
Isothioureas, Ureas, and Their N-Methyl Amides from 2-Aminobenzothiazole and Chiral Amino Acids

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
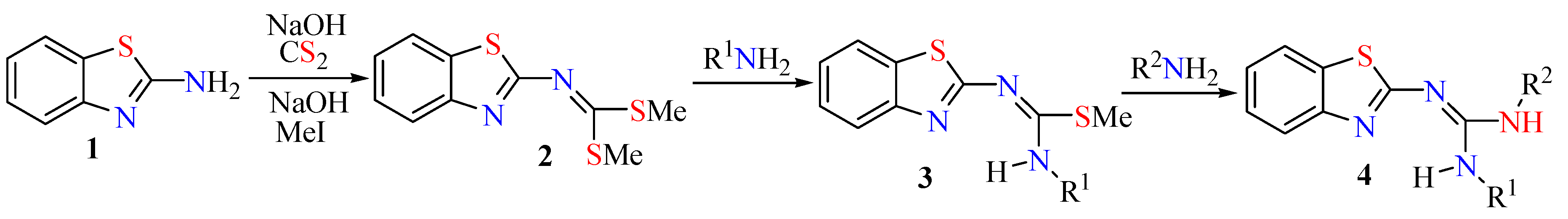{kind=link}
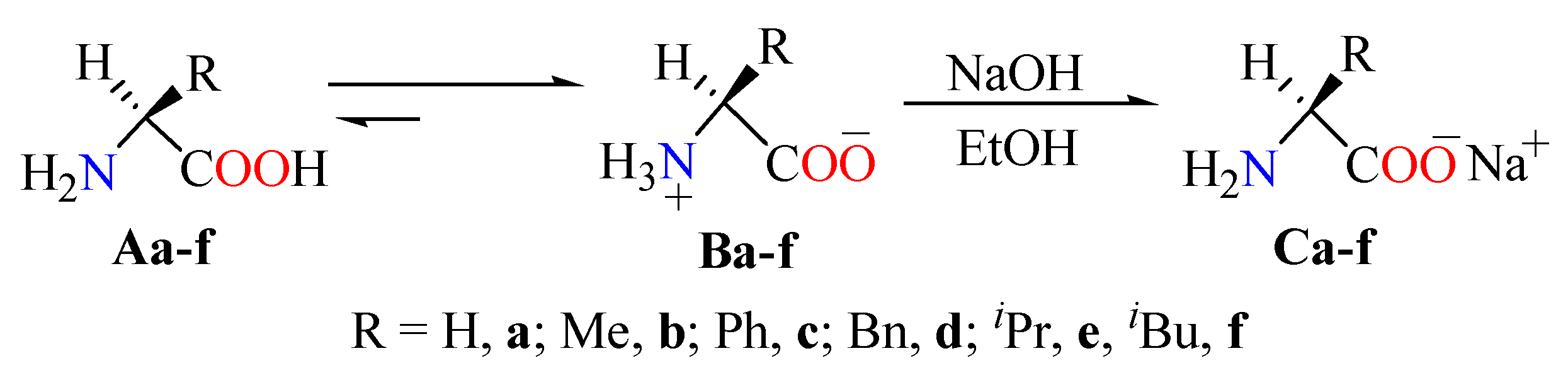{kind=link}
{kind=link}
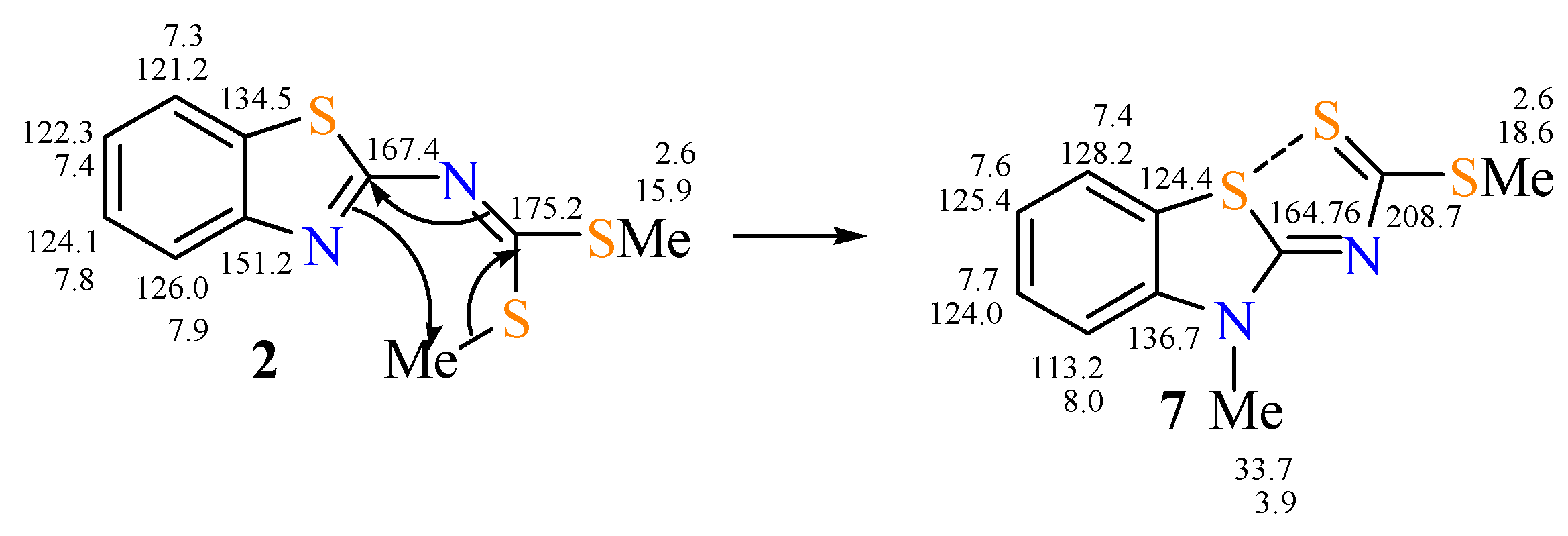{kind=link}
Abstract
1. Introduction
2. Results and Discussion
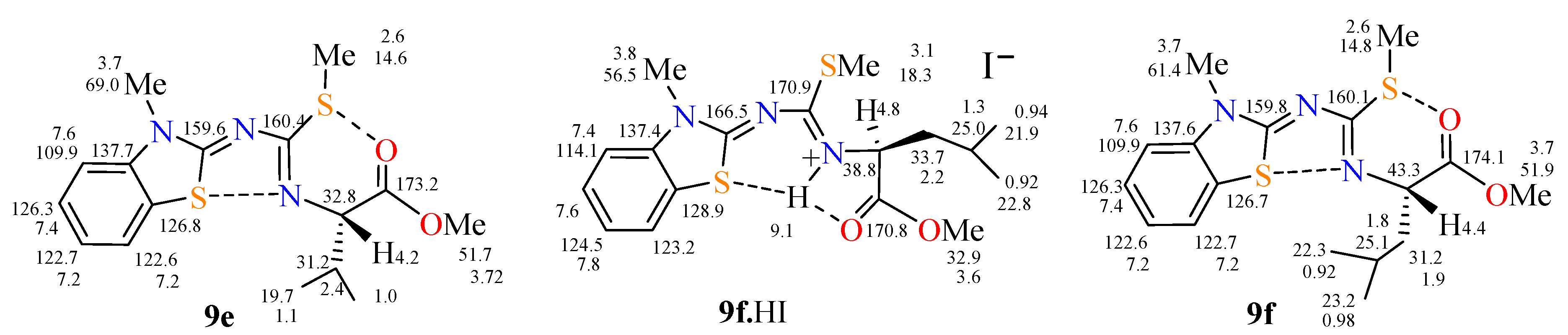
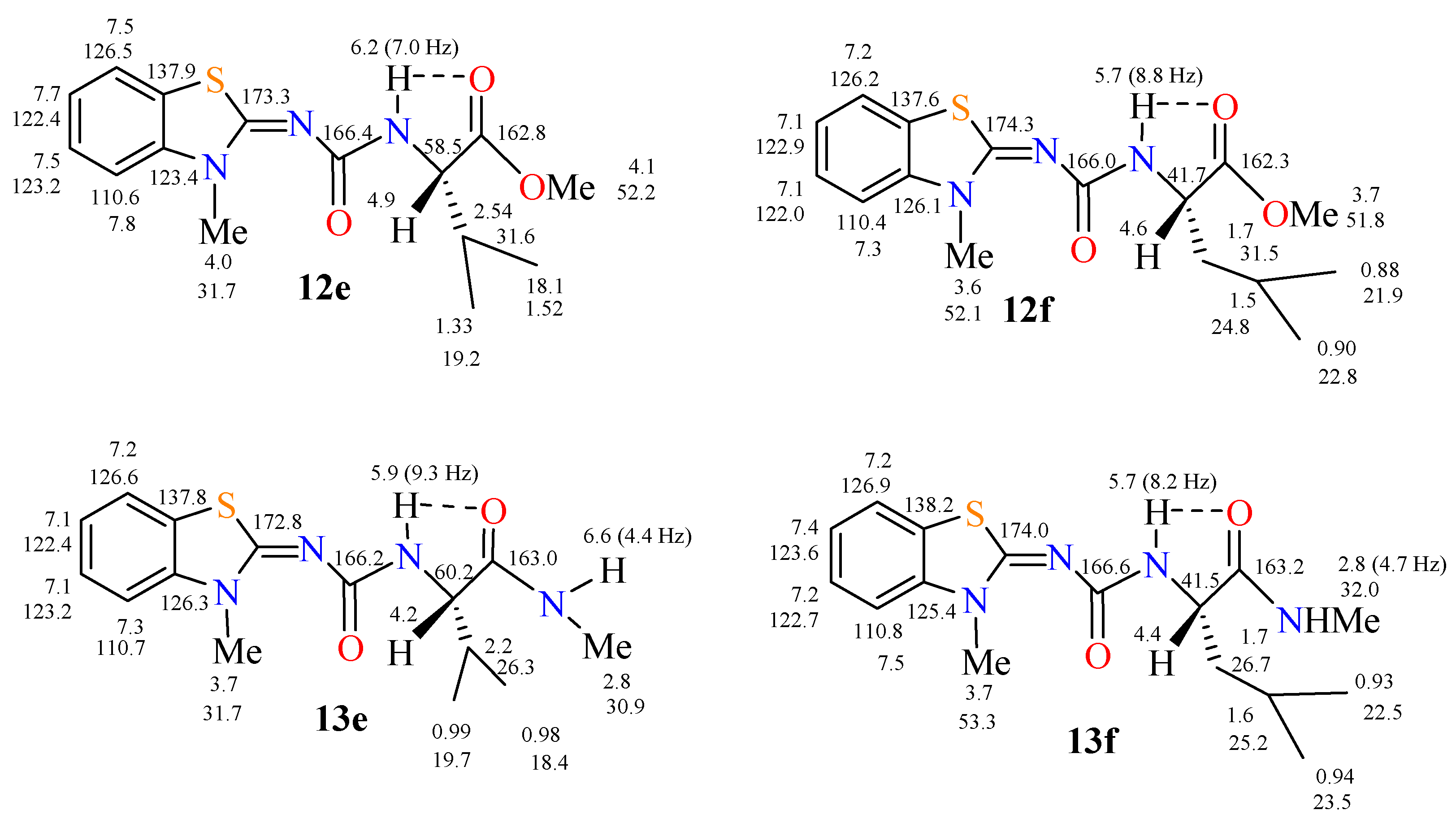
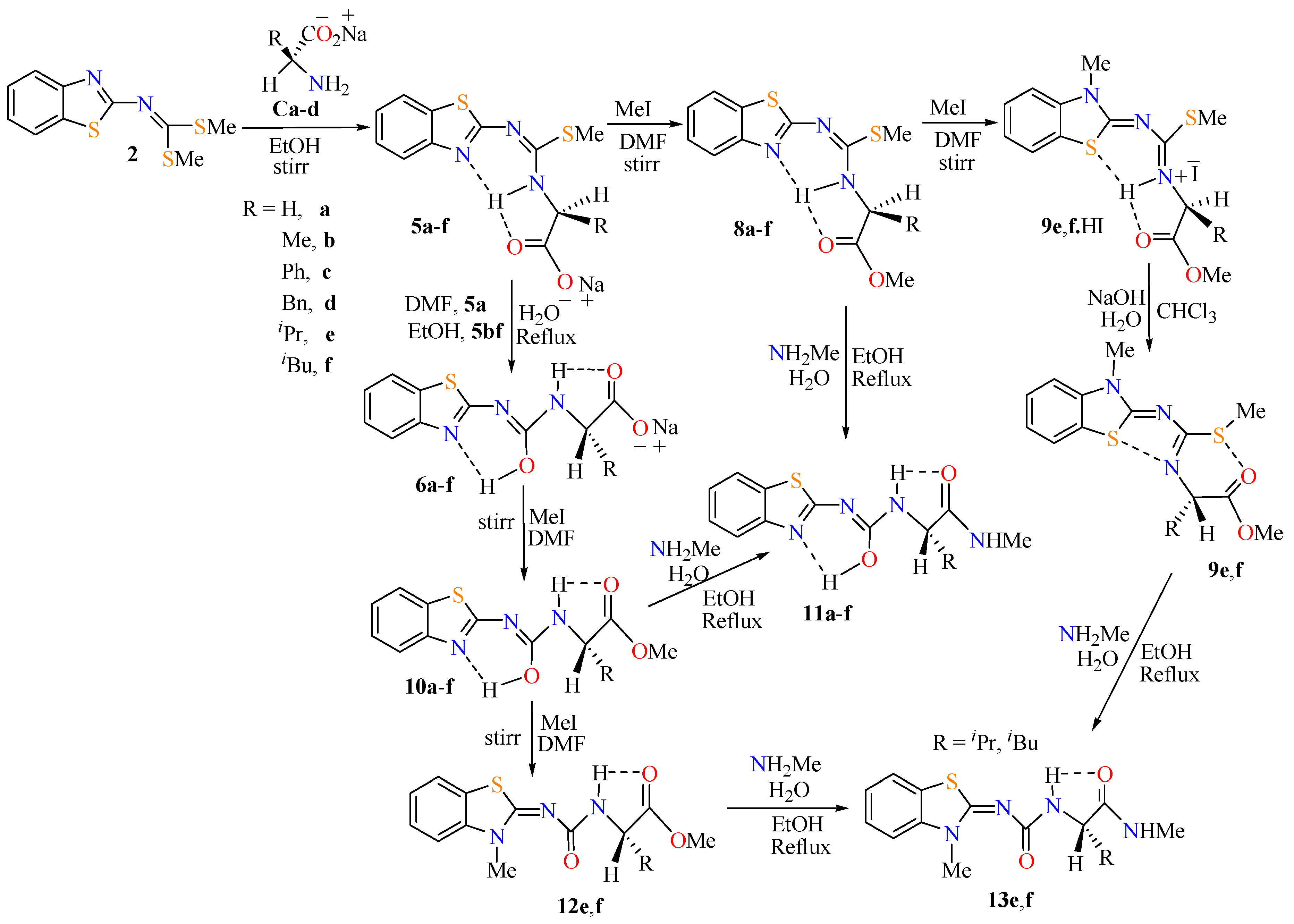
2.1. Reactions and Characterization
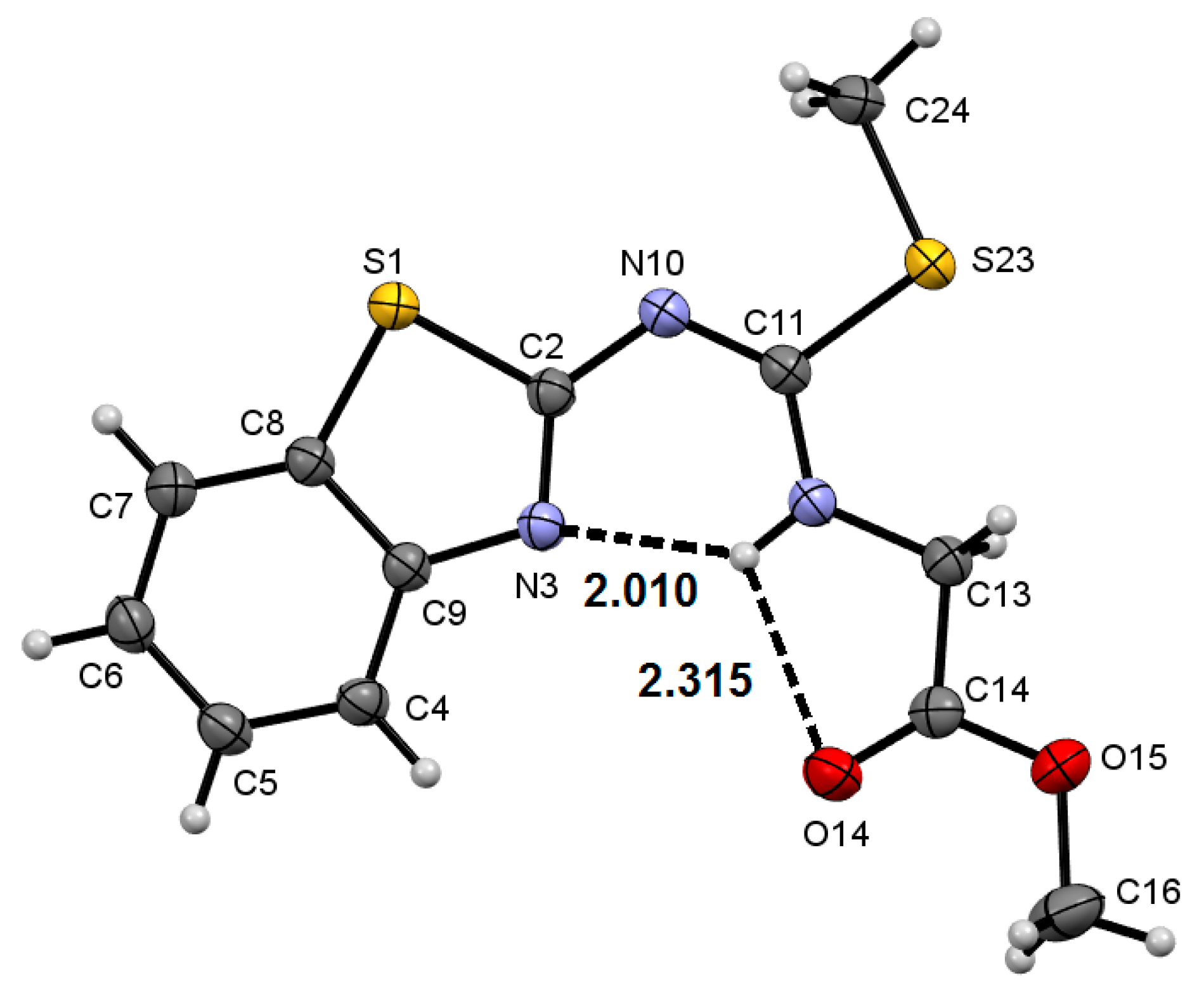
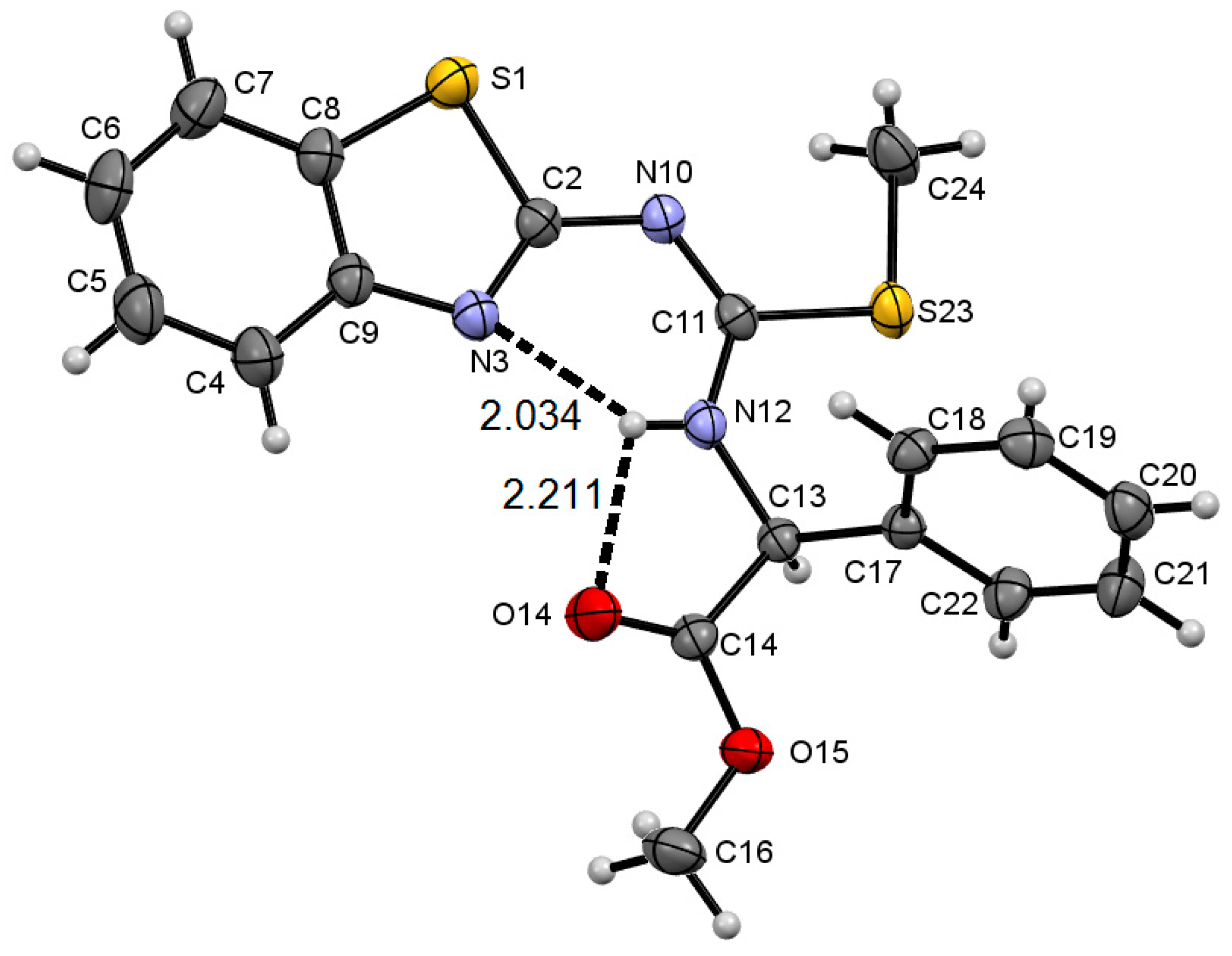
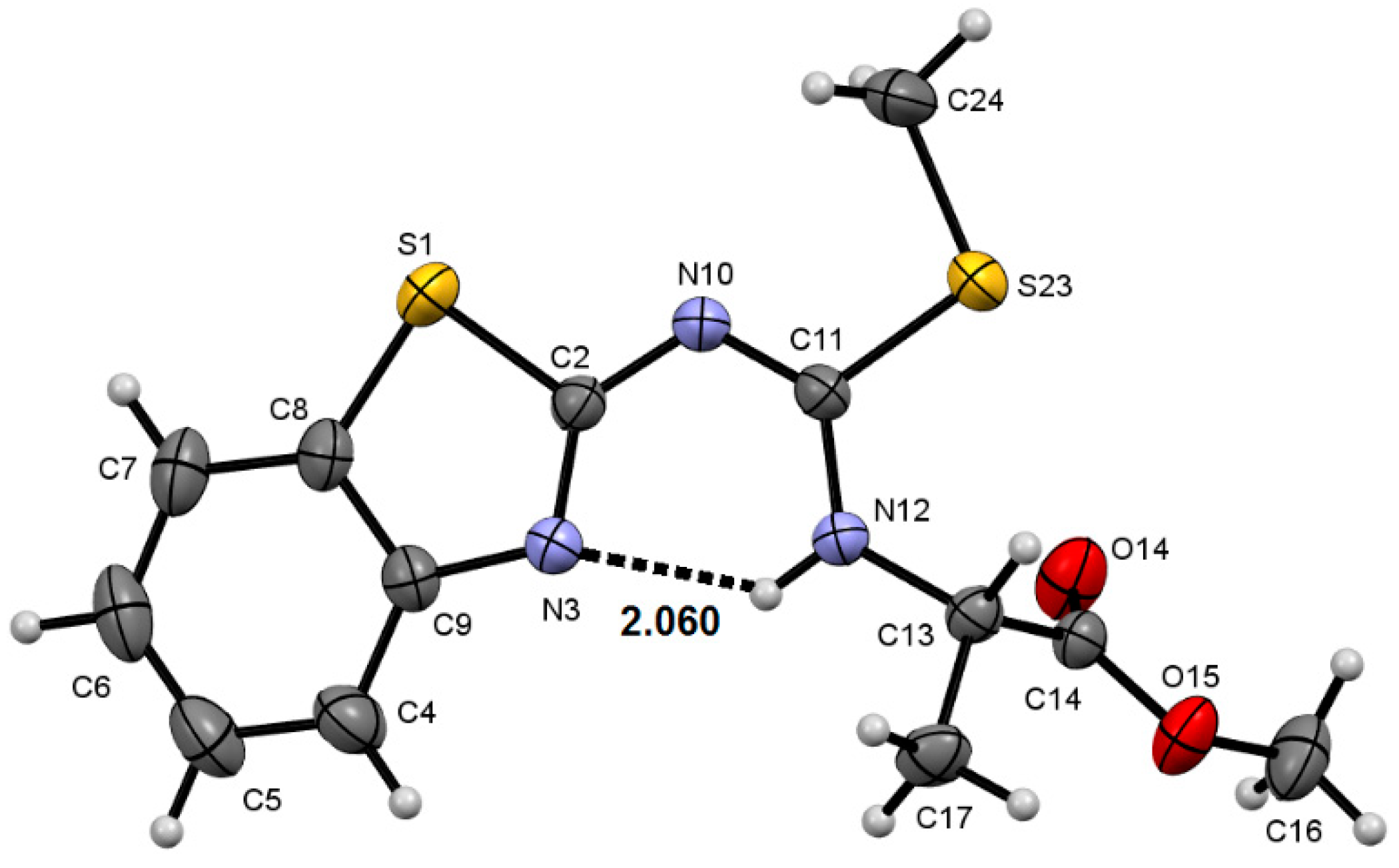
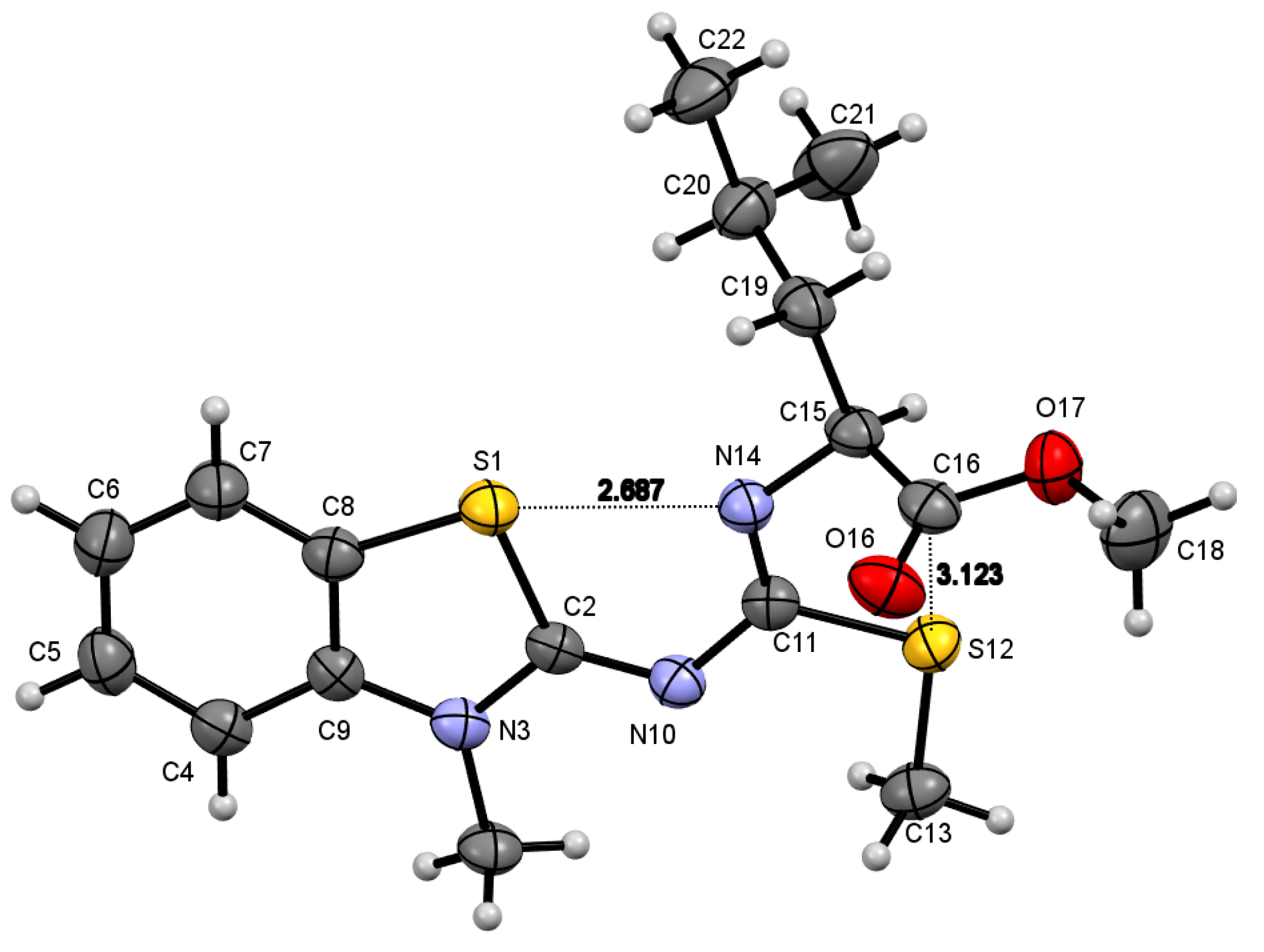
2.2. Molecular Structure of Compounds 8a–c and 9f
3. Materials and Methods
3.1. Experimental Section
3.2. General Method for Isothiourea Carboxylates 5a–f
3.2.1. Sodium (E)-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Acetate 5a
3.2.2. Sodium (S,E)-(+)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Propionate 5b
3.2.3. Sodium (R,E)-(+)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Phenyl-Acetate 5c
3.2.4. Sodium (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-3-Phenyl-Propionate 5d
3.2.5. Sodium (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-3-Methyl-Butanonate 5e
3.2.6. Sodium (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-4-Methyl-Pentanonate 5f
3.3. General Method for Isourea Carboxylates 6a–f
3.3.1. Sodium (3-Benzothiazol-2-Yl-Isoureido)-Acetate 6a
3.3.2. Sodium 2-(3-Benzothiazol-2-Yl-Isoureido)-Propionate 6b
3.3.3. Sodium (3-Benzothiazol-2-Yl-Isoureido)-Phenyl-Acetate 6c
3.3.4. Sodium 2-(3-Benzothiazol-2-Yl-Isoureido)-3-Phenyl-Propionate 6d
3.3.5. Sodium 2-(3-Benzothiazol-2-Yl-Isoureido)-3-Methyl-Butirate 6e
3.3.6. Sodium 2-(3-Benzothiazol-2-Yl-Isoureido)-4-Methyl-Pentanonate 6f
3.4. General Method for SMe-Isothiourea Carboxylate Methyl Esters 8a–f and 9e,f or Isourea Carboxylate Methyl Esters 10a–f and Urea Methyl Ester 12e,f
3.4.1. (E)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Acetic Acid Methyl Ester 8a
3.4.2. (S,E)-(+)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Propionic Acid Methyl Ester 8b
3.4.3. (S,E)-(+)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-Phenyl-Acetic Acid Methyl Ester 8c
3.4.4. (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-3-Phenyl-Propionic Acid Methyl Ester 8d
3.4.5. (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-3-Methyl-Butanoic Acid Methyl Ester 8e
3.4.6. (S,E)-(−)-2-(3-Benzothiazol-2-Yl-2-Methyl-Isothioureido)-4-Methyl-Pentanonic Acid Methyl Ester 8f
3.4.7. 3-Methyl-2-[2-Methyl-3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Isothioureido]-Butyric Acid Methyl Ester 9e
3.4.8. 4-Methyl-2-[2-Methyl-3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Isothioureido]-Pentanoic Acid Methyl Ester 9f
3.4.9. (3-Benzothiazol-2-Yl-Isoureido)-Acetic Acid Methyl Ester 10a
3.4.10. 2-(3-Benzothiazol-2-Yl-Isoureido)-Propionic Acid Methyl Ester 10b
3.4.11. (3-Benzothiazol-2-Yl-Isoureido)-Phenyl-Acetic Acid Methyl Ester 10c
3.4.12. 2-(3-Benzothiazol-2-Yl-Isoureido)-3-Phenyl-Propionic Acid Methyl Ester 10d
3.4.13. 2-(3-Benzothiazol-2-Yl-Isoureido)-3-Methyl-Butyric Acid Methyl Ester 10e
3.4.14. 2-(3-Benzothiazol-2-Yl-Isoureido)-4-Methyl-Pentanoic Acid Methyl Ester 10f
3.4.15. 3-Methyl-2-[3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Ureido]-Butyric Acid Methyl Ester 12e
3.4.16. 4-Methyl-2-[3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Ureido]-Pentanoic Acid Methyl Ester 12f
3.5. General Method for Isourea Amides 11a–f or Urea-Amides 13e,f
3.5.1. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-Acetamide 11a
3.5.2. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-Propionamide 11b
3.5.3. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-2-Phenyl-Acetamide 11c
3.5.4. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-3-Phenyl-Propionamide 11d
3.5.5. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-3-Methyl-Butiramide 11e
3.5.6. 2-[3-(3H-Benzothiazol-2-Ylidene)-Ureido]-N-Methyl-4-Methyl-Pentylamide 11f
3.5.7. 3,N-Dimethyl-2-[3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Ureido]-Butiramide 13e
3.5.8. 4-Methyl-2-[3-(3-Methyl-3H-Benzothiazol-2-Ylidene)-Ureido]-Pentanoic Acid Methylamide 13f
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borthwick, A.D.; Davies, D.E.; Ertl, P.F.; Exall, A.M.; Haley, T.M.; Hart, G.J.; Jackson, D.L.; Parry, N.R.; Patikis, A.; Trivedi, N.; et al. Design and synthesis of pyrrolidine-5,5′-trans-lactams (5-oxo-hexahydropyrrolo [3, 2-b] pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J. Med. Chem. 2003, 46, 4428–4449. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.A.; Loddo, R.; La Colla, P. In vitro antitumor and antiviral activities of new benzothiazoles and 1,3,4-oxadiazole-2-tione derivatives. Acta Pharm. 2008, 58, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Wei, Y.; Ziwen, Y.; Wang, K.; Liang, Y.; Shi, L. Novel cycloalkylthiophene–imine derivatives bearing benzothiazole scaffold: Synthesis, characterization and antiviral activity evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 5131–5134. [Google Scholar] [CrossRef] [PubMed]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R.; Baker, P.J.; Barynin, V.V.; Rice, D.W.; Sedelnikova, S.E.; et al. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Rafique, H.; Hameed, A.; Rasheed, S. Synthesis and antibacterial activity of some new 1-aroyl-3-(substituted-2-benzothiazolyl)thio-ureas. Pharm. Chem. J. 2008, 42, 191–195. [Google Scholar]
- Haydon, D.J.; Bennett, J.M.; Brown, D.; Collins, I.; Galbraith, G.; Lancett, P.; Macdonald, R.; Stokes, N.R.; Pramod, K.; Chauhan, J.K.S.; et al. Creating an antibacterial with in vivo efficacy: Synthesis and characterization of potent inhibitors of the bacterial cell division protein FtsZ with improved pharmaceutical properties. J. Med. Chem. 2010, 53, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Scheich, C.; Puetter, V.; Schade, M. Novel small molecule inhibitors of MDR mycobacterium tuberculosis by NMR fragment screening of antigen 85C. J. Med. Chem. 2010, 53, 8362–8367. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Tilak, R.; Nath, G.; Awasthi, S.K.; Agarwal, A. Design, synthesis and antimicrobial activity of novel benzothiazole analogs. Eur. J. Med. Chem. 2013, 63, 635–644. [Google Scholar] [CrossRef]
- Singh, M.; Gangwar, M.; Nath, G.; Singh, S.K. Synthesis, DNA cleavage and antimicrobial activity of 4-thiazolidinones-benzothiazole conjugates. Indian J. Exp. Biol. 2014, 52, 1062–1070. [Google Scholar]
- Mittal, S.; Samota, M.K.; Kaur, J.; Seth, G.; Mittal, S.; Samota, M.K.; Kaur, J.; Seth, G. Synthesis, spectral, and antifungal evaluation of phosphorylated and thiophosphorylated benzothiazole derivatives. Phosphorus Sulfur 2007, 182, 2105–2113. [Google Scholar] [CrossRef]
- Huang, W.; Yang, G.-F. Microwave-assisted, one-pot syntheses and fungicidal activity of polyfluorinated 2-benzylthiobenzothiazoles. Bioorg. Med. Chem. 2006, 14, 8280–8285. [Google Scholar] [CrossRef] [PubMed]
- Yevich, J.P.; Temple, D.L.; Covington, R.R.; Owens, D.A.; Seidehamel, R.J.; Dungan, K.W. Antiallergics: 3-(1H-Tetrazol-5-yl)-4H-pyrimido[2,1-b ]benzo-thiazol-4-ones. J. Med. Chem. 1982, 25, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Musser, J.H.; Brown, R.E.; Love, B.; Baily, K.; Jones, H.; Kahen, R. Synthesis of 2-(2,3-dihydro-2-oxo-1,3,4-oxadiazol-5-yl) benzo heterocycles. A novel series of orally active antiallergic agents. J. Med. Chem. 1984, 27, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Ager, I.R.; Barnes, A.C.; Danswan, G.W.; Hairsine, P.W.; Kay, D.P.; Kennewell, P.D.; Matharu, S.S.; Miller, P.; Robson, P. Synthesis and oral antiallergic activity of carboxylic acids derived from imidazo[2,1-c][1,4]benzoxazines, imidazo[1,2-a]quinolines, imidazo[1,2-a]quinoxalines, imidazo[1,2-a]quinoxalinones, pyrrolo[1,2-a]quinoxalinones, pyrrolo[2,3-a]quinoxalinones, and imidazo[2,1-b]benzothiazoles. J. Med. Chem. 1988, 31, 1098–1115. [Google Scholar] [PubMed]
- Pattan, S.R.; Suresh, C.; Pujar, V.D.; Reddy, V.V.K.; Rasal, V.P.; Kotti, B.C. Synthesis and antidiabetic activity of 2-amino [5’(4-sulphonylbenzylidine)-2,4-thiazolidinedione]-7-chloro-6-fluorobenzothiazole. Indian J. Chem. 2005, 44, 2404–2408. [Google Scholar]
- Moreno-Díaz, H.; Villalobos-Molina, R.; Ortiz-Andrade, R.; Díaz-Coutiño, D.; Medina-Franco, J.L.; Webster, S.P.; Binnie, M.; Estrada-Soto, S.; Ibarra-Barajas, M.; León-Rivera, I.; et al. Antidiabetic activity of N-(6-substituted-1,3- benzothiazol-2-yl)benzenesulfonamides. Bioorg. Med. Chem. Lett. 2008, 18, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, G.; Prabhat, P.; Sutharson, L.; Banerjee, J.; Patangia, U.; Nath, S. Synthesis and antidiabetic evaluation of benzothiazole derivatives. J. Korean Chem. Soc. 2012, 56, 251–256. [Google Scholar] [CrossRef]
- Yan, Y.; Xie, X.; Zhu, N.; Liu, G. Benzothiazoles exhibit broad-spectrum antitumor activity: Their potency structure-activity and structure-metabolism relationships. Eur. J. Med. Chem. 2014, 76, 67–78. [Google Scholar]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M. Synthesis and in vitro antitumor activity of new series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives. Eur. J. Med. Chem. 2014, 85, 576–592. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R. El-Kerdawy, M.M. New series of benzothiazole and pyrimido[2,1-b]benzothiazole derivatives: Synthesis, antitumor activity, EGFR tyrosine kinase inhibitory activity and molecular modeling studies. Med. Chem. Res. 2015, 24, 860–878. [Google Scholar] [CrossRef]
- Yurttas, L.; Tay, F.; Demirayak, S. Synthesis and antitumor activity evaluation of new 2-(4-amino-phenyl)benzothiazole derivatives bearing different heterocyclic ring. J. Enzyme Inhib. Med. Chem. 2015, 30, 458–465. [Google Scholar] [PubMed]
- Paramashivappa, R.; Kumar, P.P.; Rao, S.P.V.; Rao, S. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657–660. [Google Scholar] [PubMed]
- Deshmukh1, V.K.; Raviprasad, P.; Kulkarni, P.A.; Kuberkar, S.V. Design, synthesis and biological activities of novel 4H-pyrimido [2, 1-b] [1,3] benzothiazole derivatives. Int. J. Chem. Tech. Res. 2011, 3, 136–142. [Google Scholar]
- Munirajasekhar, D.; Himaja, M.; Sunil, V.M. Synthesis and anthelmintic activity of 2-amino-6-substituted benzothiazoles. Int. Res. J. Pharm. 2011, 2, 114–117. [Google Scholar]
- Suresh, C.H.; Rao, J.V.; Jayaveera, K.N.; Subudhi, S.K. Synthesis and anthelmintic activity of 3-(2-hydrazinobenzothiazoles)-substituted indole-2-one. Int. Res. J. Pharm. 2011, 2, 257–261. [Google Scholar]
- Balaji, P.N.; Ranganayakulu, D.; Yadav, K.R.; Jayamma, J.; Kumar, S.; Sivaramaiah, C. Anthelmintic and anti-microbial activities of synthesized heterocyclic pyrazole and its derivatives from fluoro substituted hydrazino benzothiazole. Int. J. Pharm. Tech. Res. 2014, 6, 1970–1975. [Google Scholar]
- Reddy, D.R.S.; Raparla, L.P.; Pradeep, K.; Ahmed, S.M.; Sindhura, J. Synthesis, analytical characterization, antimicrobial, anti-oxidant and anti-convulsant evaluation of some novel 6-fluorobenzothiazole substituted pyrazole analogues. Int. J. Pharm. Chem. 2013, 3, 72–81. [Google Scholar]
- Joseph, J.; Janaki, G.B. Synthesis, structural characterization and biological studies of copper complexes with 2-aminobenzothiazole derivatives. J. Mater. Environ. Sci. 2014, 5, 693–704. [Google Scholar] [CrossRef]
- Fen, R.; Christian, S. Antioxidant potential of novel 2-(4-amino-2-arylaminothiazol-5-oyl) benzothiazoles. Int. J. Pharm. Bio. Sci. 2014, 5, 74–79. [Google Scholar]
- Yadav, A.G.; Patil, V.N.; Asrondkar, A.L.; Naik, A.A.; Ansulkar, P.V.; Bobade, A.S.; Chowdhary, A.S. Anti-oxidant and anti-microbial activities of pyrazolyl-benzothiazole derivatives using Vilsmeier-Haack reaction. Rayazan J. Chem. 2012, 5, 117–120. [Google Scholar]
- Nessim, M.I.; Ahmed, M.H.M.; Ali, A.M.B.; Salem, A.A.; Attia, S.K. The effect of some benzothiazole derivatives as antioxidants for base stock. Int. J. Curr. Res. 2013, 5, 111–1113. [Google Scholar]
- Choudhary, S.; Kini, S.G.; Mubeen, M. Antioxidant activity of novel coumarin substituted benzothiazole derivatives. Der. Pharm. Chem. 2013, 5, 213–222. [Google Scholar]
- Raju, G.N.; Karumudi, B.S.; Rao, N.R. Benzothiazole-Versatile heterocyclic nucleus in medicinal chemistry: A review. Int. J. Pharm. Chem. 2015, 5, 104–114. [Google Scholar]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagumpi, S. A comprehensive review in current developments of benzothiazole based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P. Bicyclic-guanidines, -guanidinates and -guanidinium salts: Wide ranging applications from a simple family of molecules. Chem. Commun. 2009, 3659–3676. [Google Scholar]
- Ishikawa, T. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts; Johon Wiley and Sons Ltd.: Chippenham, UK, 2009. [Google Scholar]
- Berlinck, R.G.S.; Burtoloso, A.C.B.; Trindade-Silva, A.E.; Romminger, S.; Morais, R.P.; Bandeira, K.; Mizuno, C.M. The chemistry and biology of organic guanidine derivatives. Nat. Prod. Rep. 2010, 27, 1871–1907. [Google Scholar] [PubMed]
- Fu, X.; Tan, C.-H. Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar]
- Cunha, S.; Rodriguez, M.T., Jr. The first bismuth(III)-catalyzed guanylation of thioureas. Tetrahedron Lett. 2006, 47, 6955–6956. [Google Scholar]
- Liu, F.; Lu, G.-Y.; He, W.-J.; Hu, J.; Mei, Y.-H.; Zhu, L.-G. Synthesis of calix[4]arene derivatives with alkyl guanidinium or chiral bicyclic guanidinium. Synthesis 2001, 607–611. [Google Scholar]
- Gers, T.; Kunce, D.; Markowski, P.; Izdebski, J. Reagents for efficient conversion of amines to protected guanidines. Synthesis 2004, 37–42. [Google Scholar]
- Ibatullin, F.M.; Selivanov, S.I.; Shavva, A.G. A General procedure for conversion of s-glycosyl isothiourea derivatives into thioglycosides, thiooligosaccharides and glycosyl thioesters. Synthesis 2001, 419–422. [Google Scholar] [CrossRef]
- Porcheddu, A.; Giacomelli, G.; Chinghine, A.; Masala, S. New cellulose-supported reagent: A sustainable approach to guanidines. Org. Lett. 2004, 6, 4925–4927. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Oplinger, J.A.; Tanoury, G.J.; Sherman, P.A.; Fowler, M.; Marshall, S.; Harmon, M.F.; Paith, J.E.; Furfine, E.S. Potent and selective inhibition of human nitric oxide synthases. Inhibition by non-amino acid isothioureas. J. Biol. Chem. 1994, 269, 26669–26676. [Google Scholar] [PubMed]
- Di Giacomo, C.; Sorrenti, V.; Salerno, L.; Cardile, V.; Guerrera, F.; Siracusa, M.A.; Avitabile, M.; Vanella, A. Novel inhibitors of neuronal nitric oxide synthase. Exp. Biol. Med. 2003, 228, 486–490. [Google Scholar]
- Harusawaa, S.; Sawada, K.; Magata, T.; Yoneyama, H.; Araki, L.; Usami, Y.; Hatano, K.; Yamamoto, K.; Yamamoto, D.; Yamatodani, A. Synthesis and evaluation of N-alkyl-S-[3-(piperidin-1-yl)propyl]isothioureas: High affinity and human/rat species-selective histamine H3 receptor antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 6415–6420. [Google Scholar] [CrossRef] [PubMed]
- Paesano, N.; Marzocco, S.; Vicidomini, C.; Carmela, S.; Autore, G.; De Martino, G.; Sbardella, G. Synthesis and biological evaluation of 3-benzyl-1-methyl- and 1-methyl-3-phenyl-isothioureas as potential inhibitors of iNOS. Bioorg. Med. Chem. Lett. 2005, 15, 539–543. [Google Scholar] [PubMed]
- Cruz, A.; Padilla-Martínez, I.I.; García-Báez, E. A synthetic method to access symmetric and non-symmetric 2-(N,N’-disubstituted)guanidinebenzothiazoles. Molecules 2012, 17, 10178–10191. [Google Scholar]
- Cruz, A.; Padilla-Martínez, I.I.; García-Báez, E.V.; Juárez, J.M. S-Methyl-(-N-aryl and-N-alkyl) isothioureas derived from 2-aminobenzothiazole. Arkivoc 2008, 200, 209. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padilla-Martínez, I.I.; González-Encarnación, J.M.; García-Báez, E.V.; Cruz, A.; Ramos-Organillo, Á.A. Isothioureas, Ureas, and Their N-Methyl Amides from 2-Aminobenzothiazole and Chiral Amino Acids. Molecules 2019, 24, 3391. https://doi.org/10.3390/molecules24183391
Padilla-Martínez II, González-Encarnación JM, García-Báez EV, Cruz A, Ramos-Organillo ÁA. Isothioureas, Ureas, and Their N-Methyl Amides from 2-Aminobenzothiazole and Chiral Amino Acids. Molecules. 2019; 24(18):3391. https://doi.org/10.3390/molecules24183391
Chicago/Turabian StylePadilla-Martínez, Itzia I., José Miguel González-Encarnación, Efrén V. García-Báez, Alejandro Cruz, and Ángel Andrés Ramos-Organillo. 2019. "Isothioureas, Ureas, and Their N-Methyl Amides from 2-Aminobenzothiazole and Chiral Amino Acids" Molecules 24, no. 18: 3391. https://doi.org/10.3390/molecules24183391
APA StylePadilla-Martínez, I. I., González-Encarnación, J. M., García-Báez, E. V., Cruz, A., & Ramos-Organillo, Á. A. (2019). Isothioureas, Ureas, and Their N-Methyl Amides from 2-Aminobenzothiazole and Chiral Amino Acids. Molecules, 24(18), 3391. https://doi.org/10.3390/molecules24183391