3.1.2. Experimental Procedures and Analytical Data
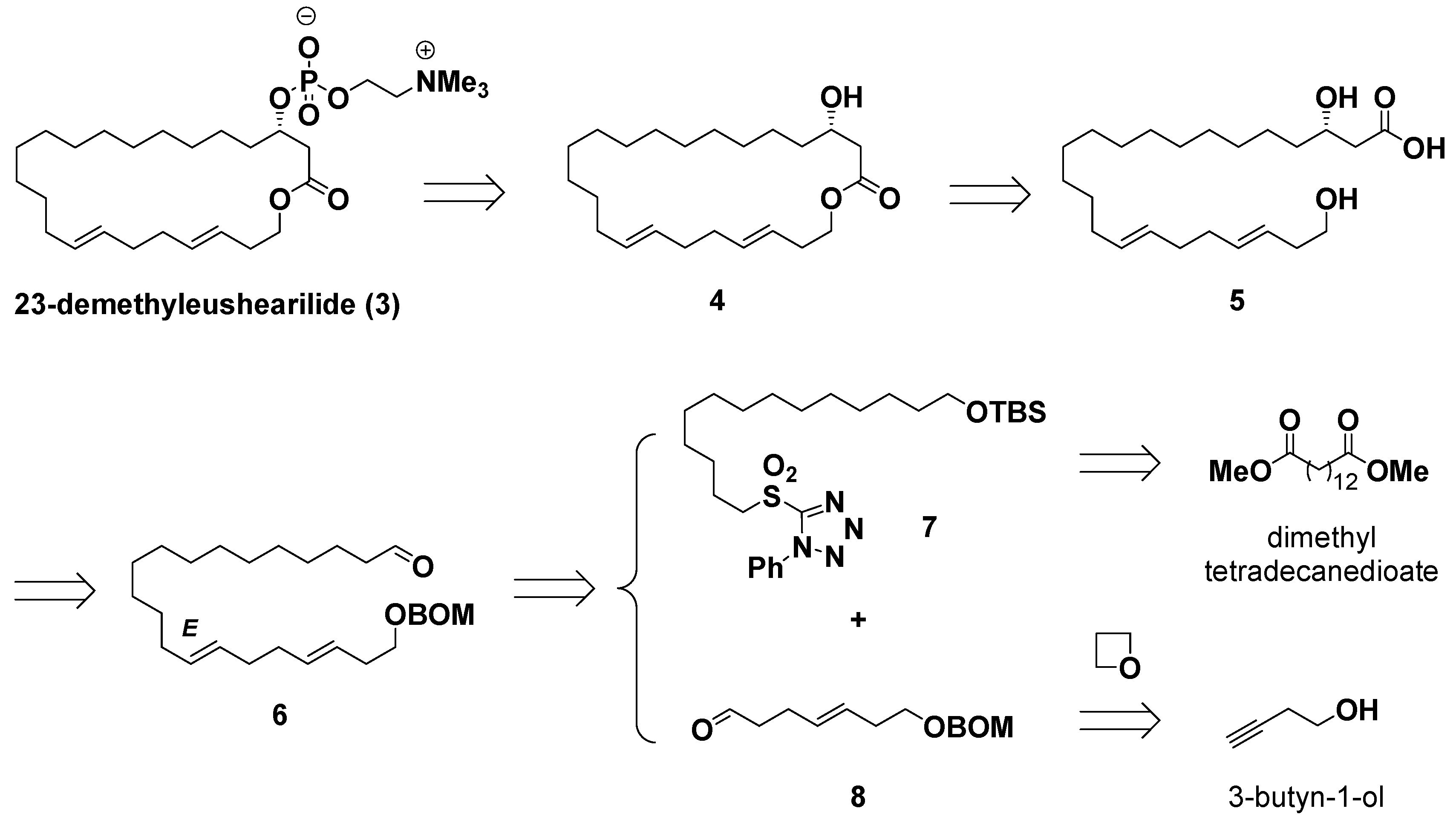
(3E,7E)-21-tert-Butyldimethylsilyloxy-1-benzyloxymethoxyhenicosa-3,7-diene (9): To a cooled (−78 °C) solution of sulfone 7 (120 mg, 0.225 mmol) in anhydrous DME (3.5 mL) was added KHMDS (1.0 M in THF, 0.31 mL, 0.311 mmol), and this was stirred for 5 min. Then, aldehyde 8 (42.9 mg, 0.173 mmol) in anhydrous DME (3.5 mL) was added dropwise via cold cannula to a solution for over 5 min at −78 °C. After the completion of the dripping, the mixture was stirred for 30 min. Then, the reaction mixture was quenched with H2O. The organic layer was separated, and the aqueous layer was extracted with diethyl ether three times. The combined organic layers were washed with brine and dried over sodium sulfate. After evaporation of the solvent, the crude product was purified by thin layer chromatography on silica gel (hexane/ethyl acetate = 8/1) to afford an E/Z mixture of 9 (75.1 mg, 78%, E/Z = 93/7) and then purified by column chromatography on silica gel impregnated with silver nitrate (hexane/ethyl acetate = 40/1) to afford 9 (67.1 mg, 70%, E/Z > 99/1). IR (neat) 2924, 2854 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.36–7.27 (m, 5H, BOM), 5.54–5.38 (m, 4H, 3-H, 4-H, 7-H, 8-H), 4.76 (s, 2H, BOM), 4.60 (s, 2H, BOM), 3.61 (t, J = 6.7 Hz, 2H, 1-H), 3.60 (t, J = 6.7 Hz, 2H, 21-H), 2.30 (q, J = 6.7 Hz, 2H, 2-H), 2.05 (br s, 4H, 5-H, 6-H), 1.96 (dd, J = 7.2, 12.3 Hz, 2H, 9-H), 1.50 (tt, J = 6.7 Hz, 2H, 20-H), 1.34–1.25 (m, 20H, 10-H to 19-H), 0.894 (s, 9H, TBS), 0.05 (s, 6H, TBS); 13C NMR (125 MHz, CDCl3) δ 137.9 (BOM), 132.2 (C4), 130.8 (C8), 129.5 (C7), 128.4 (BOM), 127.8 (BOM), 127.6 (BOM), 126.4 (C3), 94.5 (BOM), 69.2 (BOM), 67.8 (C1), 63.3 (C21), 33.0 (C20), 32.9 (C2), 32.8 (C5), 32.6, 32.5 (C6, C9), 29.6, 29.6, 29.5, 29.4, 29.2 (C10 to C18), 26.0 (TBS), 25.8 (C19), 18.4 (TBS), −5.28 (TBS); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C35H62O3SiNa [M + Na]+ 581.4366, found 581.4360.
(14E,18E)-21-Benzyloxymethoxyhenicosa-14,18-dien-1-ol (10): To a cooled (0 °C) solution of 9 (550 mg, 0.984 mmol) in THF (9.8 mL) was added TBAF (1.0 M in THF, 3.0 mL, 2.95 mmol), and this was stirred at room temperature for 2 h. After the starting material was consumed, the reaction was quenched with saturated aqueous sodium hydrogen carbonate at 0 °C. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with brine and dried over sodium sulfate. After evaporation of the solvent, the crude product was purified by column chromatography on silica gel (hexane/ethyl acetate = 10/1 to 3/1) to afford 10 (418 mg, 95%). IR (neat) 3363, 2916, 2846 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.36–7.27 (m, 5H, BOM), 5.54–5.35 (m, 4H, 14-H, 15-H, 18-H, 19-H), 4.76 (s, 2H, BOM), 4.60 (s, 2H, BOM), 3.62 (t, J = 6.8 Hz, 2H, 1-H), 3.61 (t, J = 6.8 Hz, 2H, 21-H), 2.30 (q, J = 6.7 Hz, 2H, 20-H), 2.05 (br s, 4H, 16-H,17-H), 1.98–1.94 (m, 2H, 13-H), 1.56 (qn, J = 6.9 Hz, 2H, 2-H), 1.33–1.26 (m, 20H, 3-H to 12-H); 13C NMR (125 MHz, CDCl3) δ 137.9 (BOM), 132.2 (C18), 130.8 (C14), 129.5 (C15), 128.3 (BOM), 127.8 (BOM), 127.6 (BOM), 126.4 (C19), 94.5 (BOM), 69.2 (BOM), 67.8 (C21), 63.0 (C1), 33.0 (C20), 32.8, 32.85 (C2, C17), 32.6, 32.5 (C13, C16), 29.6, 29.6, 29.5, 29.4, 29.1 (C4 to C12), 25.7 (C3); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C29H48O3Na [M+Na]+ 467.3501, found 367.3502.
(14E,18E)-21-Benzyloxymethoxyhenicosa-14,18-dienal (6): To a cooled (0 °C) solution of 10 (220 mg, 0.494 mmol) in dichloromethane (4.0 mL) and DMSO (1.0 mL) were added Et3N (0.55 mL, 3.96 mmol) and SO3·Py (315 mg, 1.98 mmol). After the solution was stirred at room temperature for 2.5 h, the reaction mixture was quenched with saturated aqueous ammonium chloride at 0 °C. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate. The combined organic layer was washed with brine and dried over sodium sulfate. After evaporation of the solvent, the crude product was purified by thin layer chromatography on silica gel (hexane/ethyl acetate = 5/1) to afford 6 (191 mg, 87%). IR (neat) 3425, 2916, 2846, 1712 cm−1; 1H NMR (500 MHz, CDCl3) δ 9.76 (t, J = 1.7 Hz, 1H, 1-H), 7.39–7.27 (m, 5H, BOM), 5.55–5.35 (m, 4H, 14-H, 15-H, 18-H, 19-H), 4.76 (s, 2H, BOM), 4.60 (s, 2H, BOM), 3.60 (t, J = 6.9 Hz, 2H, 21-H), 2.41 (td, J = 1.7, 7.4 Hz, 2H, 2-H), 2.30 (q, J = 6.5 Hz, 2H, 20-H), 2.05 (br s, 4H, 16-H, 17-H), 1.98–1.94 (m, 2H, 13-H), 1.62 (tt, J = 7.3 Hz, 2H, 3-H), 1.32–1.27 (m, 18H, 4-H to 12-H); 13C NMR (125 MHz, CDCl3): δ 202.9 (C1), 137.9 (BOM), 132.2 (C18), 130.8 (C14), 129.5 (C15), 128.4 (BOM), 127.8 (BOM), 127.6 (BOM), 126.4 (C19), 94.5 (BOM), 69.2 (BOM), 67.8 (C21), 43.9 (C2), 33.0 (C20), 32.8 (C17), 32.6, 32.5 (C13, C16), 29.6, 29.5, 29.5, 29.4, 29.3, 29.1 (C4 to C12), 22.1 (C3); HRMS (ESI/TOF) m/z [M+Na]+ calcd for C29H46O3Na [M + Na]+ 465.3345, found 465.3330.
(3S,16E,20E)-Ethyl 23-benzyloxymethoxy-3-hydroxytricosa-16,20-dienethioate (11): To a solution of Sn(OTf)2 (268 mg, 0.643 mmol) in dichloromethane (4.4 mL) were added solutions of (R)-1-methyl-2-(1-naphthylaminomethyl)pyrrolidine (168 mg, 0.700 mmol) in dichloromethane (1.4 mL) and nBu3SnF (199 mg, 0.644 mmol). The mixture was cooled to −78 °C. To the reaction mixture were added solutions of KSA (114 mg, 0.643 mmol) in dichloromethane (1.4 mL) and the aldehyde 6 (190 mg, 0.429 mmol) in dichloromethane (1.0 mL) at −78 °C, successively. The mixture was stirred for 1 h at that temperature and then quenched with saturated aqueous sodium hydrogen carbonate. The organic layer was separated, and the aqueous layer was extracted with dichloromethane three times. The combined organic layer was washed with brine and dried over sodium sulfate. After evaporation of the solvent, the crude product was purified by thin layer chromatography on silica gel (hexane/ethyl acetate = 3/1) to afford the aldol product 11 (187 mg, 80%, dr = 96/4). +9.06 (c 0.78, CHCl3); IR (neat) 3464, 3433, 2924, 2854, 1682 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.38–7.27 (m, 5H, BOM), 5.55–5.35 (m, 4H, 16-H, 17-H, 20-H, 21-H), 4.76 (s, 2H, BOM), 4.60 (s, 2H, BOM), 4.06–4.02 (m, 1H, 3-H), 3.60 (t, J = 6.9 Hz, 2H, 23-H), 2.90 (q, J = 7.4 Hz, 2H, Et), 2.73 (dd, J = 3.4, 15.5 Hz, 1H, 2-H), 2.65 (dd, J = 8.6, 15.5 Hz, 1H, 2-H), 2.30 (q, J = 6.7 Hz, 1H, 22-H), 2.04 (br s, 4H, 18-H, 19-H), 1.98–1.94 (m, 2H, 15-H), 1.53–1.39 (m, 4H, 4-H, 5-H), 1.32–1.26 (m, 21H, 6-H to 14-H, Et); 13C NMR (125 MHz, CDCl3) δ 199.7 (C1), 137.9 (BOM), 132.2 (C20), 130.8 (C16), 129.5 (C17), 128.4 (BOM), 127.9 (BOM), 127.6 (BOM), 126.4 (C21), 94.5 (BOM), 69.3 (BOM), 68.7 (C3), 67.8 (C23), 50.6 (C2), 36.5 (C4), 33.0 (C22), 32.8 (C19), 32.6, 32.5 (C15, C18), 29.6, 29.6, 29.5, 29.5, 29.5, 29.2 (C6 to C14), 25.4 (C5), 23.4 (Et), 14.6 (Et); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C33H54O4SNa [M + Na]+ 569.3635, found 569.3640.
(3S,16E,20E)-Ethyl 23-benzyloxymethoxy-3-hydroxytricosa-16,20-dienoate (12): To a cooled (0 °C) solution of 11 (172 mg, 0.315 mmol) in EtOH (3.1 mL) were added iPr2NEt (0.22 mL, 1.26 mmol) and AgOCOCF3 (139 mg, 0.629 mmol). After the solution was stirred at room temperature for 7 h, the reaction mixture was filtered through Celite. After evaporation of the solvent, the crude product was purified by thin layer chromatography on silica gel (hexane/ethyl acetate = 3/1) to afford 57 (168 mg, quant.). +6.91 (c 1.03, CHCl3); IR (neat) 3433, 2916, 2846, 1712 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.36–7.28 (m, 5H, BOM), 5.54–5.35 (m, 4H, 16-H, 17-H, 20-H, 21-H), 4.76 (s, 2H, BOM), 4.60 (s, 2H, BOM), 4.17 (q, J = 7.3 Hz, 2H, Et), 3.99 (brs, 1H, 3-H), 3.60 (t, J = 6.9, 2H, 23-H), 2.93 (s, 1H, OH), 2.50 (dd, J = 2.9, 16.6 Hz, 1H, 2-H), 2.39 (dd, J = 9.2, 16.6 Hz, 1H, 2-H), 2.30 (q, J = 6.5 Hz, 2H, 22-H), 2.04 (br s, 4H, 18-H, 19-H), 1.96 (dd, J = 7.4, 12.0 Hz, 2H, 15-H), 1.53–1.39 (m, 4H, 4-H, 5-H), 1.32–1.25 (m, 21H, 6-H to 14-H, Et); 13C NMR (125 MHz, CDCl3) δ 173.0 (C1), 137.9 (BOM), 132.1 (C20), 130.8 (C16), 129.4 (C17), 128.3 (BOM), 127.8 (BOM), 127.6 (BOM), 126.4 (C21), 94.5 (BOM), 69.2 (BOM), 68.0 (C3), 67.8 (C23), 60.6 (Et), 41.3 (C2), 36.5 (C4), 33.0 (C22), 32.7 (C19), 32.5, 32.5 (C15, C18), 29.6, 29.6, 29.5, 29.5, 29.1 (C6 to C14), 25.4 (C5), 14.1 (Et); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C33H54O5Na [M + Na]+ 553.3869, found 553.3866.
(3S,16E,20E)-3,23-Dihydroxytricosa-16,20-dienoic acid (5): A solution of 12 (168 mg, 0.317 mmol) in 2.0 M HCl (EtOH/HCl = 5/1, 6.3 mL) was stirred at room temperature for 10 h. After the starting material was consumed, 4.0 M aqueous LiOH was added at 0 °C. Distilled water (0.91 mL) was poured into the reaction mixture, followed by the addition of 4.0 M aqueous LiOH (0.16 mL, 0.633 mmol). While being stirred at room temperature for 20 h, 1.0 M aqueous HCl was added until the solution became acidic. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate five times. The combined organic layer was washed with water and brine and dried over sodium sulfate. After evaporation of the solvent, the organic phase was added (10% aqueous NaOH solution) and washed with ethyl acetate five times. The aqueous phase was separated, 1.0 M aqueous HCl solution was added, and it was washed with ethyl acetate five times. The combined organic layers were washed with water and brine and dried over sodium sulfate. After evaporation of the solvent, the desired product 5 (115 mg, 95%) was obtained as a white solid. M.P. 81.3–82.1 °C; −2.44 (c 0.23, CH3OH); IR (neat): 3402, 3379, 2916, 2846 cm−1; 1H NMR (500 MHz, CD3OD) δ 5.47–5.39 (m, 4H, 16-H, 17-H, 20-H, 21-H), 3.98–3.93 (m, 1H, 3-H), 3.52 (t, J = 6.9 Hz, 23-H), 2.43 (dd, J = 4.9, 15.2 Hz, 1H, 2-H), 2.35 (dd, J = 8.3, 15.2 Hz, 1H, 2-H), 2.20 (q, J = 6.7 Hz, 2H, 22-H), 2.04 (br s, 4H, 18-H, 19-H), 1.97 (dd, J = 6.9, 11.5 Hz, 2H, 15-H), 1.47–1.46 (m, 4H, 4-H, 5-H), 1.33–1.28 (m, 18H, 6-H to 14-H); 13C NMR (125 MHz, CD3OD) δ 175.9 (C1), 133.3 (C20), 131.9 (C16), 130.9 (C17), 127.8 (C21), 69.4 (C3), 63.1 (C23), 43.4 (C2), 38.1 (C4), 37.1 (C22), 34.0 (C19), 33.7, 33.6 (C15, C18), 30.8, 30.8, 30.6, 30.2 (C6 to 14), 26.7 (C5); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C23H42O4Na [M + Na]+ 405.2981, found 405.2979.
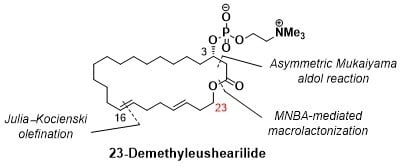
(3S,16E,20E)-3-Hydroxytricosa-16,20-dienolide (4): To a solution of MNBA (65.5 mg, 0.190 mmol) and DMAP (107 mg, 0.878 mmol) in dichloromethane (58 mL) at room temperature was slowly added a solution of the seco acid 5 (56.0 mg, 146 mmol) in dichloromethane (15 mL) with a mechanically driven syringe over a 12 h period. After cooling to 0 °C, saturated aqueous sodium hydrogen carbonate was added. The organic layer was separated, the aqueous layer was extracted with dichloromethane, and the organic layer was washed with brine and dried over sodium sulfate. After evaporation of the solvent, the crude product was purified by thin layer chromatography on silica gel (hexane/ethyl acetate = 3/1) to afford 41 (45.6 mg, 85%). +9.71 (c 0.97, CHCl3); IR (neat) 3448, 2924, 2854, 1728 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.53–5.37 (m, 4H, 16-H, 17-H, 20-H, 21-H), 4.20–4.10 (m, 2H, 23-H), 3.96 (brs, 1H, 3-H), 2.79 (s, 1H, OH), 2.52 (dd, J = 4.0, 15.5 Hz, 1H, 2-H), 2.42 (dd, J = 7.7, 15.8 Hz, 1H, 2-H), 2.33 (q, J = 5.9 Hz, 2H, 22-H), 2.05 (br s, 4H, 18-H, 19-H), 1.99 (dd, J = 6.3, 10.3 Hz, 2H, 15-H), 1.54–1.42 (m, 2H, 4-H), 1.39–1.27 (m, 20 H, 5-H to 14-H); 13C NMR (125 MHz, CDCl3) δ 172.6 (C1), 133.6 (C20), 130.8 (C16), 129.8 (C17), 125.7 (C21), 68.1 (C3), 63.9 (C23), 41.3 (C2), 36.1 (C4), 32.8 (C18), 32.5 (C19), 32.0, 31.9 (C15, C22), 28.6, 28.5, 28.4, 28.3, 28.2, 28.1, 28.0, 27.4 (C6 to C14), 24.7 (C5); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C23H40O3Na [M + Na]+ 387.2875, found 387.2868.
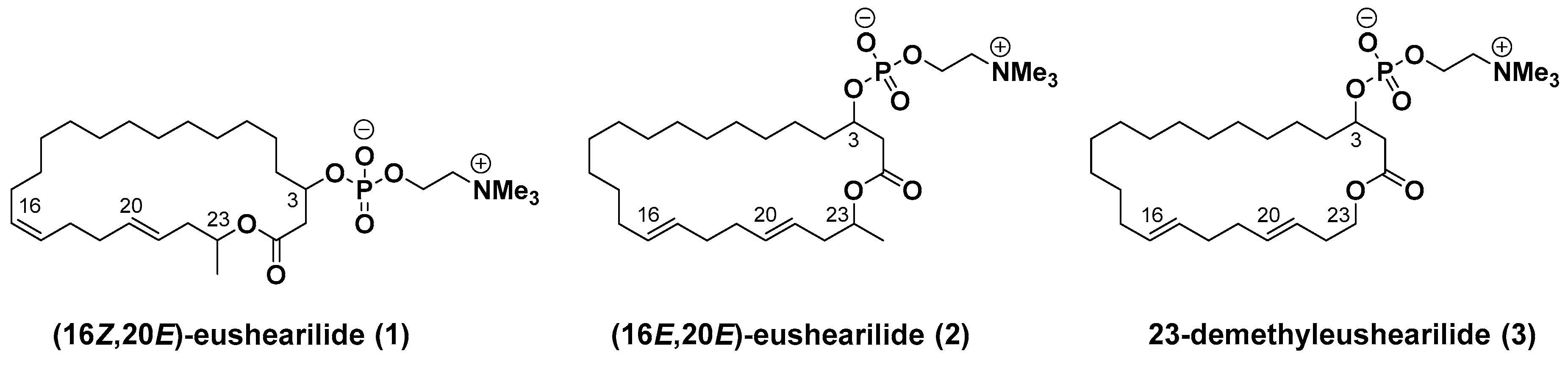
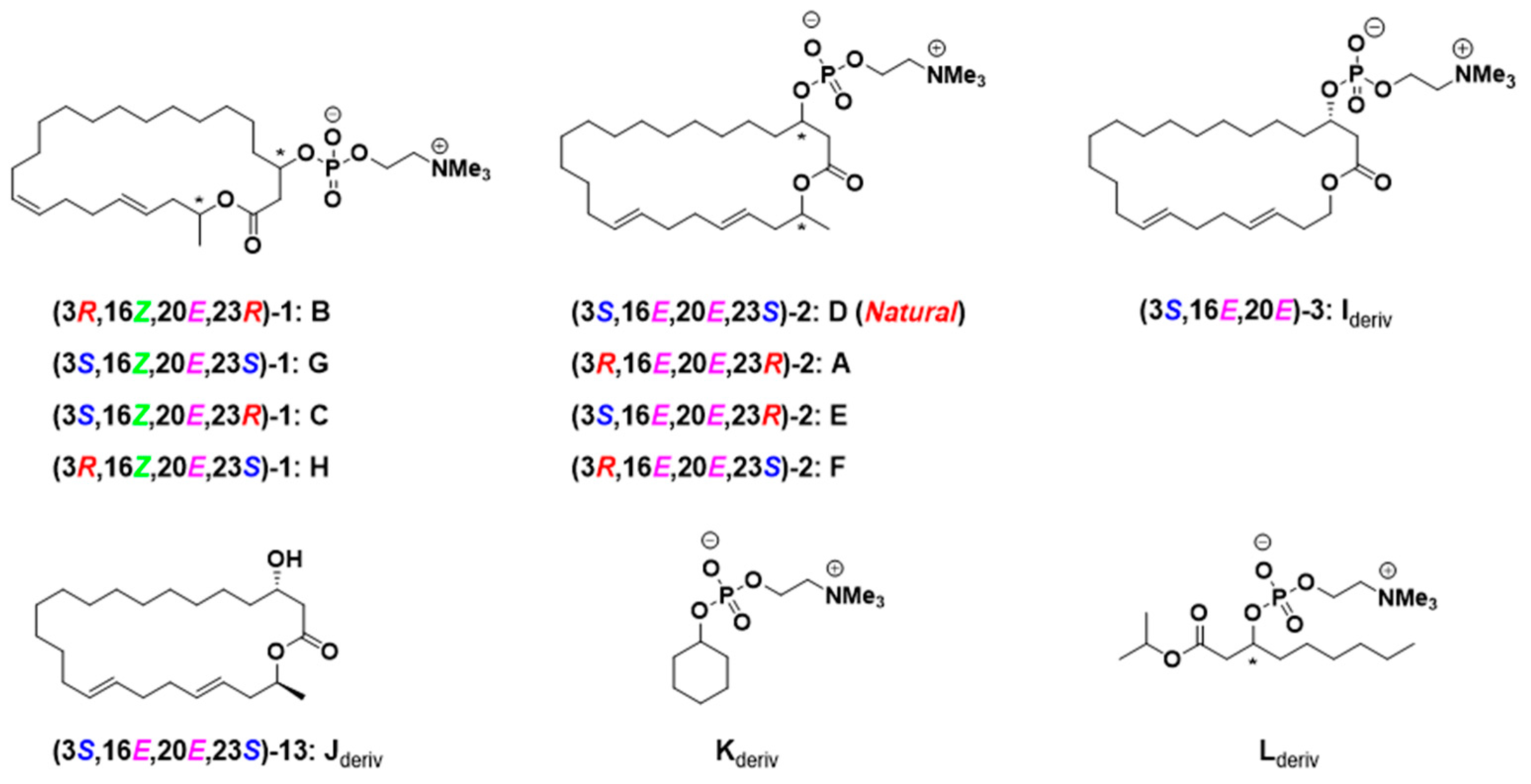
23-Demethyleushearilide (3) (Ideriv): To a cooled (0 °C) solution of 4 (45 mg, 0.123 mmol) in toluene (2.5 mL) were added Et3N (69 μL, 0.493 mmol) and 2-chloro-2-oxo-1,3,2-dioxaphospholane (34 μL, 0.370 mmol). After the solution was stirred at room temperature for 4.5 h, the crude product obtained after the separation of amine salt by filtration was used in the next step without further purification. To a cooled (−15 °C) solution of the crude product in acetonitrile (2.5 mL) was added an excess amount of Me3N. The solution was stirred at 70 °C for 14 h in a stirred autoclave of stainless steel. After cooling to room temperature, the solvent was removed, and the crude product was purified by thin layer chromatography on silica gel (Chromatorex NH-DM1020; chloroform/MeOH = 6/1) to afford 23-demethyleushearilide (3) (23.9 mg, 37%). +8.66 (c 1.14, CH3OH); IR (neat) 3433, 3402, 2924, 2862, 1728 cm−1; 1H NMR (500 MHz, CD3OD) δ 5.49–5.37 (m, 4H, 16-H, 17-H, 20-H, 21-H), 4.56-4.53 (m, 1H, 3-H), 4.26 (m, 2H, 24-H), 4.12–4.03 (m, 2H, 23-H), 3.63–3.62 (m, 2H, 25-H), 3.22 (s, 9H, 26-H), 2.80 (dd, J = 4.6, 14.9 Hz, 1H, 2-H), 2.56 (dd, J = 8.0, 14.9 Hz, 1H, 2-H), 2.31 (q, 2H, J = 6.5 Hz, 22-H), 2.06 (br s, 4H, 18-H, 19-H), 2.00 (dd, J =5.4, 11.7 Hz, 2H, 15-H), 1.68–1.63 (m, 2H, 4-H), 1.44–1.30 (m, 20H, 5-H to 14-H); 13C NMR (125 MHz, CD3OD) δ 172.4 (C1), 134.0 (C20), 131.8 (C16), 131.2 (C17), 127.0 (C21), 74.1 (d, J = 6.0 Hz, C3), 67.5 (C25), 65.3 (C23), 60.3 (d, J = 6.0 Hz, C24), 54.7 (t, J = 3.6 Hz, C26), 41.4 (d, J = 2.4 Hz, C2), 36.2 (d, J = 4.8 Hz, C4), 34.0, 33.6 (C18, C19), 33.1 (C20), 32.7 (C15), 30.1 (C6), 29.8, 29.8, 29.7, 29.6, 29.5, 29.4, 29.2, 28.3 (C7 to C14), 25.4 (C5); HRMS (ESI/TOF) m/z [M + Na]+ calcd for C28H52NO6PNa [M + Na]+ 552.3430, found 552.3446.
Phosphate Kderiv: To a cooled (0 °C) solution of cyclohexanol (30 mg, 0.30 mmol) in toluene (3.0 mL) were added Et3N (71 μL, 0.51 mmol) and 2-chloro-2-oxo-1,3,2-dioxaphospholane (36 μL, 0.39 mmol). After the solution was stirred at room temperature for 2.5 h, the crude product obtained after the separation of amine salt by filtration was used in the next step without further purification. To a cooled (−15 °C) solution of the crude product in acetonitrile (3.0 mL) was added an excess amount (ca. 20 eq.) of Me3N. The solution was stirred at 70 °C for 14 h in a stirred autoclave of stainless steel. After cooling to room temperature, the solvent was removed, and the crude product was purified by thin layer chromatography on silica gel (Chromatorex NH-DM1020; chloroform/MeOH = 5/1) to afford the desired product (Kderiv) (36.9 mg, 46%). IR (neat) 2931, 2854, 1226, 1087, 1049 cm−1; 1H NMR (500 MHz, CD3OD) δ 4.26–4.21 (m, 2H, 7-H), 4.17–4.09 (m, 1H, 1-H), 3.64–3.59 (m, 2H, 8-H), 3.21 (s, 9H, 9-H), 1.98–1.91 (m, 2H, 2-H, 6-H), 1.78–1.71 (m, 2H, 2-H, 6-H), 1.56–1.20 (m, 6H, 3-H to 4-H); 13C NMR (125 MHz, CD3OD) δ 75.8 (d, J = 6.0 Hz), 67.6–67.4 (m), 60.2 (d, J = 4.8 Hz), 54.8–54.6 (m), 34.9 (d, J = 3.6 Hz), 26.5, 24.9; HRMS (ESI/TOF) m/z [M + Na]+ calcd for C11H24NO4PNa 288.1335, found 288.1328.
Phosphate Lderiv: To a cooled (0 °C) solution of isopropyl 3-hydroxynonanoate (40 mg, 0.18 mmol) in toluene (3.7 mL) were added Et3N (0.13 mL, 0.92 mmol) and 2-chloro-2-oxo-1,3,2-dioxaphospholane (68 μL, 0.74 mmol). After the solution was stirred at room temperature for 3 h, the crude product obtained after the separation of amine salt by filtration was used in the next step without further purification. To a cooled (−15 °C) solution of the crude product in acetonitrile (3.7 mL) was added an excess amount (ca. 20 eq.) of Me3N. The solution was stirred at 70 °C for 15 h in a stirred autoclave of stainless steel. After cooling to room temperature, the solvent was removed, and the crude product was purified by thin layer chromatography on silica gel (Chromatorex NH-DM1020; chloroform/MeOH = 5/1) to afford the desired product (Lderiv) (32.3 mg, 46%). M.P. 164.3–164.9 °C; IR (KBr) 2931, 2862, 1720, 1643, 1227, 1088 cm−1; 1H NMR (500 MHz, CD3OD); δ 4.96 (sep, J = 6.0 Hz, 1H, iPr CH), 4.52 (dtd, J = 13.0, 6.5, 6.5 Hz, 1H, 3-H), 4.25 (m, 2H, 10-H), 3.62 (m, 2H, 11-H), 3.22 (s, 9H, 12-H), 2.73 (dd, J =15.0, 6.0 Hz, 1H, 2-H), 2.52 (dd, J =15.0, 7.0 Hz, 1H, 2-H), 1.71–1.60 (m, 2H, 4-H), 1.47–1.26 (m, 8H, 5-H to 8-H), 1.23 (d, J = 2.0 Hz, 3H, iPr CH3), 1.22 (d, J = 5.9 Hz, 3H, iPr CH3), 0.90 (t, J = 7.0 Hz, 3H, 9-H); 13C NMR (125 MHz, CD3OD) δ 172.1, 74.3 (d, J = 5.9 Hz), 69.1, 67.5, 60.3 (d, J = 4.8 Hz), 54.7, 41.8 (d, J = 3.6 Hz), 36.6 (d, J = 3.5 Hz), 32.9, 30.4, 26.0, 23.7, 22.11, 22.07, 14.4; HRMS (ESI/TOF) m/z [M+Na]+ calcd for C17H37NO6PNa 382.2353, found 382.2360.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







