
Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas

 ,
,
Abstract
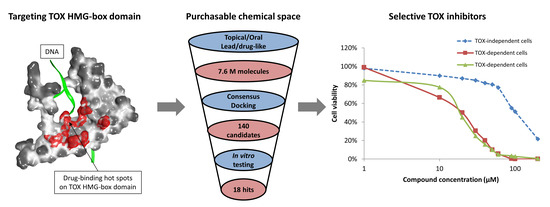
1. Introduction
2. Results
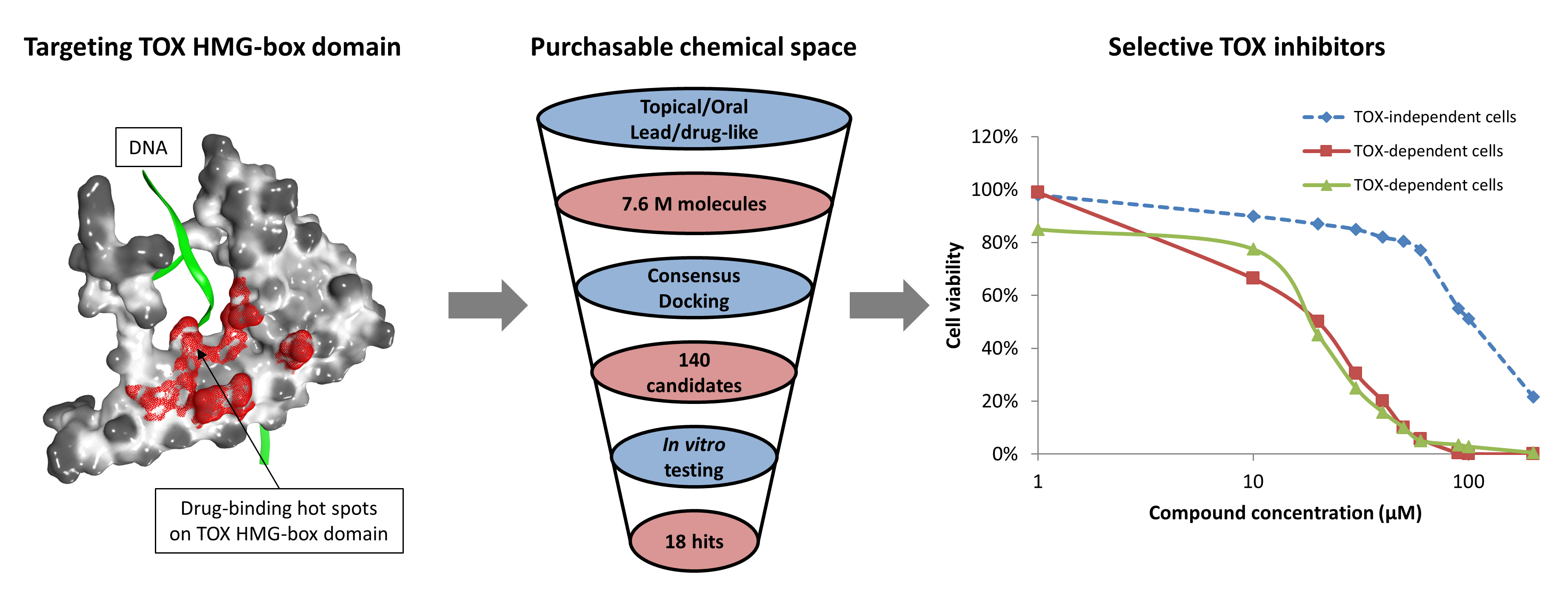
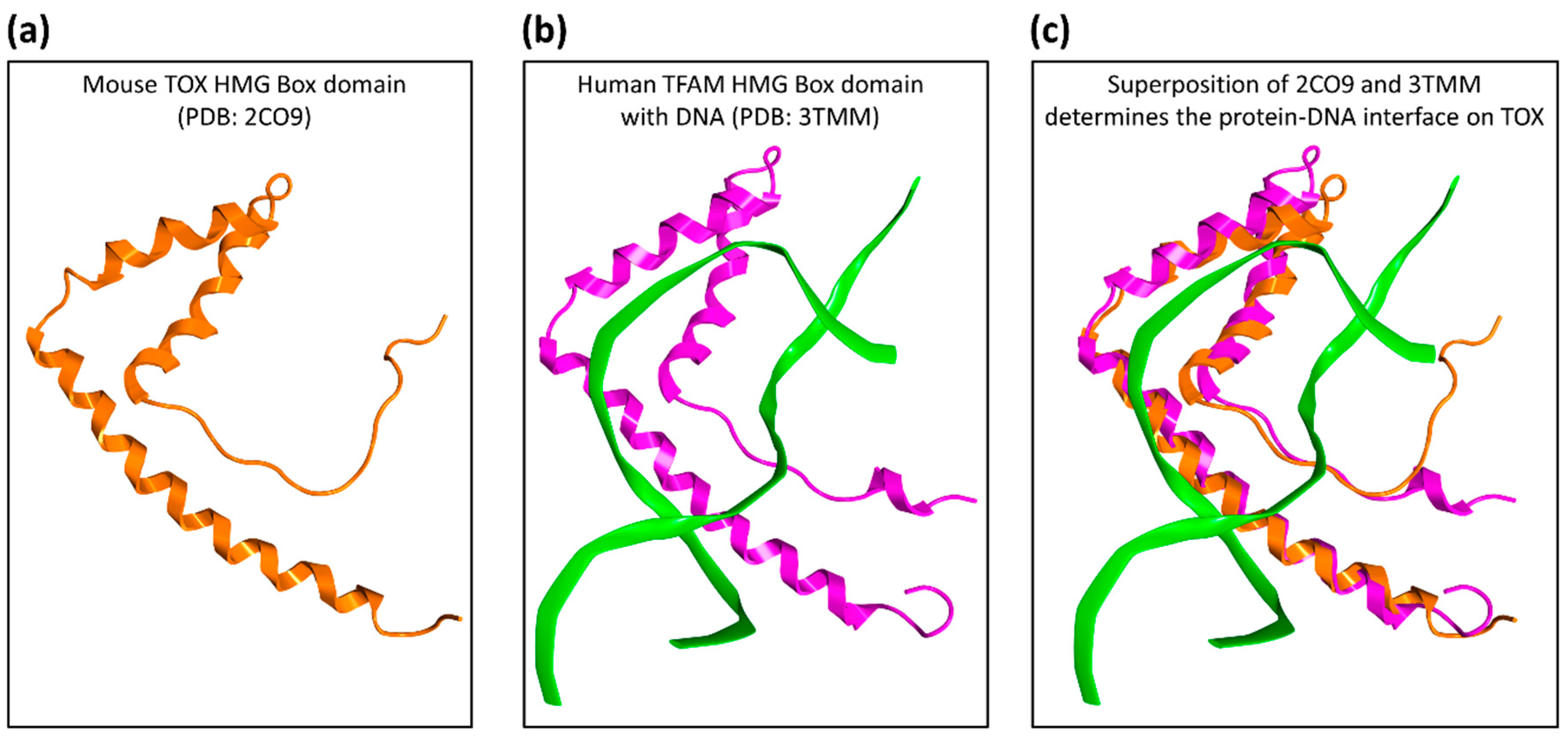
2.1. Druggability Assessment of the TOX HMG-Box Domain
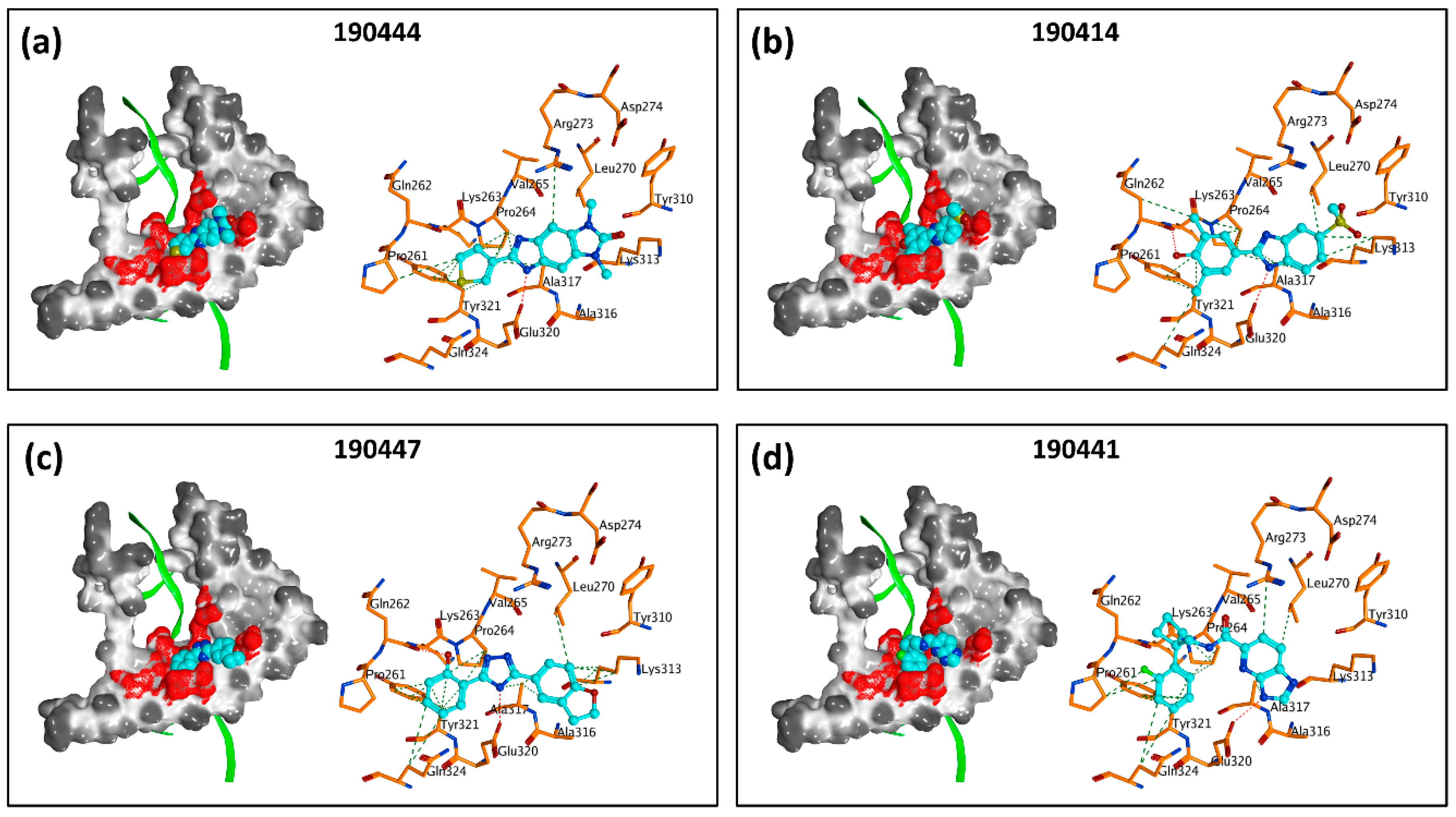
2.2. Large-Scale In Silico Screening
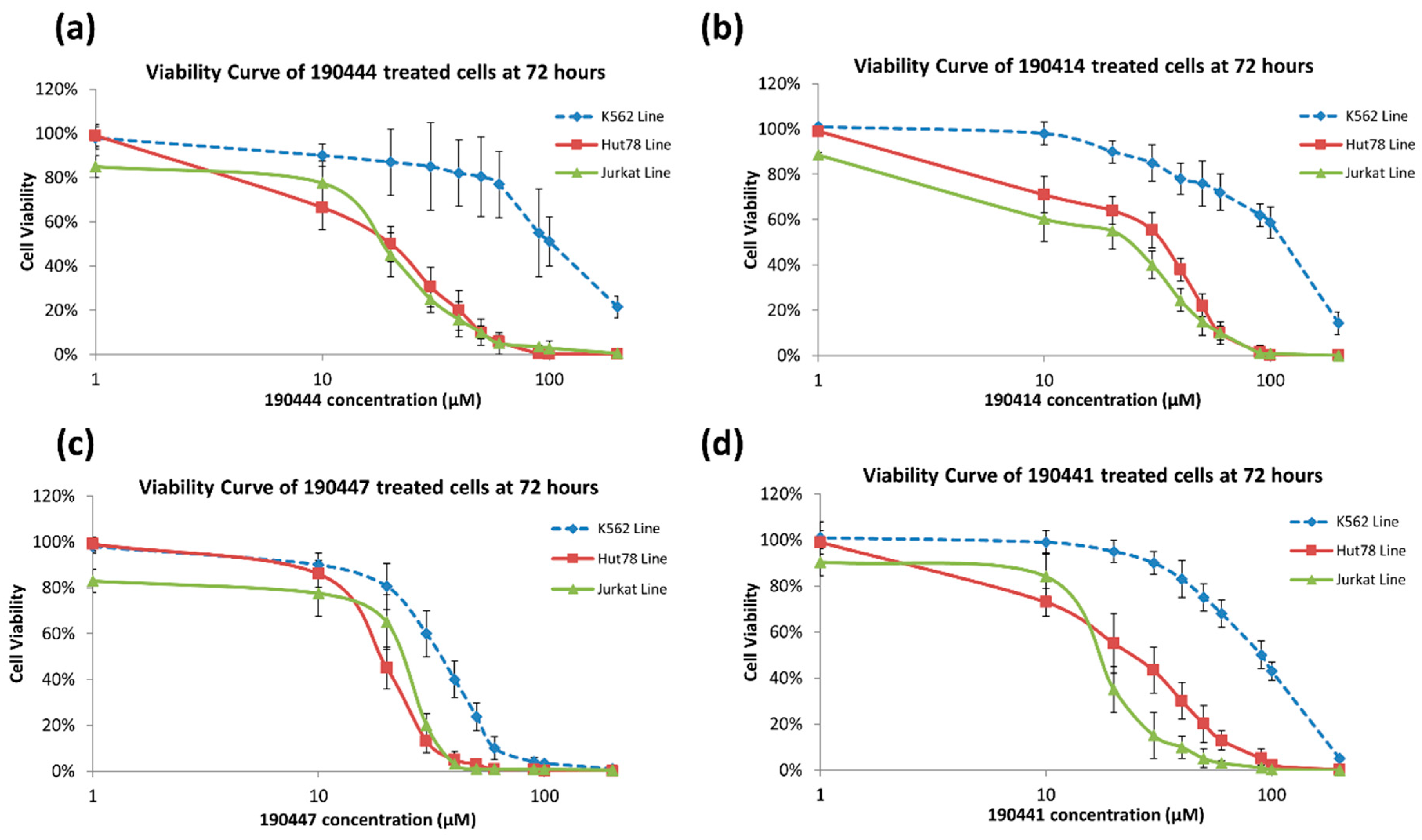
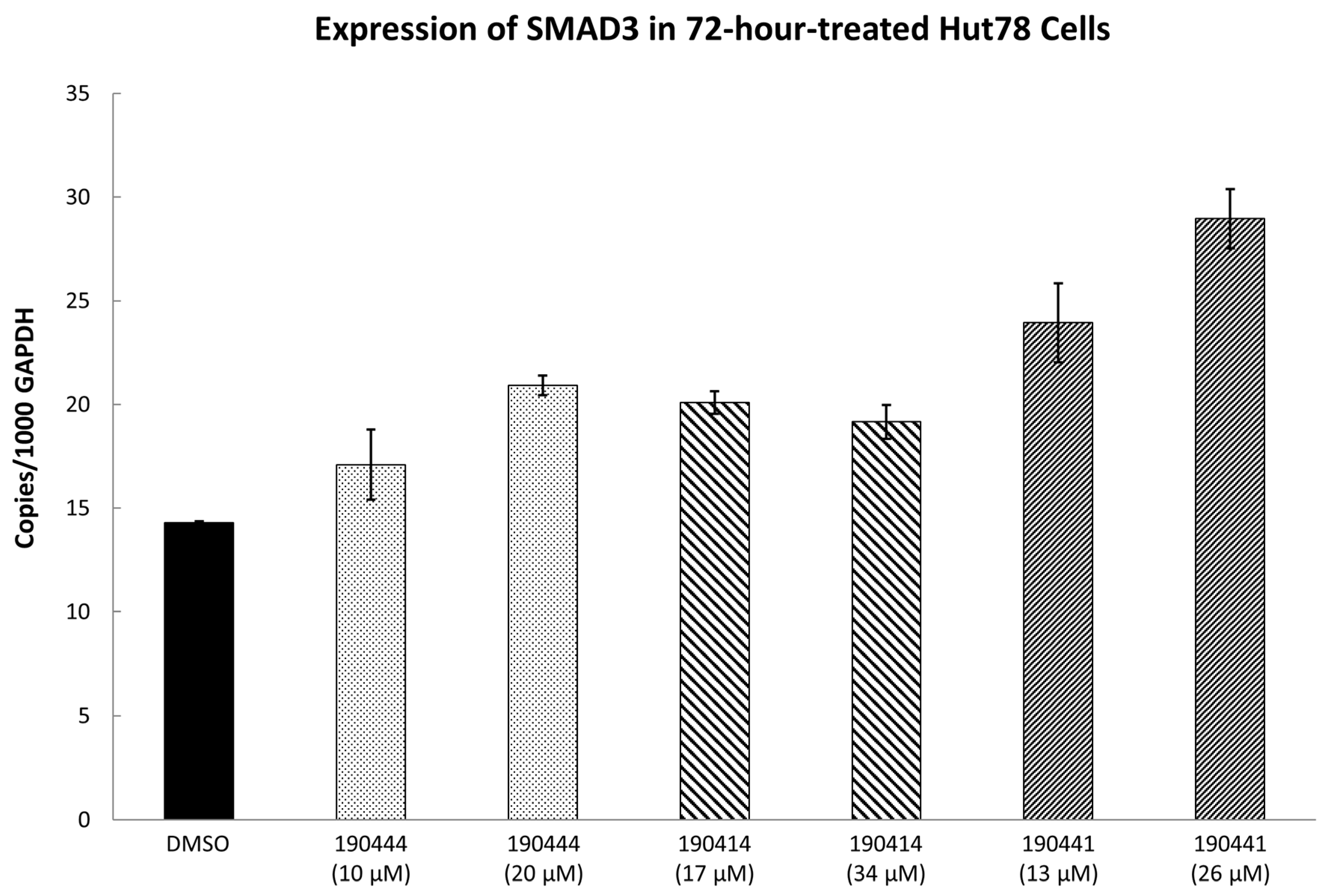
2.3. In Vitro Experimental Validation
3. Discussion
4. Materials and Methods
4.1. Structural Evaluation of TOX Druggability
4.2. In Silico Screening (Initial)
4.3. In Silico Screening (Large-Scale)
4.4. In Vitro Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and Sezary syndrome. Lancet 2008, 371, 945–957. [Google Scholar] [CrossRef]
- Wilkinson, B.; Chen, J.Y.; Han, P.; Rufner, K.M.; Goularte, O.D.; Kaye, J. TOX: An HMG box protein implicated in the regulation of thymocyte selection. Nat. Immunol. 2002, 3, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Aliahmad, P.; Seksenyan, A.; Kaye, J. The many roles of TOX in the immune system. Curr. Opin. Immunol. 2012, 24, 173–177. [Google Scholar] [CrossRef]
- Aliahmad, P.; Kadavallore, A.; de la Torre, B.; Kappes, D.; Kaye, J. TOX is required for development of the CD4 T cell lineage gene program. J. Immunol. 2011, 187, 5931–5940. [Google Scholar] [CrossRef] [PubMed]
- Aliahmad, P.; de la Torre, B.; Kaye, J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat. Immunol. 2010, 11, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Aliahmad, P.; O’Flaherty, E.; Han, P.; Goularte, O.D.; Wilkinson, B.; Satake, M.; Molkentin, J.D.; Kaye, J. TOX provides a link between calcineurin activation and CD8 lineage commitment. J. Exp. Med. 2004, 199, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Zargham, H.; Pehr, K.; Gilbert, M.; Zhou, Y.; Moreau, L.; Woetmann, A.; Odum, N.; et al. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology 2014, 3, e970025. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Su, M.W.; Jiang, X.; Zhou, Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T-cell lymphoma. Blood 2015, 125, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Litvinov, I.V.; Wang, Y.; Su, M.W.; Tu, P.; Jiang, X.; Kupper, T.S.; Dutz, J.P.; Sasseville, D.; Zhou, Y. Thymocyte selection-associated high mobility group box gene (TOX) is aberrantly over-expressed in mycosis fungoides and correlates with poor prognosis. Oncotarget 2014, 5, 4418–4425. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Yu, R.; Huang, Y.; Su, M.; Xiao, C.; Martinka, M.; Dutz, J.P.; Zhang, X.; Zheng, Z.; et al. Molecular markers of early-stage mycosis fungoides. J. Invest. Dermatol 2012, 132, 1698–1706. [Google Scholar] [CrossRef]
- Ban, F.; Dalal, K.; Li, H.; LeBlanc, E.; Rennie, P.S.; Cherkasov, A. Best Practices of Computer-Aided Drug Discovery: Lessons Learned from the Development of a Preclinical Candidate for Prostate Cancer with a New Mechanism of Action. J. Chem. Inf. Model. 2017, 57, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Roshan-Moniri, M.; Sharma, A.; Li, H.; Ban, F.; Hassona, M.D.; Hsing, M.; Singh, K.; LeBlanc, E.; Dehm, S.; et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J. Biol. Chem. 2014, 289, 26417–26429. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Munuganti, R.S.; Leblanc, E.; Lin, Y.L.; Leung, E.; Lallous, N.; Butler, M.; Cherkasov, A.; Rennie, P.S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor alpha. Breast Cancer Res. 2015, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Roshan-Moniri, M.; Hsing, M.; Lau, D.; Kim, A.; Yen, P.; Mroczek, M.; Nouri, M.; Lien, S.; Axerio-Cilies, P.; et al. Discovery and characterization of small molecules targeting the DNA-binding ETS domain of ERG in prostate cancer. Oncotarget 2017, 8, 42438–42454. [Google Scholar] [CrossRef]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef]
- Rose, P.W.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dimitropoulos, D.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Prlic, A.; Quesada, M.; et al. The RCSB Protein Data Bank: New resources for research and education. Nucleic Acids Res. 2013, 41, D475–D482. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided. Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- ADMET Predictor; Simulations Plus: Lancaster, CA, USA, 2018.
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- ZINC15. Available online: http://zinc15.docking.org/ (accessed on 09 July 2019).
- Cherkasov, A.; Ban, F.; Li, Y.; Fallahi, M.; Hammond, G.L. Progressive docking: A hybrid QSAR/docking approach for accelerating in silico high throughput screening. J. Med. Chem. 2006, 49, 7466–7478. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef]
- Maestro; Schrodinger, LLC: New York, NY, USA, 2016.
- Molecular Operating Environment; Chemical Computing Group: Montréal, Canada, 2018.
- Ngo, H.B.; Kaiser, J.T.; Chan, D.C. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat. Struct. Mol. Biol. 2011, 18, 1290–1296. [Google Scholar] [CrossRef]
- FAFDrugs4. Details of physico-chemical property filters. Available online: http://fafdrugs4.mti.univ-paris-diderot.fr/filters.html (accessed on 23 July 2018).
- Paudel, K.S.; Milewski, M.; Swadley, C.L.; Brogden, N.K.; Ghosh, P.; Stinchcomb, A.L. Challenges and opportunities in dermal/transdermal delivery. Ther. Deliv. 2010, 1, 109–131. [Google Scholar] [CrossRef]
- Bennion, B.J.; Be, N.A.; McNerney, M.W.; Lao, V.; Carlson, E.M.; Valdez, C.A.; Malfatti, M.A.; Enright, H.A.; Nguyen, T.H.; Lightstone, F.C.; et al. Predicting a Drug’s Membrane Permeability: A Computational Model Validated With in Vitro Permeability Assay Data. J. Phys. Chem. B 2017, 121, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- Netchiporouk, E.; Gantchev, J.; Tsang, M.; Thibault, P.; Watters, A.K.; Hughes, J.M.; Ghazawi, F.M.; Woetmann, A.; Ødum, N.; Sasseville, D.; et al. Analysis of CTCL cell lines reveals important differences between mycosis fungoides/Sézary syndrome. Oncotarget 2017, 8, 95981–95998. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound VPC-ID | Chemical Structure 1 | Average IC50 (µM) (TOX-High Cells) 2 | Average IC50 (µM) (TOX-Low Cells) 3 | TOX-Selectivity Index 4 |
|---|---|---|---|---|
| 190444 |  | 16.28 ± 1.64 | 69.94 ± 14.66 | 4.30 |
| 190414 |  | 20.64 ± 5.59 | 69.55 ± 20.62 | 3.37 |
| 190350 |  | 12.01 ± 2.65 | 33.69 ± 7.15 | 2.80 |
| 190447 |  | 16.68 ± 3.32 | 41.13 ± 3.59 | 2.47 |
| 190410 |  | 15.35 ± 1.77 | 37.45 ± 5.13 | 2.44 |
| 190358 |  | 15.13 ± 4.34 | 32.98 ± 5.35 | 2.18 |
| 190441 |  | 34.76 ± 12.67 | 64.69 ± 13.83 | 1.86 |
| 190327 |  | 16.55 ± 1.96 | 30.29 ± 8.09 | 1.83 |
| 190343 |  | 33.26 ± 5.57 | 56.29 ± 5.09 | 1.69 |
| 190341 |  | 52.44 ± 3.96 | 85.82 ± 8.36 | 1.64 |
| 190325 |  | 39.77 ± 6.20 | 64.63 ± 7.39 | 1.63 |
| 190323 |  | 43.79 ± 4.94 | 68.68 ± 9.51 | 1.57 |
| 190339 |  | 34.02 ± 4.14 | 51.83 ± 3.19 | 1.52 |
| 190322 |  | 24.64 ± 4.74 | 36.19 ± 7.14 | 1.47 |
| 190349 |  | 52.88 ± 11.76 | 70.57 ± 10.05 | 1.33 |
| 190301 |  | 38.75 ± 1.94 | 47.29 ± 3.97 | 1.22 |
| 190354 |  | 58.59 ± 3.80 | 66.12 ± 11.85 | 1.13 |
| 190010 |  | 37.70 ± 8.11 | n/a 5 | n/a 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agrawal, V.; Su, M.; Huang, Y.; Hsing, M.; Cherkasov, A.; Zhou, Y. Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas. Molecules 2019, 24, 3459. https://doi.org/10.3390/molecules24193459
Agrawal V, Su M, Huang Y, Hsing M, Cherkasov A, Zhou Y. Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas. Molecules. 2019; 24(19):3459. https://doi.org/10.3390/molecules24193459
Chicago/Turabian StyleAgrawal, Vibudh, Mingwan Su, Yuanshen Huang, Michael Hsing, Artem Cherkasov, and Youwen Zhou. 2019. "Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas" Molecules 24, no. 19: 3459. https://doi.org/10.3390/molecules24193459
APA StyleAgrawal, V., Su, M., Huang, Y., Hsing, M., Cherkasov, A., & Zhou, Y. (2019). Computer-Aided Discovery of Small Molecule Inhibitors of Thymocyte Selection-Associated High Mobility Group Box Protein (TOX) as Potential Therapeutics for Cutaneous T-Cell Lymphomas. Molecules, 24(19), 3459. https://doi.org/10.3390/molecules24193459