Titanocene Selenide Sulfides Revisited: Formation, Stabilities, and NMR Spectroscopic Properties

 and
and 
Abstract

1. Introduction
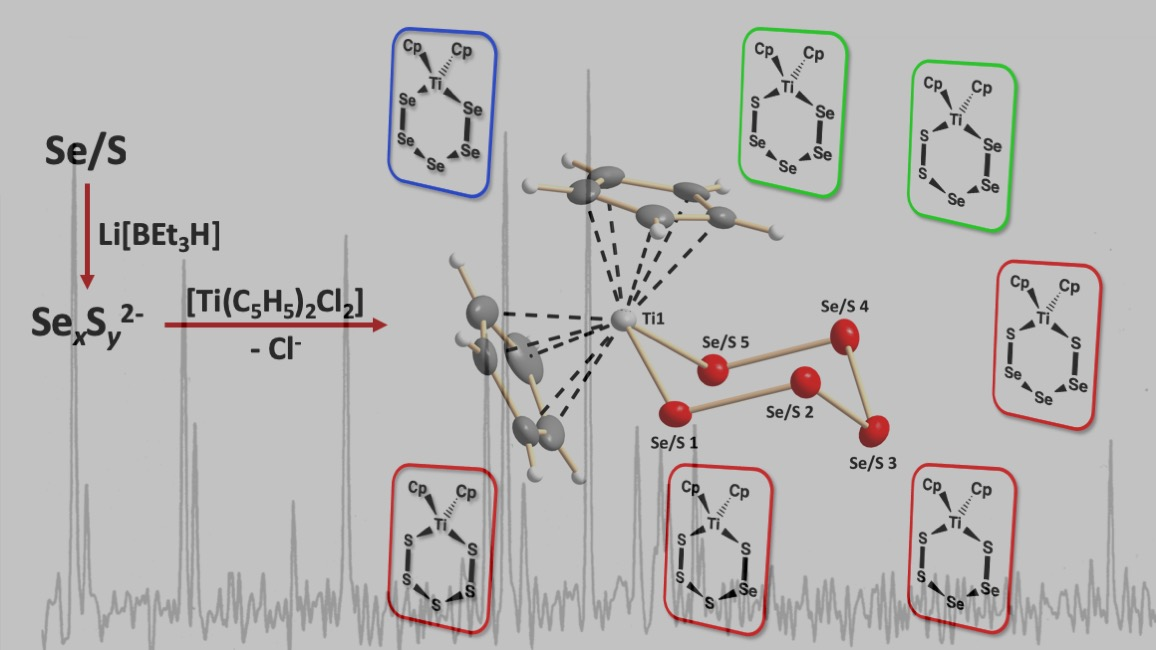
2. Results and Discussion
2.1. Crystal Structures of Phases B, C, and E
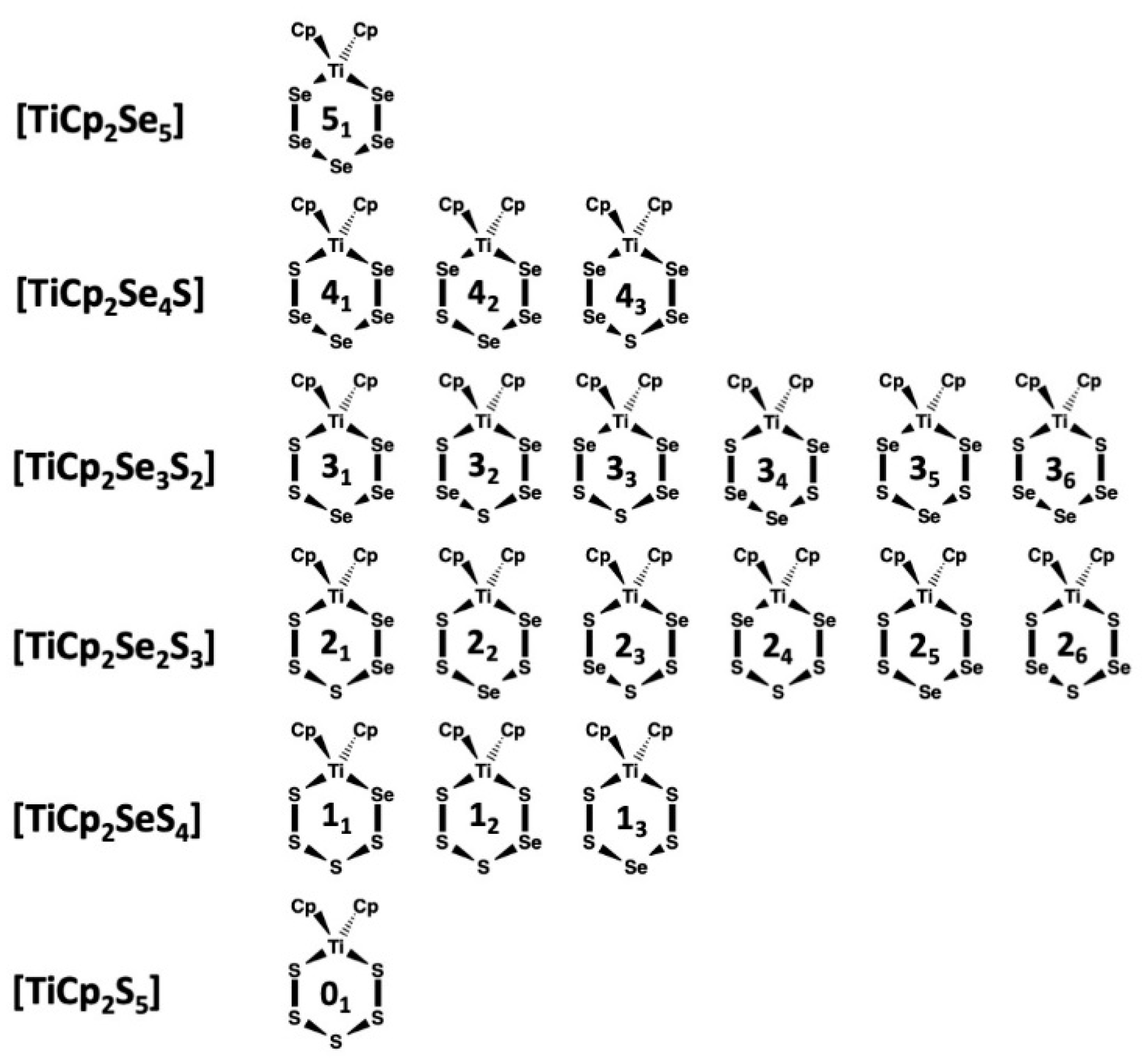
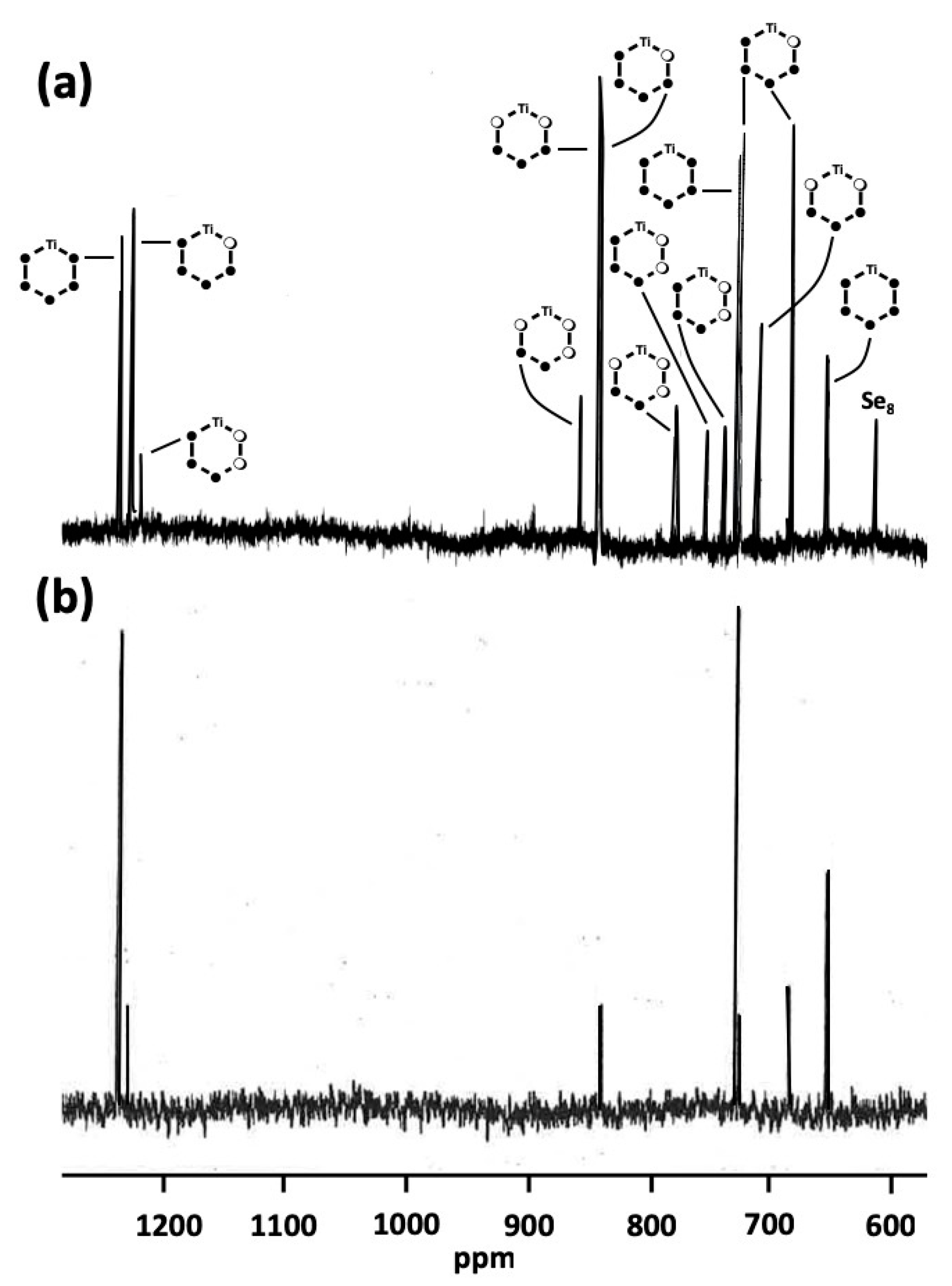
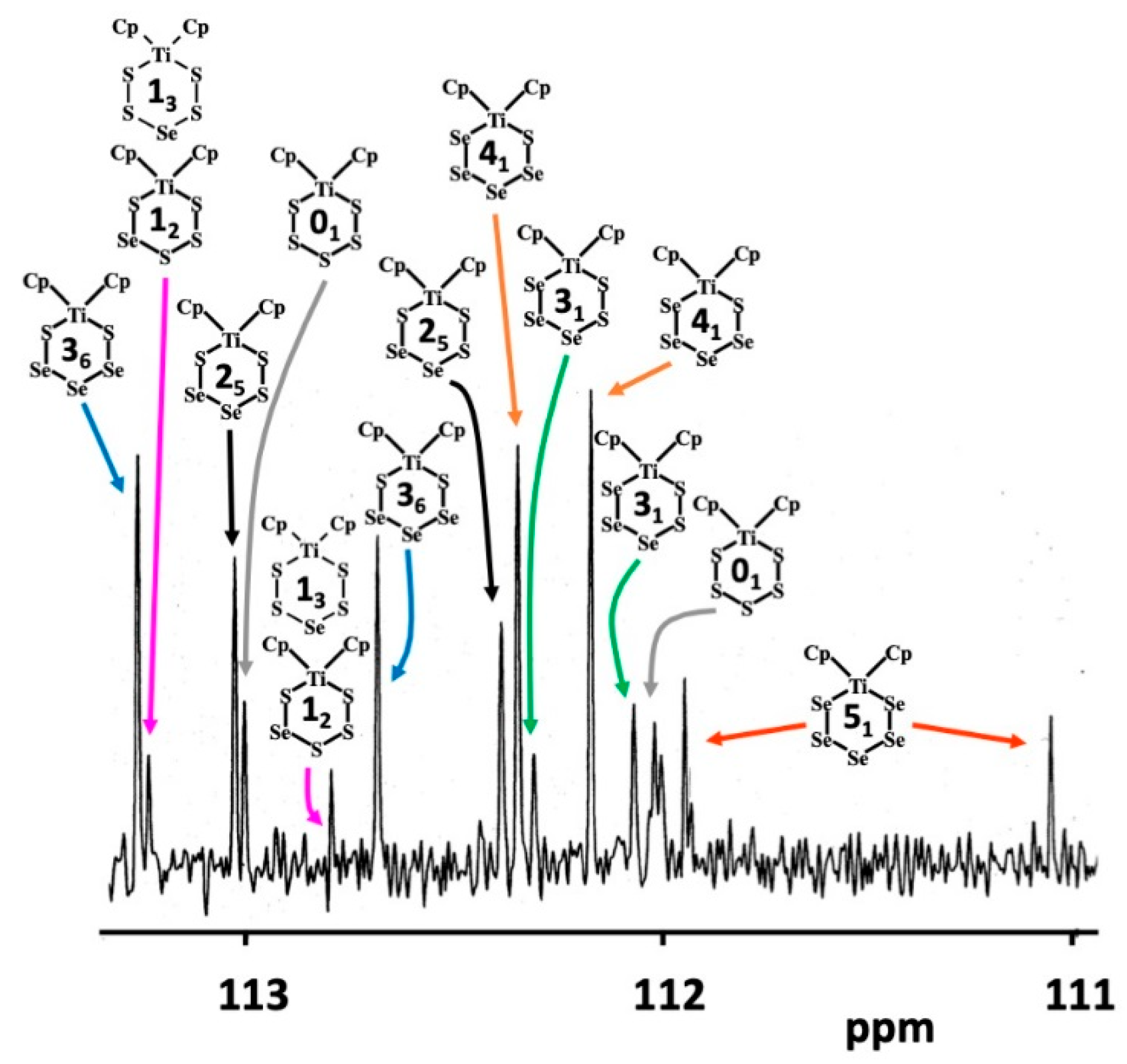
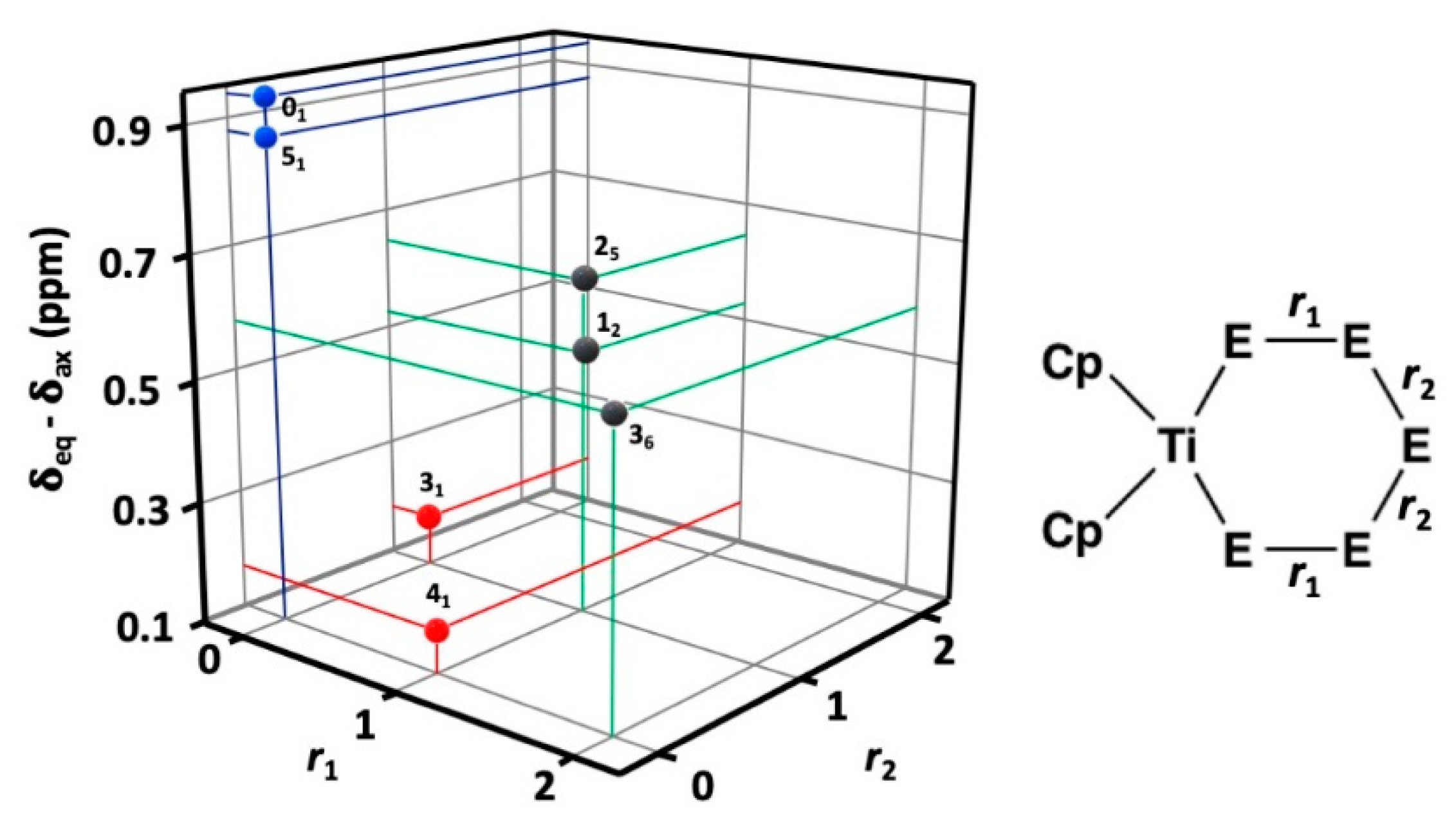
2.2. 77Se and 13C-NMR Spectra of the [TiCp2SexS5−x] (x = 0–5) Phases A–F
2.3. Composition of [TiCp2SexS5−x] Phases B–E
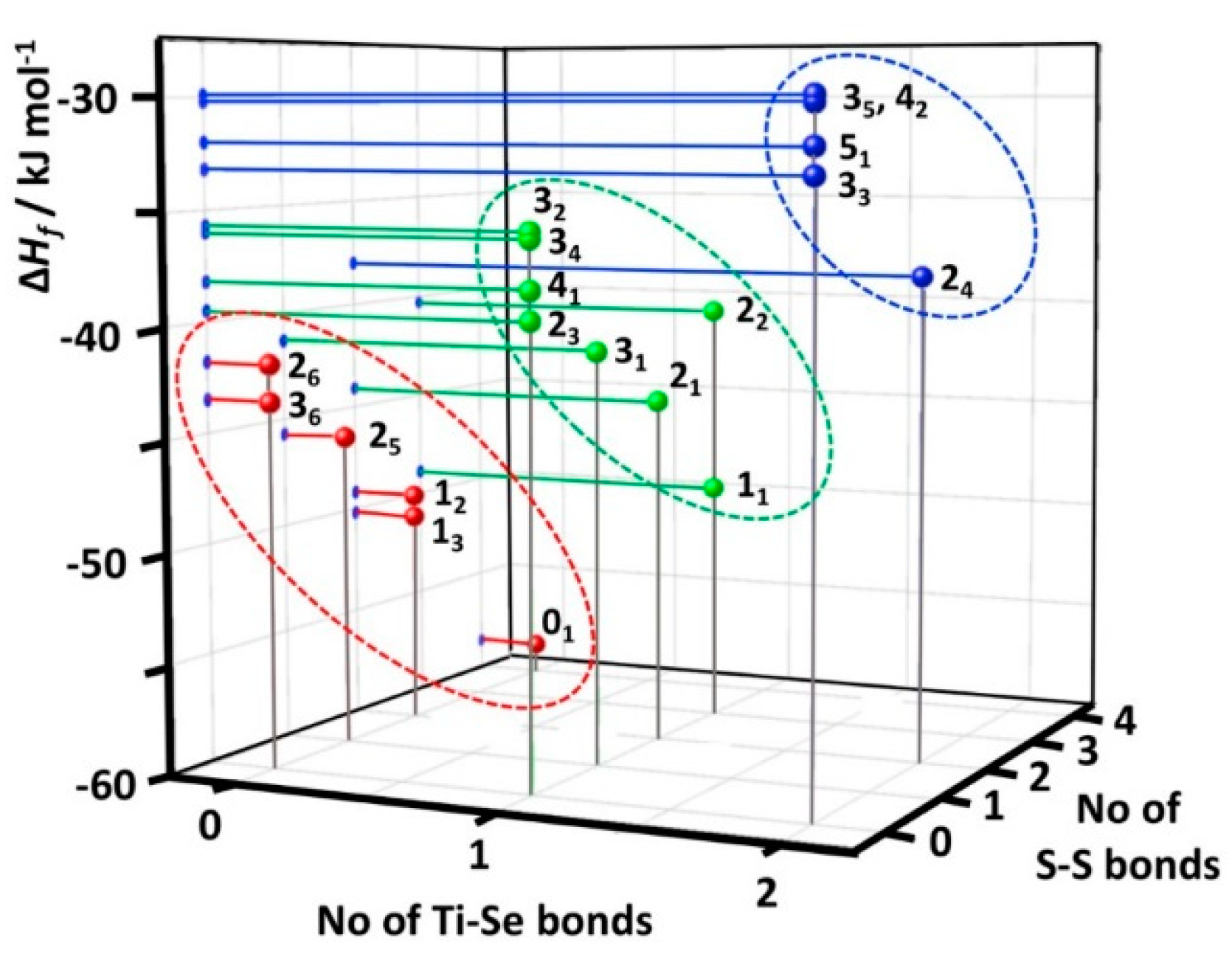
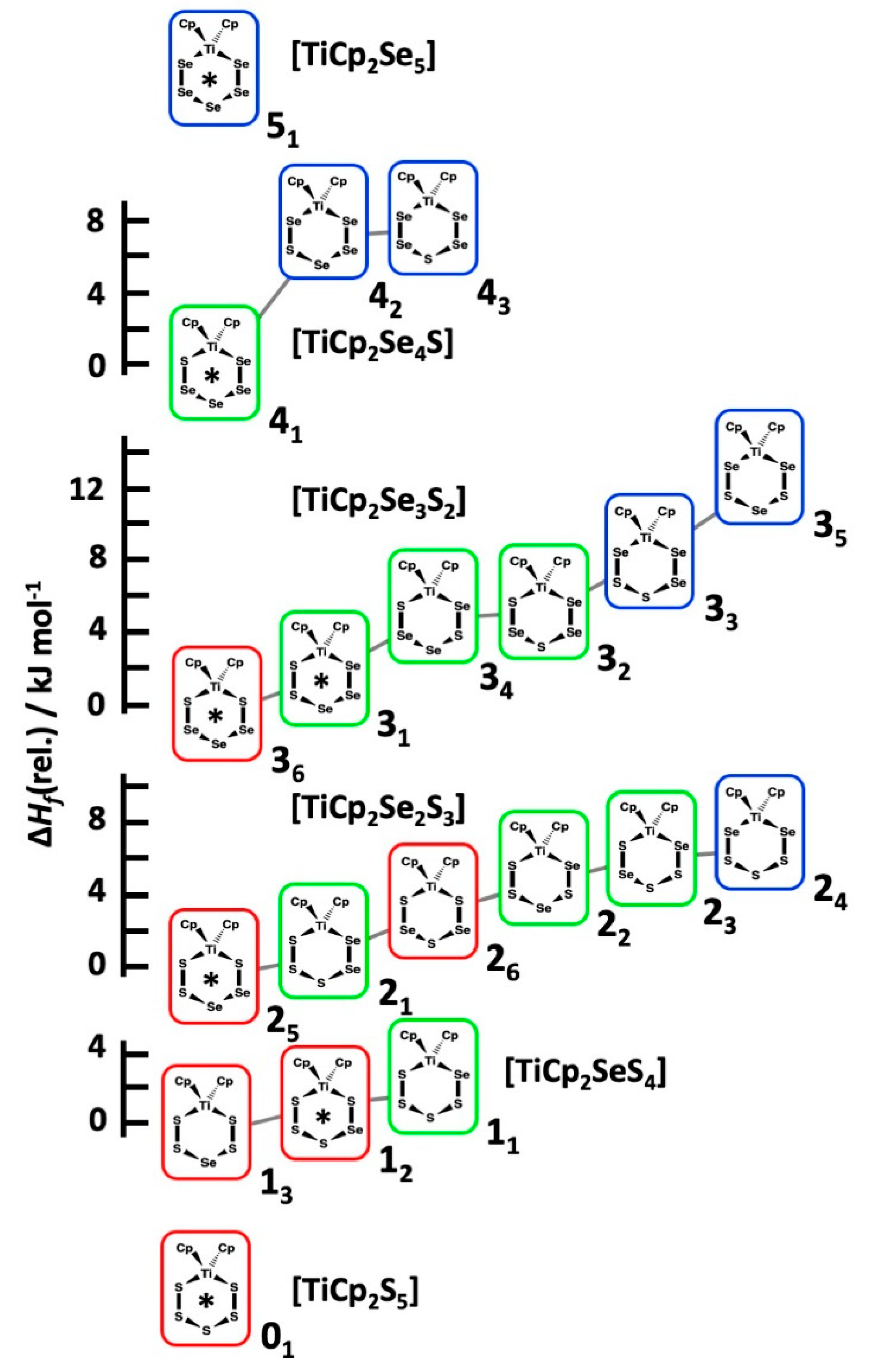
2.4. Relative Stabilities of Individual [TiCp2SexS5−x] (x = 0–5) Complexes
3. Experimental
3.1. Preparation of [TiCp2SexS5−x]
3.2. NMR Spectroscopy
3.3. X-ray Crystallography
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roof, L.C.; Kolis, J.W. New developments in the coordination chemistry of inorganic selenide and telluride ligands. Chem. Rev. 1993, 93, 1037–1080. [Google Scholar] [CrossRef]
- Laitinen, R.S.; Pekonen, P.; Suontamo, R.J. Homo- and heteroatomic chalcogen rings. Coord. Chem. Rev. 1994, 130, 1–62. [Google Scholar] [CrossRef]
- Kanatzidis, M.G.; Huang, S.P. Coordination chemistry of heavy polychalcogenide ligands. Coord. Chem. Rev. 1994, 130, 509–621. [Google Scholar] [CrossRef]
- Steudel, R.; Kustos, M.; Pridöhl, M.; Westphal, U. The utilization of titanocene polysulfide complexes for the synthesis of organic polysulfanes. Phosphorus Sulfur Silicon Relat. Elem. 1994, 93–94, 61–72. [Google Scholar] [CrossRef]
- Steudel, R.; Eckert, B. Solid sulfur allotropes. Top. Curr. Chem. 2003, 230, 1–79. [Google Scholar] [CrossRef]
- Takeda, N.; Tokitoh, N.; Okazaki, R. Polysulfido complexes of main group and transition metals. Top. Curr. Chem. 2003, 231, 153–202. [Google Scholar]
- Laitinen, R.S.; Oilunkaniemi, R. Catenated Compounds: Group 16 (Se, Te). In Comprehensive Inorganic Chemistry II, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 197–231. [Google Scholar]
- Horácek, M. Titanocene sulfide chemistry. Coord. Chem. Rev. 2016, 314, 83–102. [Google Scholar] [CrossRef]
- Muller, E.G.; Petersen, J.L.; Dahl, L.F. Synthesis and characterization of Di-π-cyclopentadienyl-metal pentasulfides of Titanium(IV) and Vanadium(IV): An operational test of the influence of an unpaired electron on the molecular geometry. J. Organomet. Chem. 1976, 111, 91–112. [Google Scholar] [CrossRef]
- Epstein, E.F.; Bernal, I. Pentachalcogenide dianions in transition-metal complexes: Crystal structure of Bis(π-cyclopentedienyl)titanium pentasulfide. J. Chem. Soc. Chem. Commun. 1970, 0, 410–411. [Google Scholar] [CrossRef]
- Epstein, E.F.; Bernal, I.; Kopf, H. The crystal and molecular structures of di-π-cyclopentadienyltitanium pentachalcogenides (C5H5)2TiS5−xSex: I. The structural properties of π-(η5-C5H5)2TiS5. J. Organomet. Chem. 1971, 26, 229–245. [Google Scholar] [CrossRef]
- Klouras, N.; Demakopoulos, I.; Terzis, A.; Raptopoulou, C.P. Crystal and molecular structure of Bis(η5-methylcyclopentadienyl)titana(IV)cyclohexasulfane Ti(η5-C5H4CH3)2S5. Z. Anorg. Allg. Chem. 1995, 621, 113–116. [Google Scholar] [CrossRef]
- Fenske, D.; Adel, J.; Dehnicke, K. Die Kristallstruktur von Bis(η-cyclopentadienyl)titana(TiIV)cyclohexaselenan, Cp2TiSe5. Z. Naturforsch. 1987, 42, 931–933. [Google Scholar] [CrossRef]
- Albrecht, N.; Weiss, E. Bis(η-cyclopentadienyl)(pentaselenido)metall-Komplexe Cp2MSe5 des Ti, Zr, Hf und (μ2-O) (μ2-Se4) (Cp2Hf)2, eine zweikernige Verbindung mit Se4- und O-Brücken. J. Organomet. Chem. 1988, 355, 89–98. [Google Scholar] [CrossRef]
- Pekonen, P.; Hiltunen, Y.; Laitinen, R.S.; Valkonen, J. 77Se NMR spectroscopic and X-ray crystallographic characterization of Bis(cyclopentadienyl)titanium selenide sulfides mixtures [Ti(C5H5)2SexS5−x]. Inorg. Chem. 1991, 30, 1874–1878. [Google Scholar] [CrossRef]
- Papavassiliou, M.; Pickardt, J.; Steudel, R. Synthesis, structures, and reactions of Cp2′TiSe3S2 and Cp2′TiSe4S (Cp′ = η5-C5H4CH3). Phosphorus Sulfur Silicon Relat. Elem. 1992, 65, 161–164. [Google Scholar] [CrossRef]
- Raptopoulou, C.P.; Terzis, A.; Tzavellas, N.; Klouras, N. Sulfure-selenium chelates: Crystal structure of the first titanocene derivative [Ti(η5-C5H5)2SSe4]. Z. Anorg. Allg. Chem. 1995, 621, 1800–1802. [Google Scholar] [CrossRef]
- Bolinger, C.M.; Rauchfuss, T.B.; Wilson, S.R. Synthesis and characterization of a cyclic bimetallic complex of the trisulfide ion. J. Am. Chem. Soc. 1981, 103, 5620–5621. [Google Scholar] [CrossRef]
- Bolinger, C.M.; Hoots, J.E.; Rauchfuss, T.B. Intermetallic chalcogenide atom transfer and the synthesis of 1,4-[(CH3C5H4)2Ti]2S4. Organometallics 1982, 1, 223–225. [Google Scholar] [CrossRef]
- Giolando, D.M.; Rauchfuss, T.B.; Rheingold, A.L.; Wilson, S.R. Chemistry of (RC5H4)2TiS5. New information on its reactions with nucleophiles, syntheses and reactions of 1,4-[(RC5H4)2Ti]2S4, and a second isomer of (RC5H4)2TiS2C2(CO2Me)2. Organometallics 1987, 6, 667–675. [Google Scholar] [CrossRef]
- Giolando, D.M.; Papavassiliou, M.; Pickardt, J.; Rauchfuss, T.B.; Steudel, R. Synthesis and structure of 1,4-[(RCp)2Ti]2Se4 and its applications to the “Chalcogenospecific” synthesis of 1,2,5,6-Se4S4. Inorg. Chem. 1988, 27, 2596–2600. [Google Scholar] [CrossRef]
- Chandra, K.; Soni, P.; Garg, B.S.; Singh, R.P. Pentachalcogenide dianions in titanium complexes. J. Ind. Chem. Soc. 1981, 58, 10–12. [Google Scholar]
- Koepf, H.; Block, B.; Schmidt, M. Di-p-cyclopentadienyltitanium(IV)pentaselenide and pentasulfide, two heterocyclohexachalcogens in fixed conformation. Chem. Ber. 1968, 101, 272–276. [Google Scholar]
- Abel, E.W.; Booth, M.; Orrell, K.G. The inversion of the titanium pentasulfide ring in Di((η5-cyclopentadienyl)titanium pentasulfide. J. Organomet. Chem. 1978, 160, 75–79. [Google Scholar] [CrossRef]
- Pekonen, P.; Hiltunen, Y.; Laitinen, R.S. The Se-Se coupling in Bis(cyclopentadienyl)titanium pentaselenide [Ti(C5H5)2Se5]. Acta Chem. Scand. 1989, 43, 914–916. [Google Scholar] [CrossRef]
- Diaz, A.; Radzewich, C.; Wicholas, M. Synthesis and variable-temperature 1H NMR conformational analysis of Bis (η5-cyclopentadienyl)titanium pentasulfide. J. Chem. Ed. 1995, 72, 937–938. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Dohm, S.; Hansen, A.; Pisarek, J.; Pracht, P.; Seibert, J.; Neese, F. Fully automated quantum-chemistry-based computation of spin-spin-coupled nuclear magnetic resonance spectra. Angew. Chem. Int. Ed. 2017, 56, 14763–14769. [Google Scholar] [CrossRef]
- Laitinen, R.S.; Pakkanen, T.A. 77Se NMR spectroscopic characterization of selenium sulfide ring molecules SenS8−n. Inorg. Chem. 1987, 26, 2598–2603. [Google Scholar] [CrossRef]
- Pekonen, P.; Taavitsainen, J.; Laitinen, R.S. New routes to heterocyclic selenium sulfides. Acta Chem. Scand. 1998, 52, 1188–1193. [Google Scholar] [CrossRef]
- Komulainen, J.; Laitinen, R.S.; Suontamo, R.J. A theoretical study of the 77Se NMR and vibrational spectroscopic properties of SenS8−n ring molecules. Can. J. Chem. 2002, 80, 1435–1443. [Google Scholar] [CrossRef]
- Pekonen, P.; Laitinen, R.S.; Hiltunen, Y. Selenium-77 nuclear magnetic resonance identification of seven-membered selenium sulfide ring molecules. J. Chem. Soc. Dalton Trans. 1992, 0, 2885–2887. [Google Scholar] [CrossRef]
- Karhu, A.J.; Pakkanen, O.J.; Rautiainen, J.M.; Oilunkaniemi, R.; Chivers, T.; Laitinen, R.S. Experimental and computational 77Se NMR investigations of the cyclic eight-membered selenium imides 1,3,5,7-Se4(NR)4 (R = Me, tBu) and 1,5-Se6(NMe)2. Inorg. Chem. 2015, 54, 4990–4997. [Google Scholar] [CrossRef] [PubMed]
- Drowart, J.; Smoes, S. Determination by the mass spectrometric knudsen cell method and discussion of the dissociation energies of the molecules Se2(g), SSe(g) and SeTe(g). J. Chem. Soc. Faraday Trans. II 1977, 73, 1755–1767. [Google Scholar] [CrossRef]
- Garvin, D.; White, H.J. Comprehensive, Consistent Thermodynamic Tables, CODATA Bulletin No. 58: Thermodynamic Databases; Pergamon Press: Oxford, UK, 1985; pp. 1–5. [Google Scholar]
- Minenkov, Y.; Bistoni, G.; Riplinger, C.; Auer, A.A.; Neese, F.; Cavallo, L. Pair natural orbital and canonical coupled cluster reaction enthalpies involving light to heavy alkali and alkaline earth metals: The importance of sub-valence correlation. Phys. Chem. Chem. Phys. 2017, 19, 9374–9391. [Google Scholar] [CrossRef] [PubMed]
- DeYonker, N.J.; Peterson, K.A.; Wilson, A.K. Systematically convergent correlation consistent basis sets for molecular core-valence correlation effects: The third-row atoms gallium through krypton. J. Phys Chem. A 2007, 111, 11383–11393. [Google Scholar] [CrossRef]
- Gladysz, J.A.; Hornby, J.L.; Garbe, J.E. Convenient one-flask synthesis of dialkyl selenides and diselenides via lithium triethylborohydride reduction of Sex. J. Org. Chem. 1978, 43, 1204–1208. [Google Scholar] [CrossRef]
- Burns, R.C.; Collins, M.J.; Gillespie, R.J.; Schrobilgen, G.J. 77Se nuclear magnetic resonance study of Se82+ and Se102+: Examples of rigid and fluxional species. Disproportionation of Se102+ at low temperatures. Inorg. Chem. 1986, 25, 4465–4469. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- McWeeny, R. Perturbation theory for the fock-dirac density matrix. Phys. Rev. 1962, 126, 1028–1034. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 24109. [Google Scholar] [CrossRef]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Paulechka, E.; Kazakov, A. Efficient DLPNO–CCSD(T)-based estimation of formation enthalpies for C-, H-, O-, and N-containing closed-shell compounds validated against critically evaluated experimental data. J. Phys. Chem. A 2017, 121, 4379–4387. [Google Scholar] [CrossRef]
- Minenkov, Y.; Wang, H.; Wang, Z.; Sarathy, S.M.; Cavallo, L. Heats of formation of medium-sized organic compounds from contemporary electronic structure methods. J. Chem. Theory Comput. 2017, 13, 3537–3560. [Google Scholar] [CrossRef] [PubMed]
- Liakos, D.G.; Sparta, M.; Kesharwani, M.K.; Martin, J.M.L.; Neese, F. Exploring the accuracy limits of local pair natural orbital coupled-cluster theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hattig, C.; Hofener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Karton, A.; Martin, J.M.L. Estimating the Hartree–Fock limit from finite basis set calculations. Theor. Chem. Acc. 2006, 115, 330–333. [Google Scholar] [CrossRef]
- Truhlar, D.G. Basis-set extrapolation. Chem. Phys. Lett. 1998, 294, 45–48. [Google Scholar] [CrossRef]
- Neese, F.; Valeev, E.F. Revisiting the atomic natural orbital approach for basis sets: Robust systematic basis sets for explicitly correlated and conventional correlated ab initio methods? J. Chem. Theory. Comput. 2011, 7, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.M.L.; Sundermann, A.; Fast, P.L.; Truhlar, D.G. Thermochemical analysis of core correlation and scalar relativistic effects on molecular atomization energies. J. Chem. Phys. 2000, 113, 1348–1358. [Google Scholar] [CrossRef]
- Chan, B.; Radom, L. W1X-1 and W1X-2: W1-quality accuracy with an order of magnitude reduction in computational cost. J. Chem. Theory Comput. 2012, 8, 4259–4269. [Google Scholar] [CrossRef] [PubMed]
- Pinski, P.; Riplinger, C.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. I. An efficient and simple linear scaling local MP2 method that uses an intermediate basis of pair natural orbitals. J. Chem. Phys. 2015, 143, 34108. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Dunning, T.H., Jr. Accurate correlation consistent basis sets for molecular core–valence correlation effects: The second row atoms Al–Ar, and the first row atoms B–Ne revisited. J. Chem. Phys. 2002, 117, 10548–10560. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Chem. Phys. 2005, 123, 64107. [Google Scholar] [CrossRef] [PubMed]
- Basis Sets Contracted for Relativistic DKH Calculations were Obtained from the Molpro Basis Set Library. Available online: http://www.molpro.net/info/basis.php (accessed on 21 September 2018).
- Moore, C.E. Atomic Energy Levels; U.S. Department of Commerce: Washington, DC, USA, 1971. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Intensity Ratio | E1 | E2 | E3 | E4 | E5 |
|---|---|---|---|---|---|---|
| [TiCp2Se5] (51) Obs. | 2:2:1 | 1265 1238 | 730 728 | 696 654 | 730 728 | 1265 1238 |
| [TiCp2Se4S] (41) Obs. | 1:1:1:1 | 1235 1229 | 723 680 | 728 725 | 832 840 | |
| [TiCp2Se3SSe] (42) | 1:1:1:1 | 1248 | 750 | 729 | 1326 | |
| [TiCp2Se2SSe2] (43) | 1:1 | 1247 | 784 | 784 | 1247 | |
| [TiCp2Se3S2] (31) Obs. | 1:1:1 | 1228 1221 | 736 737 | 764 752 | ||
| [TiCp2Se2SSeS] (32) | 1:1:1 | 1214 | 779 | 875 | ||
| [TiCp2Se2S2Se] (33) | 1:1:1 | 1231 | 828 | 1300 | ||
| [TiCp2SeSSe2S] (34) | 1:1:1 | 1296 | 761 | 852 | ||
| [TiCp2SeSSeSSe] (35) | 2:1 | 1311 | 753 | 1311 | ||
| [TiCp2SSe3S] (36) Obs. | 2:1 | 825 841 | 745 710 | 825 841 | ||
| [TiCp2Se2S3] (21) | 1:1 | 1209 | 812 | |||
| [TiCp2SeSSeS2] (22) | 1:1 | 1285 | 790 | |||
| [TiCp2SeS2SeS] (23) | 1:1 | 1267 | 917 | |||
| [TiCp2SeS3Se] (24) | 1 | 1288 | 1288 | |||
| [TiCp2SSe2S2] (25) Obs. | 1:1 | 850 858 | 789 778 | |||
| [TiCp2SSeSSeS] (26) | 1 | 870 | 870 | |||
| [TiCp2SeS4] (11) | 1 | 1255 | ||||
| [TiCp2SSeS3] (12) Obs. | 1 | 916 936 | ||||
| [TiCp2S2SeS2] (13) | 1 | 818 |
| Complex | 77Se Chemical Shifts (ppm) | 13C Chemical Shifts (ppm) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ba | Ca | Da | E | B | C | D | E | |||
| 51 | 1238, 728, 654 (2:2:1) | - | 5 | 19 | 65 | 113.93, 111.06 | - | 5 | 19 | 65 |
| 41 | 1229, 840, 725, 680 (1:1:1:1) | 6 | 24 | 40 | 31 | 112.35, 112.17 | 8 | 27 | 41 | 35 |
| 31 | 1221, 752, 737 (1:1:1) | - | 6 | 4 | 4 | 112.31, 112.07 | - | 6 | 7 | - |
| 36 | 841, 710 (2:1) | 14 | 28 | 24 | - | 113.26, 112.68 | 15 | 27 | 21 | - |
| 25 | 858, 778 (1:1) | 20 | 16 | 6 | - | 113.03, 112.39 | 23 | 13 | 5 | - |
| 12 | 936 | 15 | 6 | - | - | 113.24, 112.80 | 9 | - | - | - |
| 01b | 45 | 15 | 7 | - | 113.01, 112.02 | 45 | 15 | 7 | - |
| Complex | Phase B | Phase C | Phase D a | Phase E |
|---|---|---|---|---|
| 51 | - | 12 | 25 | 65 |
| 41 | 33 | 25 | 23 | 35 |
| 31 | - | 9 | 4 | - |
| 36 | 1 | 18 | 24 | - |
| 25 | 23 | 21 | 6 | - |
| 12 | 2 | 6 | - | - |
| 01 | 41 | 9 | 18 | - |
| Phase B | Phase C | Phase E | |
|---|---|---|---|
| Empirical formula | C10H10S3.17Se1.83Ti | C10H10S2.12Se2.88Ti | C10H10S0.35Se4.65Ti |
| Formula weight | 424.21 | 473.57 | 556.46 |
| Temperature (K) | 120 | 120 | 120 |
| Crystal colour, habit | Dark red, Needle | Dark red, Block | Dark red, Block |
| Crystal dimensions (mm2) | 0.450 × 0.150 × 0.100 | 0.180 × 0.120 × 0.080 | 0.200 × 0.150 × 0.120 |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| a (Å) | 13.000(3) | 13.091(3) | 8.011(2) |
| b (Å) | 7.950(2) | 8.062(2) | 8.135(2) |
| c (Å) | 14.300(3) | 14.277(3) | 11.791(2) |
| α (o) | 96.46(3) | ||
| β (o) | 114.20(3) | 114.29(3) | 105.84(3) |
| γ (o) | 108.51(3) | ||
| V (Å3) | 1348.0(6) | 1373.4(6) | 684.1(3) |
| Space Group | P21/c | P21/c | P-1 |
| Z | 4 | 4 | 2 |
| Dcalc (g/cm3) | 2.090 | 2.290 | 2.702 |
| F(000) | 820 | 896 | 511 |
| μ(MoKα) (cm−1) | 6.035 | 8.553 | 13.019 |
| No. of reflections measured | 7403 | 6434 | 4558 |
| No. of unique reflections | 2337 | 2374 | 2264 |
| No. of observed reflections/No. Variables | 2109/151 | 2089/151 | 2077/148 |
| Reflection/Parameter Ratio | 13.97 | 13.83 | 14.03 |
| Min. and Max. Transmissions | |||
| RINT | 0.0299 | 0.0523 | 0.1025 |
| R1 [I > 2σ(I)] a | 0.0256 | 0.0345 | 0.0695 |
| R1 (all reflections) a | 0.0299 | 0.0421 | 0.0733 |
| wR2 (all reflections) b | 0.0638 | 0.0859 | 0.1840 |
| Goodness of fit | 1.086 | 1.192 | 1.054 |
| Max., min. residual electron density (e−/Å3) | 0.381, −0.297 | 0.756, −0.634 | 2.562, −1.829 |
| CCDC No. | 1887990 | 1887988 | 1887989 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laasonen, H.; Ikäheimonen, J.; Suomela, M.; Rautiainen, J.M.; Laitinen, R.S. Titanocene Selenide Sulfides Revisited: Formation, Stabilities, and NMR Spectroscopic Properties. Molecules 2019, 24, 319. https://doi.org/10.3390/molecules24020319
Laasonen H, Ikäheimonen J, Suomela M, Rautiainen JM, Laitinen RS. Titanocene Selenide Sulfides Revisited: Formation, Stabilities, and NMR Spectroscopic Properties. Molecules. 2019; 24(2):319. https://doi.org/10.3390/molecules24020319
Chicago/Turabian StyleLaasonen, Heli, Johanna Ikäheimonen, Mikko Suomela, J. Mikko Rautiainen, and Risto S. Laitinen. 2019. "Titanocene Selenide Sulfides Revisited: Formation, Stabilities, and NMR Spectroscopic Properties" Molecules 24, no. 2: 319. https://doi.org/10.3390/molecules24020319
APA StyleLaasonen, H., Ikäheimonen, J., Suomela, M., Rautiainen, J. M., & Laitinen, R. S. (2019). Titanocene Selenide Sulfides Revisited: Formation, Stabilities, and NMR Spectroscopic Properties. Molecules, 24(2), 319. https://doi.org/10.3390/molecules24020319