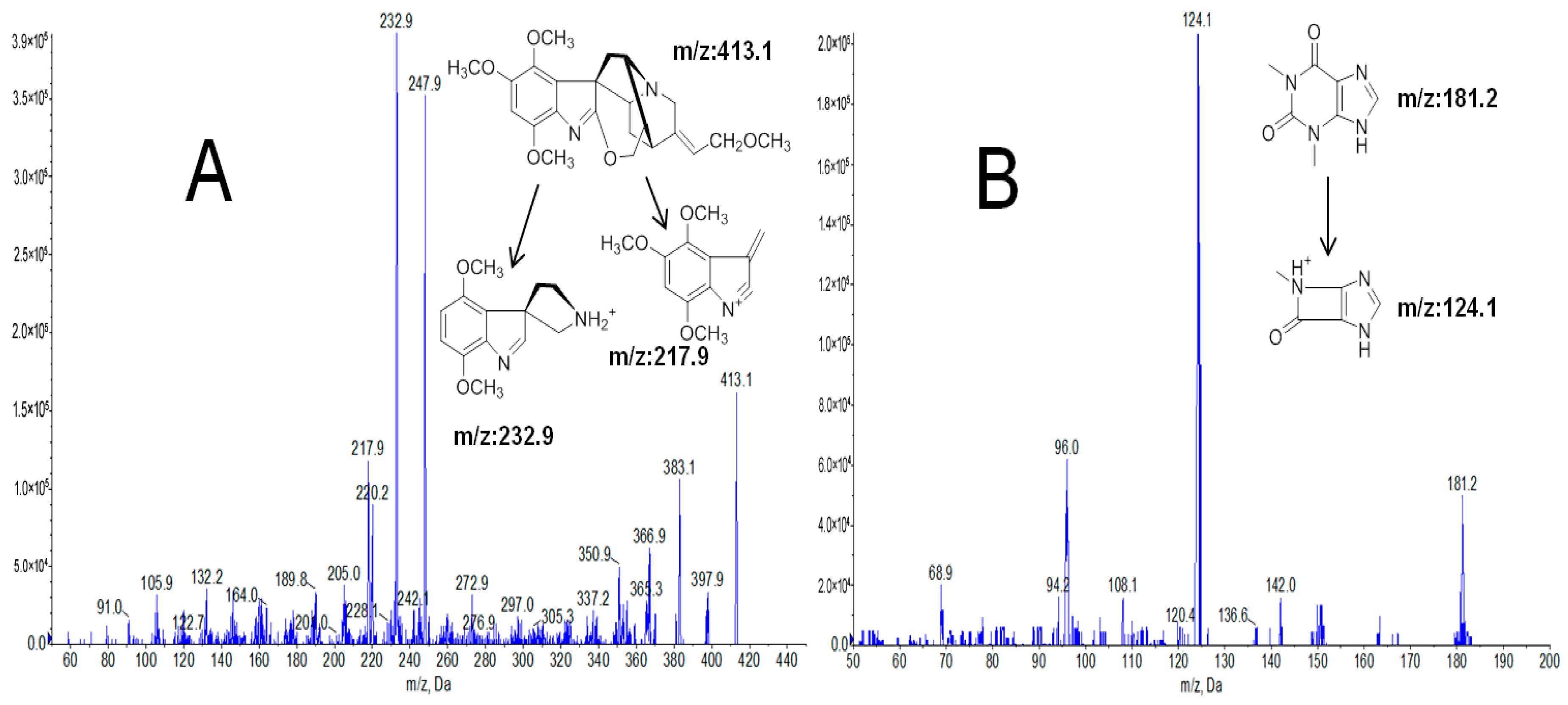
2.1. Optimization of LC-MS/MS Conditions
2.2. Optimization of the Extraction Method
2.3. Method Validation
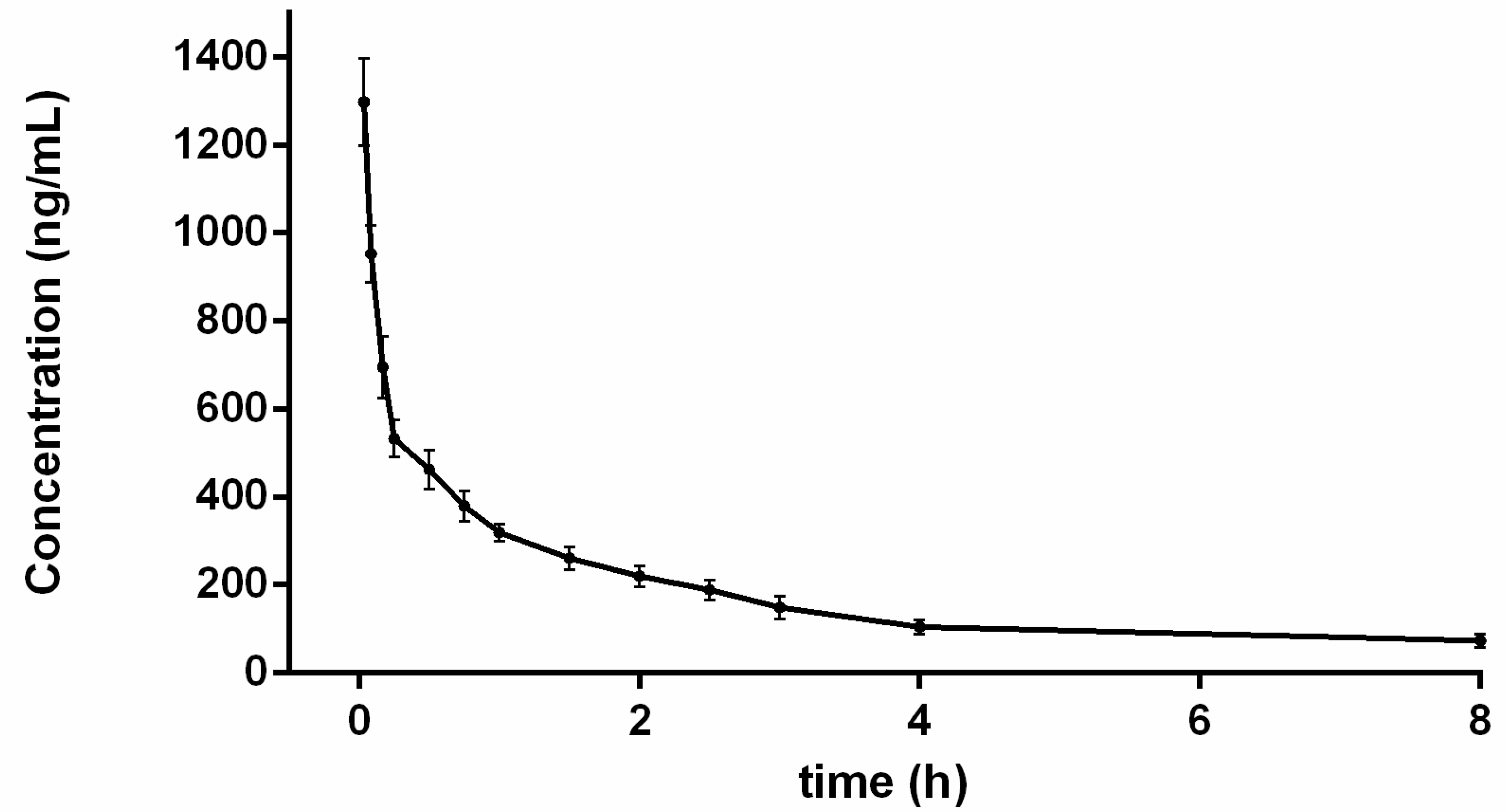
2.4. Pharmacokinetic Study
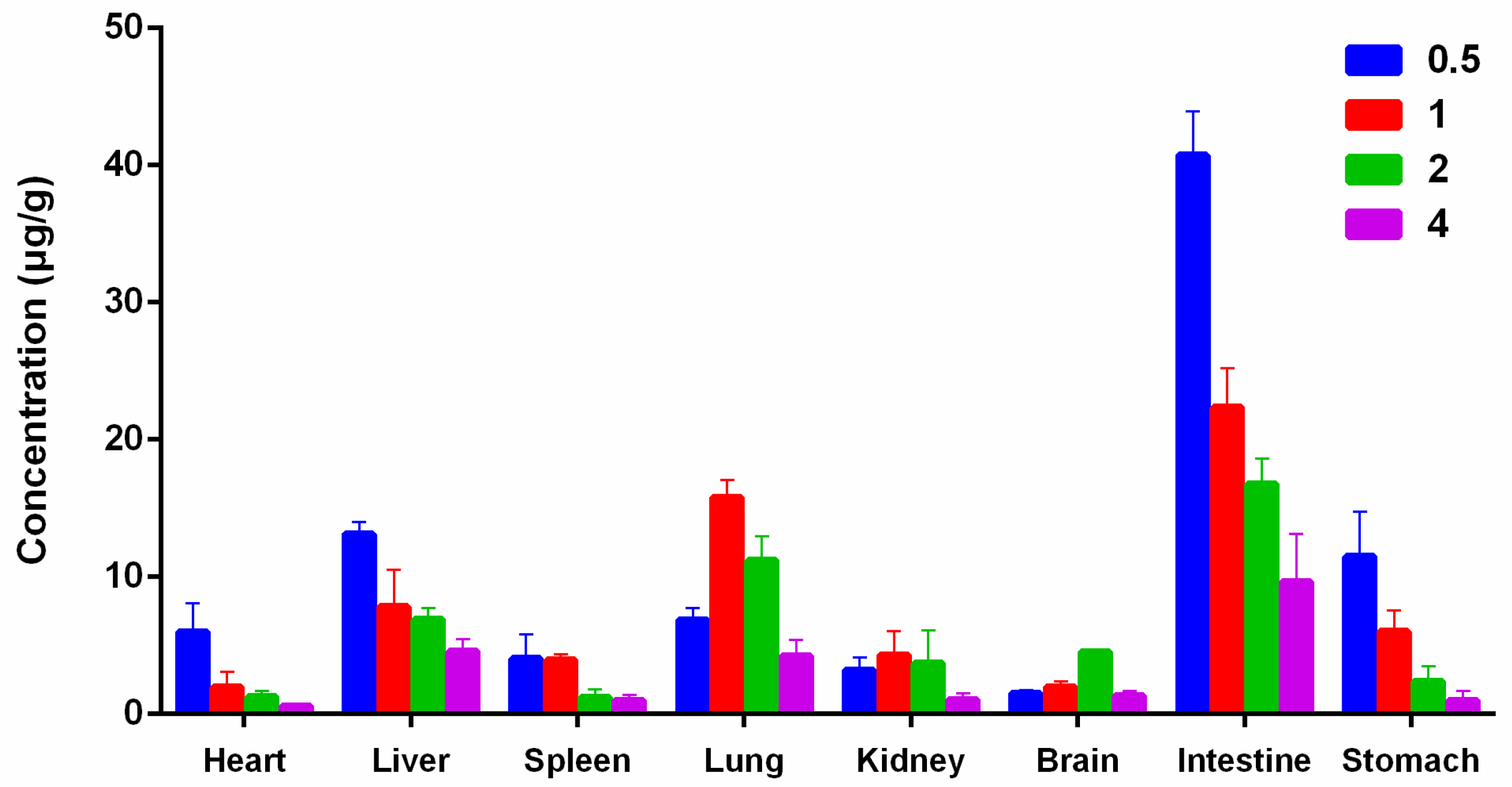
2.5. Tissue Distribution
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals
3.3. Instrumentation and Conditions
3.4. Preparation of Calibration Standards and Quality Control Samples
3.5. Sample Preparation
3.6. Method Validation
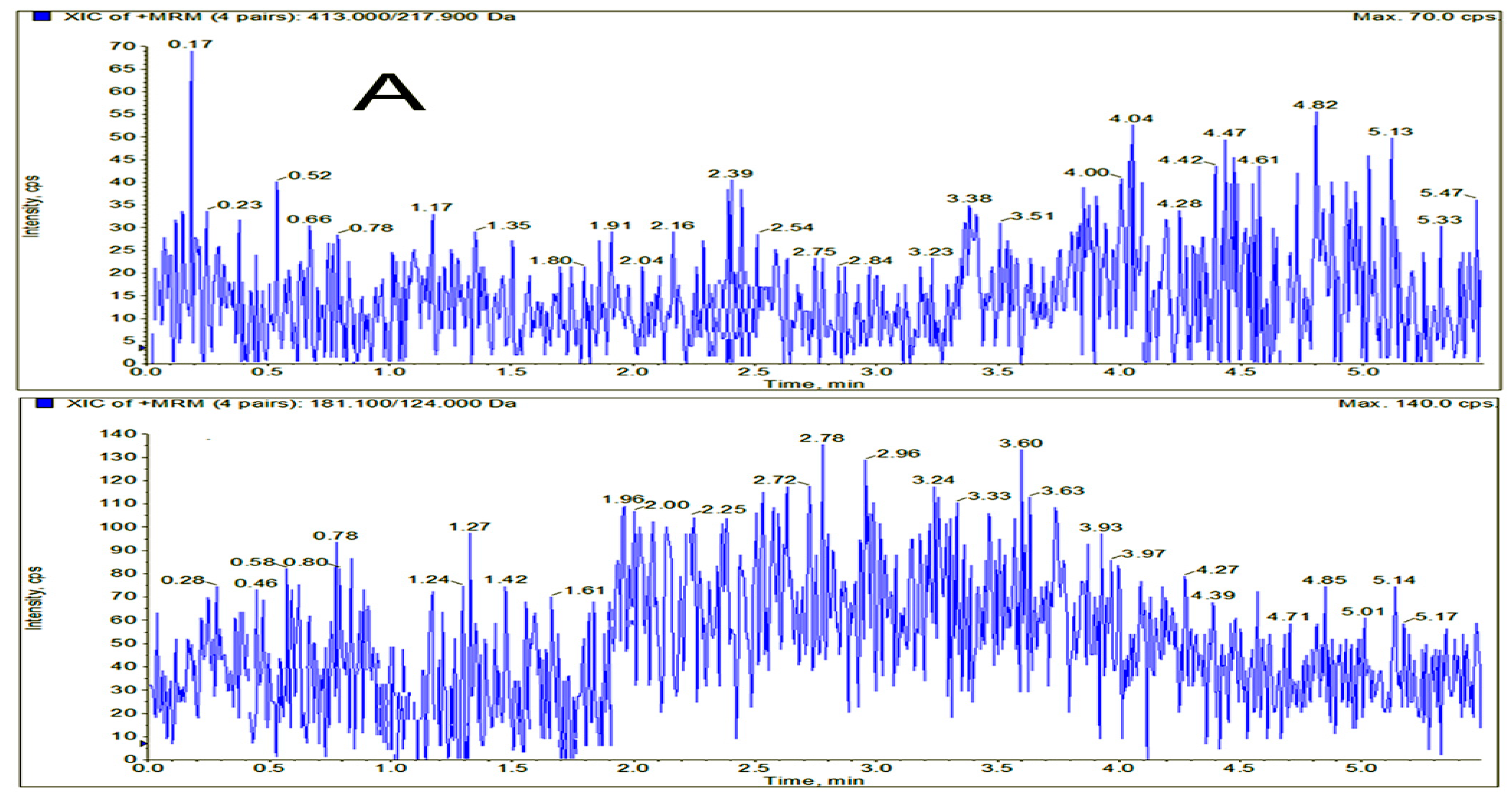
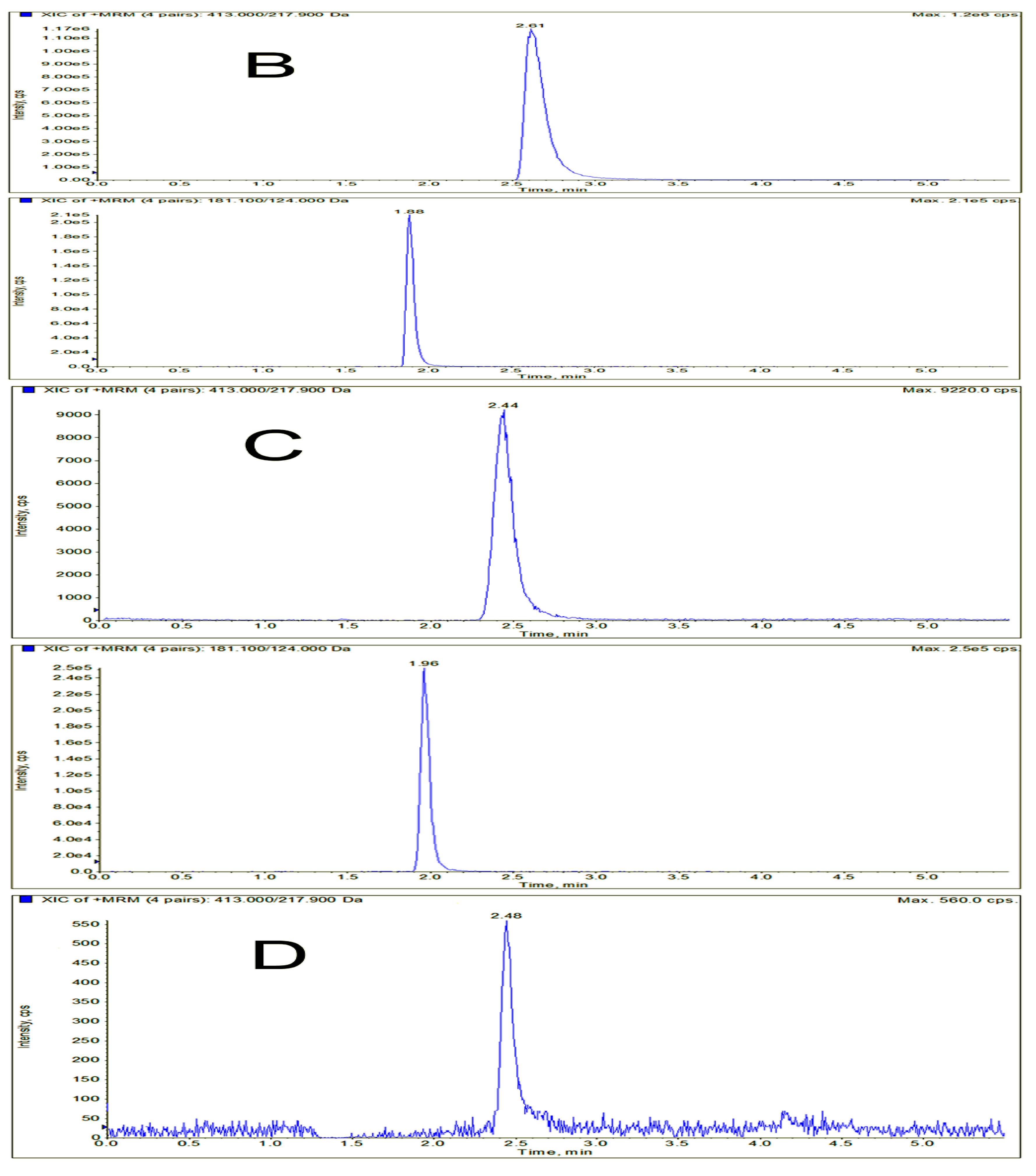
3.6.1. Selectivity
3.6.2. Linearity and Sensitivity
3.6.3. Precision and Accuracy
3.6.4. Extraction Recovery and Matrix Effects
3.6.5. Stability
3.6.6. Dilution Integrity
3.7. Pharmacokinetic Study
3.8. Tissue Distribution Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haginiwa, J.; Sakai, S.; Kubo, A.; Hamamoto, T. Alkaloids of Gardneria nutans sieb. et Zucc. Yakugaku Zasshi 1967, 87, 1484–1488. [Google Scholar] [CrossRef][Green Version]
- Sakai, S.; Aimi, N.; Yamaguchi, K.; Ohhira, H.; Hori, K.; Haginiwa, J. Gardneria alkaloids-Ⅸ structures of chitosenine and three other minor bases from Gardneria multiflora Makino. Tetrahedron Lett. 1975, 16, 715–718. [Google Scholar] [CrossRef]
- Li, X.N.; Cai, X.H.; Feng, T.; Li, Y.; Liu, Y.P.; Luo, X.D. Monoterpenoid indole alkaloids from Gardneria ovate. J. Nat. Prod. 2011, 74, 1073–1078. [Google Scholar] [CrossRef]
- Yang, W.X.; Chen, Y.F.; Yang, J.; Huang, T.; Wu, L.L.; Xiao, N.; Hao, X.J.; Zhang, Y.H. Monterpenoid indole alkaloids from Gardneria multiflora. Fitoterapia 2018, 124, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Haginiwa, J.; Sakai, S.; Kubo, A.; Takahashi, K.; Taguchi, M. Gardneria alkaloids-Ⅳ comparative study of alkaloids on Gardneria nutans sieb. et Zucc., G. multiflora Makino, G. shimadai hayata and so-called G. insularis nakai. Yakugaku Zasshi 1970, 90, 219–223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, X.M. The geographical distribution of the loganiaceae in China. J. South China Agric. Univ. 1995, 16, 92–97. [Google Scholar]
- Editorial Committee of Flora of China Chinese Academy of Sciences. Flora of China; Science Press: Beijing, China, 1999; Volume 61, p. 242. [Google Scholar]
- Wang, G.Q. National Compilation of Chinese Herbal Medicine, 3rd ed.; People’s Medical Publishing House: Beijing, China, 2014; Volume 3, p. 616. [Google Scholar]
- Frederich, M.; Jacquier, M.; Thepenier, P.; Mol, P.D.; Tits, M.; Genevieve, P.; Clement, D.; Luc, A.; Monique, Z. Antiplasmodial activity of alkaloids from various strychnos species. J. Nat. Prod. 2002, 65, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Sarah, E.O.C.; Justin, J.M. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar]
- Passos, C.D.S.; Soldi, T.C.; Abib, R.T.; Apel, M.A.; Claudia, S.P.; Marcourt, L.; Gottfried, C.; Henriques, A.T. Monoamine oxidase inhibition by monoterpene indole alkaloids and fractions obtained from Psychotria suterella and Psychotria laciniata. J. Enzyme Inhib. Med. Chem. 2013, 28, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Ozaki, Y.; Murayama, S.; Sakai, S.; Haginiwa, J. Pharmacological studies on Gardneria alkaloids.Ⅰ. central effects. Yakugaku Zasshi 1971, 91, 997–1003. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harada, M.; Ozaki, Y. Effect of indole alkaloids from Gardneria genus and Uncaria genus on neuromuscular transmission in the rat limb in situ. Chem. Pharm. Bull. 1976, 24, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Ozaki, Y. Pharmacological studies on Gardneria alkaloids.Ⅱ. peripheral effects (effects on circulatory and digestive systems). Yakugaku Zasshi 1972, 92, 1540–1546. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Ozaki, Y. Effect of Gardneria alkaloids on ganglionic transmission in the rabbit and rat superior cervical ganglia in situ. Chem. Pharm. Bull. 1978, 26, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Ozaki, Y.; Ohno, H. Effects of indole alkaloids from Gardneria nutans sieb. et Zucc. and Uncaria rhynchophylla Mio. on a guinea pig urinary bladder preparation in situ. Chem. Pharm. Bull. 1979, 27, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Harada, M. Site of the ganglion blocking action of gardneramine and hirsutine in the dog urinary bladder in situ preparation. Japan. J. Pharmacol. 1983, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behavior. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; Mccoy, K.D.; et al. The intestinal microbiota affect central levels of brain-derived neurotropic factor and behavior in mice. Gastroenterology 2011, 141, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.A.; Marcela, J.P.; Forsythe, P.; Kunze, W.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Communication between gastrointestinal bacteria and the nervous system. Curr. Opin. Pharmacol. 2012, 12, 667–672. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds gardneramine and theophylline are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Precursor Ion (m/z) | Product Ion (m/z) | Declustering Potential (V) | Collision Energy (eV) |
|---|---|---|---|---|
| gardneramine | 413.1 | 217.9 | 134.8 | 62.9 |
| 232.9 | 123.9 | 46 | ||
| I.S. | 181.2 | 124.1 | 70 | 26.6 |
| Samples | Calibration Curves | Correlation Coefficients (r) | Linear Ranges (ng/mL) | LLOQs (ng/mL) |
|---|---|---|---|---|
| Plasma | Y = 0.0720 + 0.18447x | 0.9924 | 1–2000 | 1 |
| Heart | Y = −0.0943 + 0.13776x | 0.9931 | 1–2000 | 1 |
| Liver | Y = 0.0836 + 0.19987x | 0.9906 | 1–2000 | 1 |
| Spleen | Y = −0.0286 + 0.20747x | 0.9913 | 1–2000 | 1 |
| Lung | Y = 0.0996 + 0.09197x | 0.9954 | 1–2000 | 1 |
| Kidney | Y = 0.0193 + 0.25430x | 0.9941 | 1–2000 | 1 |
| Brain | Y = −0.0749 + 0.14529x | 0.9917 | 1–2000 | 1 |
| Intestine | Y = 0.0974 + 0.19646x | 0.9962 | 1–2000 | 1 |
| Stomach | Y = 0.0432 + 0.37274x | 0.9901 | 1–2000 | 1 |
| Samples | Spiked Concentration (ng/mL) | Intraday | Interday | ||
|---|---|---|---|---|---|
| Precision (RSD, %) | Accuracy (Mean%) | Precision (RSD, %) | Accuracy (Mean%) | ||
| Plasma | 1 | 8.94 | 0.98 | 2.31 | −1.38 |
| 3 | 6.09 | 3.13 | 2.30 | 5.49 | |
| 45 | 4.89 | 1.33 | 6.27 | 4.29 | |
| 1600 | 5.80 | 3.65 | 3.47 | 4.89 | |
| Heart | 1 | 3.05 | 1.02 | 4.03 | 4.21 |
| 3 | 4.78 | −1.42 | 3.75 | −2.88 | |
| 45 | 4.53 | 1.33 | 4.08 | 3.18 | |
| 1600 | 3.88 | 3.79 | 2.81 | 3.88 | |
| Liver | 1 | 3.64 | 1.02 | 3.27 | 3.62 |
| 3 | 4.58 | 2.42 | 3.12 | 2.28 | |
| 45 | 2.57 | 0.33 | 2.13 | 0.17 | |
| 1600 | 3.96 | 0.53 | 5.07 | −1.61 | |
| Spleen | 1 | 3.34 | 0.33 | 7.10 | −2.91 |
| 3 | 3.07 | −0.78 | 4.47 | 1.23 | |
| 45 | 2.24 | −0.28 | 4.44 | 1.40 | |
| 1600 | 3.50 | −1.39 | 1.04 | −1.66 | |
| Lung | 1 | 4.67 | 1.02 | 1.03 | 2.81 |
| 3 | 3.56 | 2.84 | 2.24 | −5.76 | |
| 45 | 1.69 | −1.00 | 4.15 | −2.26 | |
| 1600 | 3.50 | 1.84 | 3.14 | 2.87 | |
| Kidney | 1 | 2.14 | 0.88 | 2.27 | 1.43 |
| 3 | 2.74 | 3.55 | 4.96 | 5.53 | |
| 45 | 3.51 | 0.76 | 5.93 | −1.72 | |
| 1600 | 9.59 | 2.32 | 4.38 | 4.77 | |
| Brain | 1 | 4.47 | 1.03 | 2.64 | 3.96 |
| 3 | 2.58 | 3.12 | 6.32 | 6.61 | |
| 45 | 4.86 | −2.06 | 1.93 | −2.77 | |
| 1600 | 10.24 | 0.47 | 4.07 | −1.27 | |
| Intestine | 1 | 4.76 | 0.92 | 7.77 | −5.36 |
| 3 | 3.26 | −0.03 | 12.32 | −4.68 | |
| 45 | 6.20 | 0.02 | 5.09 | 0.84 | |
| 1600 | 3.32 | 4.66 | 2.98 | 6.13 | |
| Stomach | 1 | 4.55 | 0.16 | 3.50 | −1.08 |
| 3 | 5.27 | 3.20 | 2.40 | 8.17 | |
| 45 | 3.58 | 0.20 | 8.53 | −2.49 | |
| 1600 | 11.08 | −6.79 | 4.05 | −8.04 | |
| Samples | Spiked Concentration (ng/mL) | Matrix Effect | Extraction Recovery | ||
|---|---|---|---|---|---|
| Mean ± SD (%) | RSD (%) | Mean ± SD (%) | RSD (%) | ||
| Plasma | 1 | 101.1 ± 5.8 | 5.8 | 101.6 ± 6.6 | 6.5 |
| 3 | 101.4 ± 7.1 | 7.0 | 99.0 ± 7.5 | 7.5 | |
| 45 | 103.1 ± 6.9 | 6.7 | 93.5 ± 6.2 | 6.6 | |
| 1600 | 99.5 ± 4.1 | 4.1 | 99.5 ± 3.3 | 3.3 | |
| Heart | 1 | 92.1 ± 3.5 | 3.8 | 94.0 ± 8.1 | 8.7 |
| 3 | 99.8 ± 5.2 | 5.2 | 101.3 ± 5.9 | 5.8 | |
| 45 | 95.8 ± 6.1 | 6.4 | 99.9 ± 6.9 | 6.9 | |
| 1600 | 106.5 ± 9.6 | 9.0 | 95.3 ± 1.1 | 1.1 | |
| Liver | 1 | 100.6 ± 1.3 | 1.3 | 99.5 ± 1.4 | 1.4 |
| 3 | 102.0 ± 4.9 | 4.8 | 104.3 ± 3.7 | 3.5 | |
| 45 | 102.9 ± 1.9 | 1.9 | 99.6 ± 2.6 | 2.6 | |
| 1600 | 95.9 ± 3.4 | 3.5 | 102.6 ± 5.6 | 5.4 | |
| Spleen | 1 | 96.3 ± 2.1 | 2.2 | 99.1 ± 2.3 | 2.4 |
| 3 | 84.0 ± 3.5 | 4.2 | 111.1 ± 6.1 | 5.5 | |
| 45 | 113.05 ± 8.5 | 7.5 | 100.8 ± 9.9 | 9.9 | |
| 1600 | 111.4 ± 6.5 | 5.8 | 102.9 ± 4.4 | 4.2 | |
| Lung | 1 | 99.9 ± 0.8 | 0.8 | 98.8 ± 5.0 | 5.1 |
| 3 | 104.3 ± 2.7 | 2.6 | 103.1 ± 9.4 | 9.2 | |
| 45 | 101.2 ± 3.8 | 3.8 | 100.6 ± 3.9 | 3.8 | |
| 1600 | 100.8 ± 0.5 | 0.5 | 102.3 ± 3.7 | 3.6 | |
| Kidney | 1 | 97.9 ± 3.6 | 3.6 | 101.5 ± 3.1 | 3.0 |
| 3 | 95.6 ± 5.4 | 5.6 | 96.9 ± 5.4 | 5.6 | |
| 45 | 104.5 ± 3.9 | 3.7 | 99.4 ± 6.1 | 6.1 | |
| 1600 | 111.3 ± 2.0 | 1.8 | 91.6 ± 3.0 | 3.2 | |
| Brain | 1 | 103.0 ± 9.1 | 8.8 | 95.0 ± 10.7 | 11.3 |
| 3 | 106.2 ± 3.3 | 3.1 | 100.7 ± 5.9 | 5.9 | |
| 45 | 93.9 ± 3.5 | 3.8 | 103.3 ± 2.6 | 2.5 | |
| 1600 | 105.2 ± 1.0 | 1.0 | 100.4 ± 1.7 | 1.7 | |
| Intestine | 1 | 100.8 ± 3.7 | 3.7 | 101.8 ± 5.5 | 5.4 |
| 3 | 98.7 ± 1.5 | 1.5 | 88.0 ± 2.0 | 2.3 | |
| 45 | 100.3 ± 5.9 | 5.9 | 98.9 ± 4.0 | 4.0 | |
| 1600 | 96.0 ± 5.3 | 5.5 | 93.0 ± 4.5 | 4.8 | |
| Stomach | 1 | 103.5 ± 3.4 | 3.3 | 100.0 ± 4.0 | 4.0 |
| 3 | 94.6 ± 4.0 | 4.2 | 107.3 ± 1.6 | 1.5 | |
| 45 | 107.4 ± 5.9 | 5.5 | 105.9 ± 4.6 | 4.4 | |
| 1600 | 99.2 ± 5.2 | 5.3 | 105.3 ± 5.5 | 5.3 | |
| Samples | Spiked CONC (ng/mL) | Short-Term (at Room Temperature for 4 h) | Autosampler 4 °C for 24 h | Three Freeze-Thaw Cycles | Long-Term (at −20 °C for 30 days) | ||||
|---|---|---|---|---|---|---|---|---|---|
| RE 1 (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | ||
| 3 | −10.2 | 6.62 | −0.37 | 8.27 | −3.45 | 5.06 | 12.8 | 4.55 | |
| Plasma | 45 | −4.78 | 1.50 | −2.49 | 5.50 | −1.72 | 5.64 | 1.37 | 4.01 |
| 1600 | −1.15 | 2.37 | −1.56 | 3.69 | −4.36 | 2.18 | −1.11 | 7.07 | |
| 3 | 4.33 | 5.94 | 2.20 | 2.11 | 3.59 | 3.60 | 0.11 | 3.81 | |
| Heart | 45 | −11.7 | 2.60 | −10.7 | 6.66 | −7.75 | 4.44 | 4.49 | 3.37 |
| 1600 | 0.77 | 3.32 | −4.75 | 6.08 | −4.89 | 2.25 | −1.86 | 5.10 | |
| 3 | −3.00 | 6.03 | 4.20 | 6.56 | 3.57 | 3.26 | −2.75 | 6.10 | |
| Liver | 45 | 1.76 | 3.19 | 0.66 | 3.75 | 5.36 | 6.22 | −5.4 | 2.31 |
| 1600 | −2.10 | 2.99 | 2.09 | 2.32 | −0.10 | 3.96 | 4.12 | 5.68 | |
| 3 | −0.92 | 2.90 | −4.67 | 5.49 | −1.69 | 5.40 | −7.62 | 2.38 | |
| Spleen | 45 | 8.75 | 2.16 | 5.20 | 3.68 | 11.4 | 3.29 | 12.5 | 3.37 |
| 1600 | 12.2 | 2.97 | 4.66 | 3.59 | 9.10 | 4.58 | 3.67 | 1.77 | |
| 3 | 7.71 | 5.70 | 7.86 | 3.79 | 9.81 | 3.23 | 3.03 | 1.13 | |
| Lung | 45 | 0.94 | 7.00 | 2.15 | 5.19 | 6.06 | 1.60 | −2.63 | 2.31 |
| 1600 | 0.47 | 1.49 | 4.32 | 4.12 | −1.53 | 2.97 | 0.59 | 5.68 | |
| 3 | 2.77 | 3.63 | −8.30 | 2.61 | −3.34 | 6.06 | −8.27 | 2.52 | |
| Kidney | 45 | 6.29 | 1.18 | −4.53 | 8.27 | −6.90 | 3.19 | −8.88 | 1.96 |
| 1600 | −0.45 | 2.08 | −10.36 | 4.59 | 6.73 | 4.85 | 0.49 | 3.49 | |
| 3 | 0.49 | 4.66 | 4.25 | 7.41 | −4.07 | 2.50 | −7.66 | 2.52 | |
| Brain | 45 | 3.50 | 4.19 | 4.25 | 2.00 | 4.02 | 5.85 | 4.91 | 5.13 |
| 1600 | 1.20 | 5.52 | −1.89 | 6.80 | 3.52 | 1.85 | 9.09 | 4.41 | |
| 3 | −4.88 | 4.45 | −1.67 | 8.33 | 0.97 | 5.99 | 6.04 | 3.51 | |
| Intestine | 45 | −2.93 | 4.54 | −6.52 | 4.54 | −5.83 | 4.03 | −9.83 | 5.02 |
| 1600 | −2.07 | 3.71 | 4.35 | 1.49 | −2.42 | 5.78 | −0.41 | 2.68 | |
| 3 | −3.04 | 2.28 | −3.46 | 1.76 | −1.04 | 4.59 | −0.18 | 6.49 | |
| Stomach | 45 | 3.71 | 2.75 | 1.39 | 4.53 | −2.04 | 4.88 | 6.23 | 2.24 |
| 1600 | −11.7 | 4.26 | 0.55 | 6.57 | −11.3 | 6.76 | −5.87 | 7.83 | |
| Parameters | Value |
|---|---|
| t1/2 (h) | 1.94 ± 0.46 |
| AUC0–t (ng h/mL) | 843.36 ± 54.02 |
| AUC0–∞ (ng h/mL) | 1443.88 ± 207.96 |
| MRTiv (h) | 2.70 ± 0.64 |
| CL (L/h/kg) | 7.07 ± 1.00 |
| Vd (L/kg) | 19.30 ± 2.88 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, N.; Tan, H.-r.; Chen, Q.-l.; Sun, Q.; Wang, L.; Song, Y.; Olounfeh, K.M.; Meng, F.-h. Development and Validation of a Sensitive UHPLC-MS/MS Method for the Measurement of Gardneramine in Rat Plasma and Tissues and Its Application to Pharmacokinetics and Tissue Distribution Study. Molecules 2019, 24, 3953. https://doi.org/10.3390/molecules24213953
Zhao N, Tan H-r, Chen Q-l, Sun Q, Wang L, Song Y, Olounfeh KM, Meng F-h. Development and Validation of a Sensitive UHPLC-MS/MS Method for the Measurement of Gardneramine in Rat Plasma and Tissues and Its Application to Pharmacokinetics and Tissue Distribution Study. Molecules. 2019; 24(21):3953. https://doi.org/10.3390/molecules24213953
Chicago/Turabian StyleZhao, Nan, Hao-ran Tan, Qi-li Chen, Qi Sun, Lin Wang, Yang Song, Kamara Mohamed Olounfeh, and Fan-hao Meng. 2019. "Development and Validation of a Sensitive UHPLC-MS/MS Method for the Measurement of Gardneramine in Rat Plasma and Tissues and Its Application to Pharmacokinetics and Tissue Distribution Study" Molecules 24, no. 21: 3953. https://doi.org/10.3390/molecules24213953
APA StyleZhao, N., Tan, H.-r., Chen, Q.-l., Sun, Q., Wang, L., Song, Y., Olounfeh, K. M., & Meng, F.-h. (2019). Development and Validation of a Sensitive UHPLC-MS/MS Method for the Measurement of Gardneramine in Rat Plasma and Tissues and Its Application to Pharmacokinetics and Tissue Distribution Study. Molecules, 24(21), 3953. https://doi.org/10.3390/molecules24213953