
Design, Synthesis, and Activity Evaluation of Novel N-benzyl Deoxynojirimycin Derivatives for Use as α-Glucosidase Inhibitors


Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.1.1. 1-DNJ Preparation
2.1.2. Synthesis of Vanillin Derivatives
2.1.3. Synthesis of NB-DNJDs
2.2. Inhibition of α-Glucosidase by NB-DNJDs
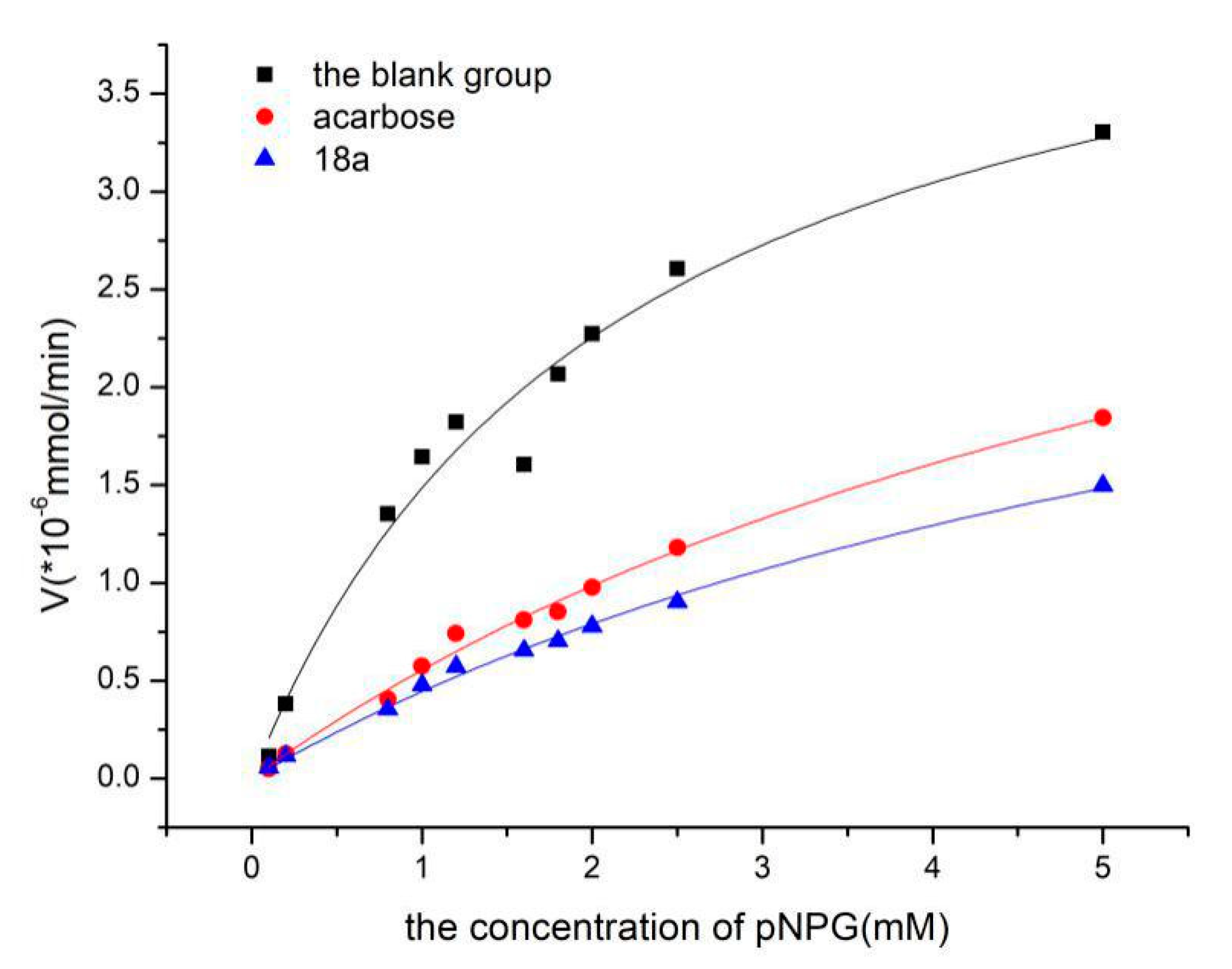
2.3. Enzyme Kinetics Study
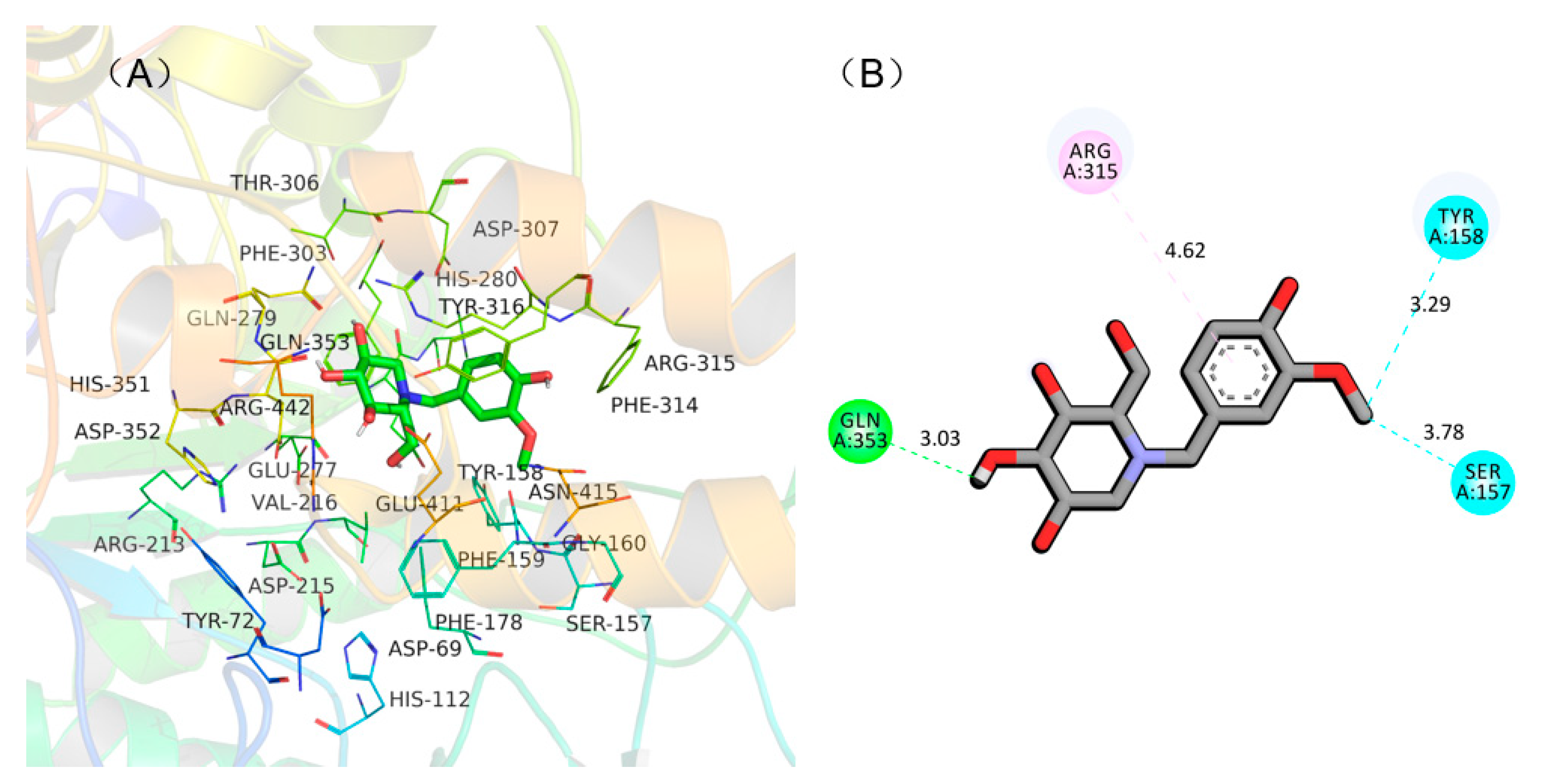
2.4. Molecular Docking Simulation of NB-DNJD and α-Glucosidase
3. Conclusions
4. Experimental Section
4.1. Chemistry
4.1.1. Synthesis of 1-(4-fluorobenzyl)-2-(hydroxymethyl) piperidine-3,4,5-triol (12a)
4.1.2. Synthesis of 1-(4-chlorobenzyl)-2-(hydroxymethyl) piperidine-3,4,5-triol (12b)
4.1.3. Synthesis of 1-(4-bromobenzyl)-2-(hydroxymethyl) Piperidine-3,4,5-triol (12c)
4.1.4. Synthesis of 2-(hydroxymethyl)-1-(4-iodobenzyl) Piperidine-3,4,5-triol (12d)
4.1.5. Synthesis of 2-(hydroxymethyl)-1-(4-methylbenzyl) Piperidine-3,4,5-triol (12e)
4.1.6. Synthesis of 2-(hydroxymethyl)-1-(4-methoxybenzyl) Piperidine-3,4,5-triol (12f)
4.1.7. Synthesis of 1-(3-fluorobenzyl)-2-(hydroxymethyl) Piperidine-3,4,5-triol (12g)
4.1.8. Synthesis of 1-(3-chlorobenzyl)-2-(hydroxymethyl) Piperidine-3,4,5-triol (12h)
4.1.9. Synthesis of 1-(3-bromobenzyl)-2-(hydroxymethyl) Piperidine-3,4,5-triol (12i)
4.1.10. Synthesis of 2-(hydroxymethyl)-1-(3-iodobenzyl) Piperidine-3,4,5-triol (12j)
4.1.11. Synthesis of 2-(hydroxymethyl)-1-(3-methylbenzyl) Piperidine-3,4,5-triol (12k)
4.1.12. Synthesis of 2-(hydroxymethyl)-1-(3-methoxybenzyl) Piperidine-3,4,5-triol (12l)
4.1.13. Synthesis of 4-(benzyloxy)-3-methoxybenzaldehyde (13)
4.1.14. Synthesis of 4-(benzyloxy)-3-bromo-5-methoxybenzaldehyde (14)
4.1.15. General Procedure for the Preparation of Alcohol Derivatives (15a–15e)
4.1.16. General Procedure for the Preparation of Benzyl Bromide Derivatives (16a–16e)
4.1.17. Synthesis of 2-(hydroxymethyl)-1-(3-methoxy-4-phenoxybenzyl) Piperidine-3,4,5-triol (17a)
4.1.18. Synthesis of 1-(3-bromo-5-methoxy-4-phenoxybenzyl)-2-(hydroxymethyl) Piperidizne-3,4,5-triol (17b)
4.1.19. Synthesis of 1-(3-bromo-4,5-dimethoxybenzyl)-2-(hydroxymethyl) Piperidine-3,4,5-triol (17c)
4.1.20. Synthesis of 1-(2,3-dibromo-4,5-dimethoxybenzyl)-2-(hydroxymethyl) Piperidine-3, 4, 5-triol (17d)
4.1.21. Synthesis of 2-(hydroxymethyl)-1-(2,3,6-tribromo-4,5-dimethoxybenzyl)-2-piperidine-3, 4, 5-triol (17e)
4.1.22. Synthesis of 1-(4-hydroxy-3-methoxybenzyl)-2-(hydroxymethyl) piperidine-3,4,5-triol (18a)
4.1.23. Synthesis of 1-(3-bromo-4-hydroxy-5-methoxybenzyl)-2-(hydroxymethyl) piperidine-3,4,5-triol (18b)
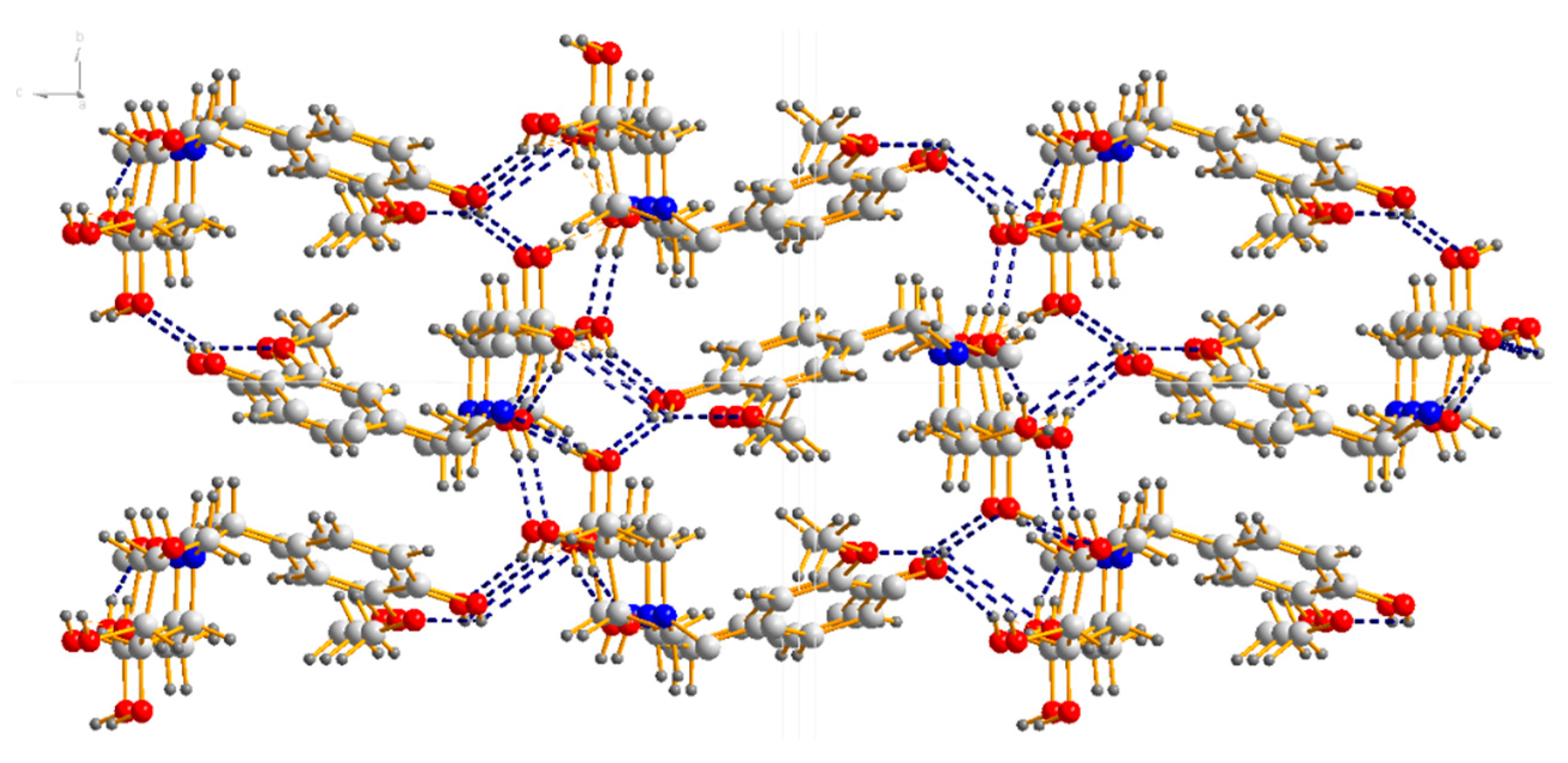
4.2. X-Ray Diffraction Experiment
4.3. In Vitro Assay of α-Glucosidase Inhibitory Activity
4.4. Molecular Docking Simulation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melo, E.B.D.; Gomes, A.D.S.; Carvalho, I. α- and β-Glucosidase inhibitors: Chemical structure and biological activity. Cheminform 2007, 38, 10277–10302. [Google Scholar]
- Kallemeijn, W.W.; Witte, M.D.; Wennekes, T. Mechanism-based inhibitors of glycosidases: Design and applications. Adv. Carbohyd. Chem. Biol. 2014, 71, 297–338. [Google Scholar]
- Junge, B.; Matzke, M.; Stoltefuss, J. Chemistry and Structure-Activity Relationships of Glucosidase Inhibitors; Springer: Heidelberg, Germany, 1996; Volume 119, pp. 411–482. [Google Scholar]
- Nakagawa, K.; Kubota, H.; Kimura, T.; Yamashita, S.; Tsuzuki, T.; Oikawa, S.; Miyazawa, T. Occurrence of orally administered mulberry 1-deoxynojirimycin in rat plasma. J. Agric. Food Chem. 2007, 55, 8928–8933. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Yang, J.; Sheng, L.; Su, C.; Zhang, M.; Jiao, F. Release speed and hypoglycemic effect of mulberry latex and 1-deoxynojirimycin slow-release granules. Sci. Sericul. 2015, 41, 120–126. [Google Scholar]
- Faber, E.D.; Oosting, R.; Neefjes, J.J.; Ploegh, H.L.; Meijer, D.K. Distribution and elimination of the glycosidase inhibitors 1-deoxymannojirimycin and N-methyl-1-deoxynojirimycin in the rat in vivo. Pharm. Res. 1992, 9, 1442–1450. [Google Scholar] [CrossRef]
- Mellor, H.R.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Membrane disruption and cytotoxicity of hydrophobic N-alkylated imino sugars is independent of the inhibition of protein and lipid glycosylation. Biochem. J. 2003, 374, 307–314. [Google Scholar] [CrossRef]
- Mellor, H.R.; Nolan, J.; Pickering, L.; Wormald, M.R.; Platt, F.M.; Gwek, R.A.; Fleet, G.W.J.; Butters, T.D. Preparation, biochemical characterization and biological properties of radiolabelled N-alkylated deoxynojirimycins. Biochem. J. 2002, 366, 225–233. [Google Scholar] [CrossRef][Green Version]
- Butters, T.D.; Van den Broek, L.A.; Fleet, G.W.; Krulle, T.M.; Wormald, M.R.; Dwek, R.A.; Platt, F.M. Molecular requirements of imino sugars for the selective control of N-linked glycosylation and glycosphingolipid biosynthesis. Tetrahedron Asymmetry 2000, 11, 113–124. [Google Scholar] [CrossRef]
- Mellor, H.R.; Neville, D.C.A.; Harvey, D.J.; Platt, F.M.; Dwek, R.A.; Butters, T.D. Cellular effects of deoxynojirimycin analogues: Uptake, retention and inhibition of glycosphingolipid biosynthesis. Biochem. J. 2004, 381, 861–866. [Google Scholar] [CrossRef]
- Weber, K.T.; Hammache, D.; Fantini, J.; Ganem, B. Synthesis of glycolipid analogues that disrupt binding of HIV-1 gp120 to galactosylceramide. Bioorg. Med. Chem. Lett. 2000, 10, 1011–1014. [Google Scholar] [CrossRef]
- Alonzi, D.S.; Dwek, R.A.; Butters, T.D. Improved cellular inhibitors for glycoprotein processing α-Glucosidases: Biological characterisation of alkyl- and arylalkyl-N-substituted deoxynojirimycins. Tetrahedron Asymmetry 2009, 20, 897–901. [Google Scholar] [CrossRef]
- Wennekes, T.; van den Berg, R.J.; Donker, W.; van der Marel, G.A.; Strijland, A.; Aerts, J.M.; Overkleeft, H.S. Development of adamantan-1-yl-methoxy-functionalized 1-deoxynojirimycin derivatives as selective inhibitors of glucosylceramide metabolism in man. J. Org. Chem. 2007, 72, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, M.; Xie, P.; Wan, S.; Ye, J.; Zhou, X.; Wu, J. Specific inhibition effects of N-pentafluorobenzyl-1-deoxynojirimycin on human CD4+ T cells. Bioorg. Med. Chem. Lett. 2004, 14, 3789–3792. [Google Scholar] [CrossRef]
- Godin, G.; Compain, P.; Martin, O.R.; Ikeda, K.; Yu, L.; Asano, N. α-1-C-Alkyl-1-deoxynojirimycin derivatives as potent and selective inhibitors of intestinal isomaltase: Remarkable effect of the alkyl chain length on glycosidase inhibitory profile. Bioorg. Med. Chem. Lett. 2004, 14, 5991–5995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Y.; Boyle, K.; Murphy, P. Hybrid angiogenesis inhibitors: Synthesis and biological evaluation of bifunctional compounds based on 1-deoxynojirimycin and aryl-1,2,3-triazoles. Bioorg. Med. Chem. Lett. 2008, 18, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; Lang, B.; Leeman, M.; van den Marel, G.A.; Smits, E.; Weber, M.; Wiltenburg, J.V.; Wolberg, M.; Aerts, J.M.F.G.; Overkleeft, H.S. Large-Scale Synthesis of the Glucosylceramide Synthase Inhibitor N-[5-(Adamantan-1-yl-methoxy)-pentyl]-1-deoxynojirimycin. Org. Proc. Res. Dev. 2008, 12, 414–423. [Google Scholar] [CrossRef]
- Matos, C.R.R.; Lopes, R.S.C.; Lopes, C.C. Synthesis of 1-Deoxynojirimycin and N-Butyl-1-Deoxynojirimycin; Thieme Stuttgart: New York, NY, USA, 1999; Volume 4, pp. 571–573. [Google Scholar]
- Jiang, B.; Shi, D.; Cui, Y.; Guo, S. Design, synthesis, and biological evaluation of bromophenol derivatives as protein tyrosine phosphatase 1B inhibitors. Arch. Der. Pharm. 2012, 345, 444–453. [Google Scholar] [CrossRef]
- Huleatt, P.B.; Choo, S.S.; Chua, S.; Chai, C.L.L. Expedient routes to valuable bromo-5,6-dimethoxyindole building blocks. Tetrahedron Lett. 2008, 49, 5309–5311. [Google Scholar] [CrossRef]
- Tsai, S.; Klinman, J.P. De novo design and utilization of photolabile caged substrates as probes of hydrogen tunneling with horse liver alcohol dehydrogenase at sub-zero temperatures: A cautionary note. Bioorg. Chem. 2003, 31, 172–190. [Google Scholar] [CrossRef]
- Jiang, B.; Guo, S.; Shi, D.; Guo, C.; Wang, T. Discovery of novel bromophenol 3,4-dibromo-5-(2-bromo-3,4-dihydroxy-6-(isobutoxymethyl)benzyl)benzene-1,2-diol as protein tyrosine phosphatase 1B inhibitor and its anti-diabetic properties in C57BL/KsJ-db/db mice. Eur. J. Med. Chem. 2013, 64, 129–136. [Google Scholar] [CrossRef]
- Akbaba, Y.; Balaydin, H.T.; Göksu, S.; Sahin, E.; Menzek, A. Total Synthesis of the Biologically Active, Naturally Occurring 3,4-Dibromo-5-[2-bromo-3,4-dihydroxy-6-(methoxymethyl)benzyl]benzene-1,2-diol and Regioselective O-Demethylation of Aryl Methyl Ethers. Helvetica Chim. Acta 2010, 93, 1127–1135. [Google Scholar] [CrossRef]
- Takenaka, Y.; Tanahashi, T.; Nagakura, N.; Hamada, N. Phenyl Ethers from Cultured Lichen Mycobionts of Graphis scripta var. serpentina and G. rikuzensis. Chem. Pharm. Bull. 2003, 51, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Jay, S.S. Diabetes mellitus:pathogenesis and treatment strategies. Med. Chem. 2004, 47, 4113–4117. [Google Scholar]
- Nik, K.N.A.Z.; Muhammad, T.; Norizan, A.; Nor, H.I.; Abdul, W.; Fazal, R. Sythesis, molecular docking studies of hybrid benzimidazole as α-Glucosidase inhibitor. Bioorg. Chem. 2017, 70, 184–191. [Google Scholar]
- Hideyuki, K.; Takeshi, M.; Jun, K.; Koretaro, T. Two New Bromophenols from the Red Alga Odonthalia corymbifera. J. Nat. Prod. 1999, 62, 882–884. [Google Scholar]
- Keun, Y.K.; The, H.N.; Hideyuki, K.; Sang, M.K. α-Glucosidase Inhibitory Activity of BromophenolPurified from the Red Alga Polyopes lancifolia. J. Food Sci. 2010, 75, 145–150. [Google Scholar]
- Hideyuki, K.; Takeshi, M.; Jun, K.; Koretaro, T. Inhibitory Potencies of Bromophenols from Rhodomelaceae Algae against α-Glucosidase Activity. Fish. Sci. 1999, 65, 300–303. [Google Scholar]
- Seino, A.K.J.; Sami, C.; Christina, T.; Gary, D.B.; Stephen, G.W.; Hiroaki, S. Rapid Discovery of Potent and Selective Glycosidase-Inhibiting De Novo Peptides. Cell Chem. Biol. 2017, 24, 381–390. [Google Scholar]
- Bruker. APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2005. [Google Scholar]
- Sheldrick, G.M. SHELXS 97, Program for Crystal Structure Solution; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Kiso, M.; Ando, K.; Inagaki, H.; Ishida, H.; Hasegawa, A. Synthetic and structural studies of α-sialyl-(2→6) and α-sialyl-(2→3) 1-deoxynojirimycin derivatives potentially useful for biomedical applications. Carbohydr. Res. 1995, 272, 159–178. [Google Scholar] [CrossRef]
- Zhu, J.; Yin, Z.; Chen, J.; Shang Guan, X.; Peng, D.; Jiang, Y. A model for screening trace amounts of α-Glucosidase inhibitors and methods for judging inhibition type. Modern Food Sci. Tech. 2016, 32, 164–170. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Ar | IC50 (mM) ± SEM | Compound | Ar | IC50 (mM) ± SEM |
|---|---|---|---|---|---|
| 12a |  | 9.8 ± 0.31 | 12k |  | 10.3 ± 0.21 |
| 12b |  | 11.22 ± 0.02 | 12l |  | 1.253 ± 0.34 |
| 12c |  | 12.55 ± 0.03 | 17a |  | 15.33 ± 0.02 |
| 12d |  | 9.88 ± 0.05 | 17b |  | 15.68 ± 0.06 |
| 12e |  | 8.65 ± 0.08 | 17c |  | 1.056 ± 0.11 |
| 12f |  | 1.017 ± 0.23 | 17d |  | 1.142 ± 0.09 |
| 12g |  | 10.23 ± 0.02 | 17e |  | 9.57 ± 0.08 |
| 12h |  | 15.32 ± 0.04 | 18a |  | 0.207 ± 0.11 |
| 12i |  | 15.66 ± 0.04 | 18b |  | 0.276 ± 0.13 |
| 12j |  | 10.54 ± 0.04 | Acarbose |  | 0.353 ± 0.09 |
| Vmax/*10−6 mmol/min | Km/mM | Ki/mM | IC50/mM | |
| blank group | 4.68 ± 0.40 | 2.15 ± 0.43 | ||
| acarbose | 4.40 ± 0.42 | 0.113 ± 0.45 | 0.353 ± 0.09 | |
| 18a | 3.56 ± 0.28 | 0.110 ± 0.30 | 0.207 ± 0.11 |
| Empirical formula | C14 H21NO6 | Z | 4 |
| Formula weight | 299.32 | Dc (g cm−3) | 1.380 |
| Crystal system | Orthorhombic | Mu (MoKa) (/mm) | 0.108 |
| Space group | P212121 | F (000) | 640 |
| a(Å) | 7.760(2) | Crystal size (mm3) | 0.160 × 0.160 × 0.140 |
| b(Å) | 9.028(2) | Θ range for date collection (°) | 2.464 to 25.492 |
| c(Å) | 20.563(6) | Reflections collected | 11,171 |
| α(°) | 90.00 | Independent reflection | 2679 [R(int) = 0.0249] |
| β(°) | 90.00 | Goodness-of-fit on F2 | 1.039 |
| γ(°) | 90.00 | Final R indices [I > 2σ(I)] a | R1 = 0.0352, wR2 = 0.0873 |
| V(Å 3) | 1440.6(7) | R indices (all date) a | R1 = 0.0386, wR2 = 0.0896 |
| Bond | Dist. | Bond | Dist. | Bond | Dist. |
| C (1)-C (2) | 1.381(4) | C (1)-C (6) | 1.396(4) | C (1)-C (7) | 1.522(3) |
| C (2)-C (3) | 1.389(4) | C(3)-C(4) | 1.380(4) | C (4)-O (1) | 1.361(3) |
| C (4)-C (5) | 1.398(4) | C(5)-O(2) | 1.370(3) | C (5)-C (6) | 1.385(4) |
| C (7)-N (1) | 1.487(3) | C(8)-O(2) | 1.406(4) | C(9)-N(1) | 1.468(3) |
| C (9)-C (10) | 1.512(3) | C(10)-O(3) | 1.426(3) | C(10)-C(11) | 1.506(4) |
| C (11)-O (4) | 1.438(3) | C(11)-C(12) | 1.507(3) | C(12)-O(5) | 1.425(3) |
| C(12)-C(13) | 1.535(3) | C(13)-N(1) | 1.478(3) | C(13)-C(14) | 1.521(3) |
| C(14)-O(6) | 1.416(3) | ||||
| Angle | (°) | Angle | (°) | Angle | (°) |
| C(2)-C(1)-C(6) | 117.8(2) | C(2)-C(1)-C(7) | 123.2(2) | C(6)-C(1)-C(7) | 118.9(2) |
| C(1)-C(2)-C(3) | 121.2(3) | C(4)-C(3)-C(2) | 120.7(3) | O(1)-C(4)-C(3) | 118.8(3) |
| O(1)-C(4)-C(5) | 122.4(3) | C(3)-C(4)-C(5) | 118.9(2) | O(2)-C(5)-C(6) | 125.2(3) |
| O(2)-C(5)-C(4) | 114.9(2) | C(6)-C(5)-C(4) | 119.9(2) | C(5)-C(6)-C(1) | 121.4(3) |
| N(1)-C(7)-C(1) | 118.1(2) | N(1)-C(9)-C(10) | 110.56(19) | O(3)-C(10)-C(11) | 106.66(19) |
| O(3)-C(10)-C(9) | 110.8(2) | C(11)-C(10)-C(9) | 111.5(2) | O(4)-C(11)-C(10) | 110.3(2) |
| O(4)-C(11)-C(12) | 107.5(2) | C(10)-C(11)-C(12) | 112.82(19) | O(5)-C(12)-C(11) | 108.51(19) |
| O(5)-C(12)-C(13) | 109.10(19) | C(11)-C(12)-C(13) | 112.8(2) | N(1)-C(13)-C(14) | 114.0(2) |
| N(1)-C(13)-C(12) | 108.88(18) | C(14)-C(13)-C(12) | 108.42(19) | O(6)-C(14)-C(13) | 112.0(2) |
| C(9)-N(1)-C(13) | 109.12(19) | C(9)-N(1)-C(7) | 112.42(19) | C(13)-N(1)-C(7) | 115.62(19) |
| C(5)-O(2)-C(8) | 117.8(2) |
| 18a | ||||
|---|---|---|---|---|
| O(6)–H(7)⋅⋅⋅O(3) #1 | 0.82 | 2.04 | 2.828(3) | 161.4 |
| O(5)–H(5)⋅⋅⋅N(1) | 0.82 | 2.11 | 2.923(3) | 171.0 |
| O(4)–H(4)⋅⋅⋅O(6) #2 | 0.082 | 2.64 | 3.096(3) | 116.6 |
| O(4)–H(4)⋅⋅⋅O(1) #3 | 0.82 | 2.17 | 2.928(3) | 154.6 |
| O(3)–H(3)⋅⋅⋅O(4) #4 | 0.82 | 1.86 | 2.670(3) | 169.5 |
| O(1)–H(1)⋅⋅⋅O(5) #5 | 0.82 | 2.06 | 2.784(3) | 146.5 |
| O(1)–H(1)⋅⋅⋅O(2) | 0.82 | 2.27 | 2.706(3) | 114.0 |
| C(10)–H(10)⋅⋅⋅O(6) #6 | 0.98 | 2.53 | 3.277(3) | 133.4 |
| C(7)–H(7B)⋅⋅⋅O(3) #1 | 0.97 | 2.60 | 3.566(3) | 173.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, F.; Yin, Z.; Chen, J.; Nie, X.; Lin, P.; Lu, T.; Wang, M.; Peng, D. Design, Synthesis, and Activity Evaluation of Novel N-benzyl Deoxynojirimycin Derivatives for Use as α-Glucosidase Inhibitors. Molecules 2019, 24, 3309. https://doi.org/10.3390/molecules24183309
Zeng F, Yin Z, Chen J, Nie X, Lin P, Lu T, Wang M, Peng D. Design, Synthesis, and Activity Evaluation of Novel N-benzyl Deoxynojirimycin Derivatives for Use as α-Glucosidase Inhibitors. Molecules. 2019; 24(18):3309. https://doi.org/10.3390/molecules24183309
Chicago/Turabian StyleZeng, Fanxin, Zhongping Yin, Jiguang Chen, Xuliang Nie, Ping Lin, Tao Lu, Meng Wang, and Dayong Peng. 2019. "Design, Synthesis, and Activity Evaluation of Novel N-benzyl Deoxynojirimycin Derivatives for Use as α-Glucosidase Inhibitors" Molecules 24, no. 18: 3309. https://doi.org/10.3390/molecules24183309
APA StyleZeng, F., Yin, Z., Chen, J., Nie, X., Lin, P., Lu, T., Wang, M., & Peng, D. (2019). Design, Synthesis, and Activity Evaluation of Novel N-benzyl Deoxynojirimycin Derivatives for Use as α-Glucosidase Inhibitors. Molecules, 24(18), 3309. https://doi.org/10.3390/molecules24183309