N…C and S…S Interactions in Complexes, Molecules, and Transition Structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN



Abstract
1. Introduction
2. Methods
3. Results and Discussion
3.1. HN(CH)SX:SCO Complexes with C1 Symmetry
3.2. HN(CH)SX:SCO Complexes with Cs Symmetry
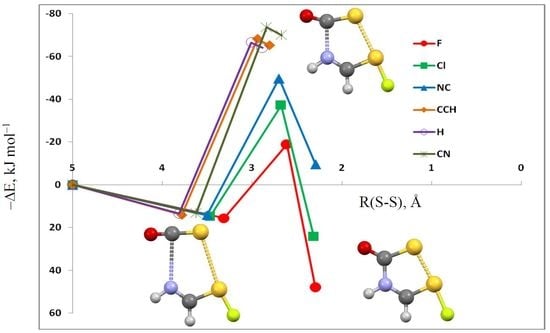
3.3. HN(CH)SX:SCO Molecules and Transition Structures
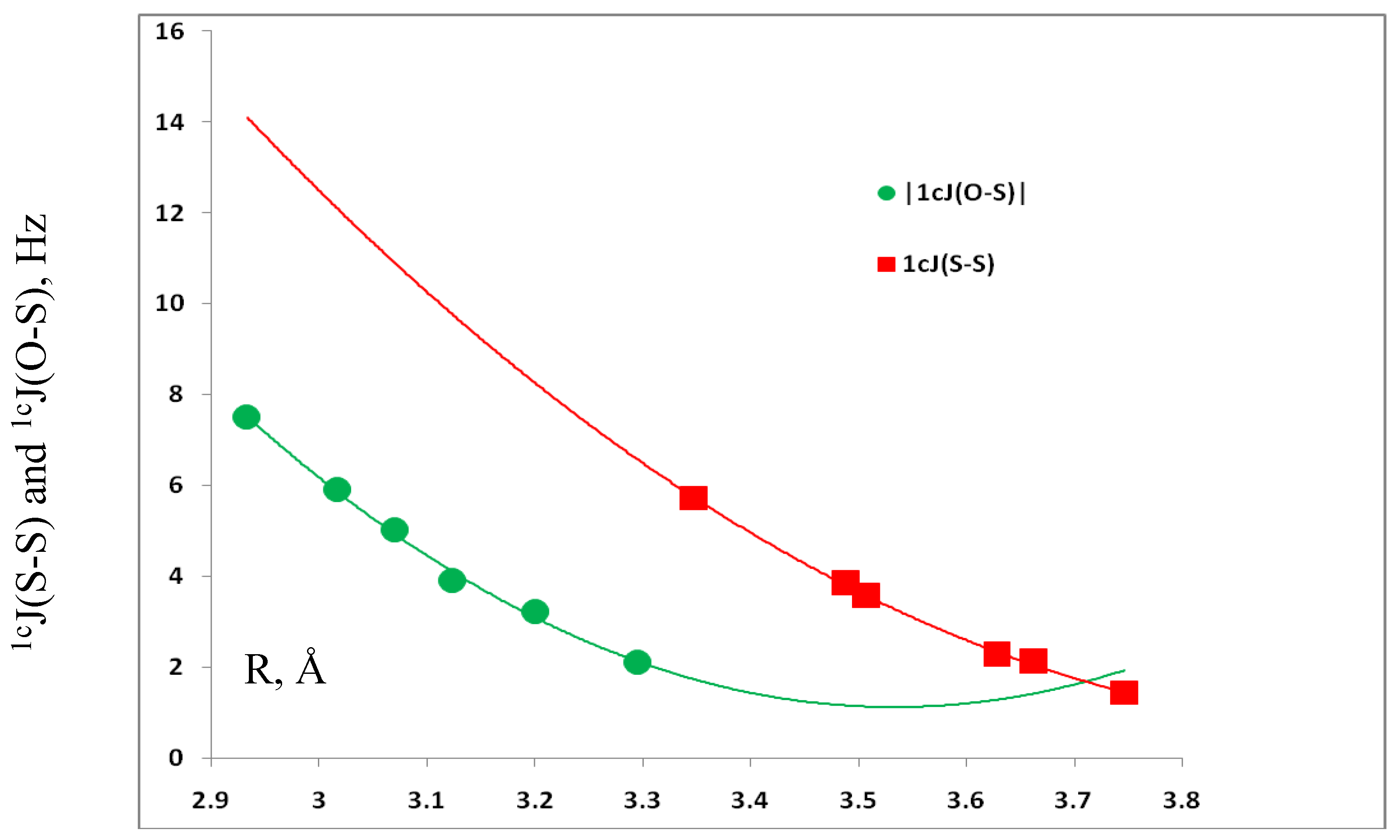
3.4. Spin-Spin Coupling Constants
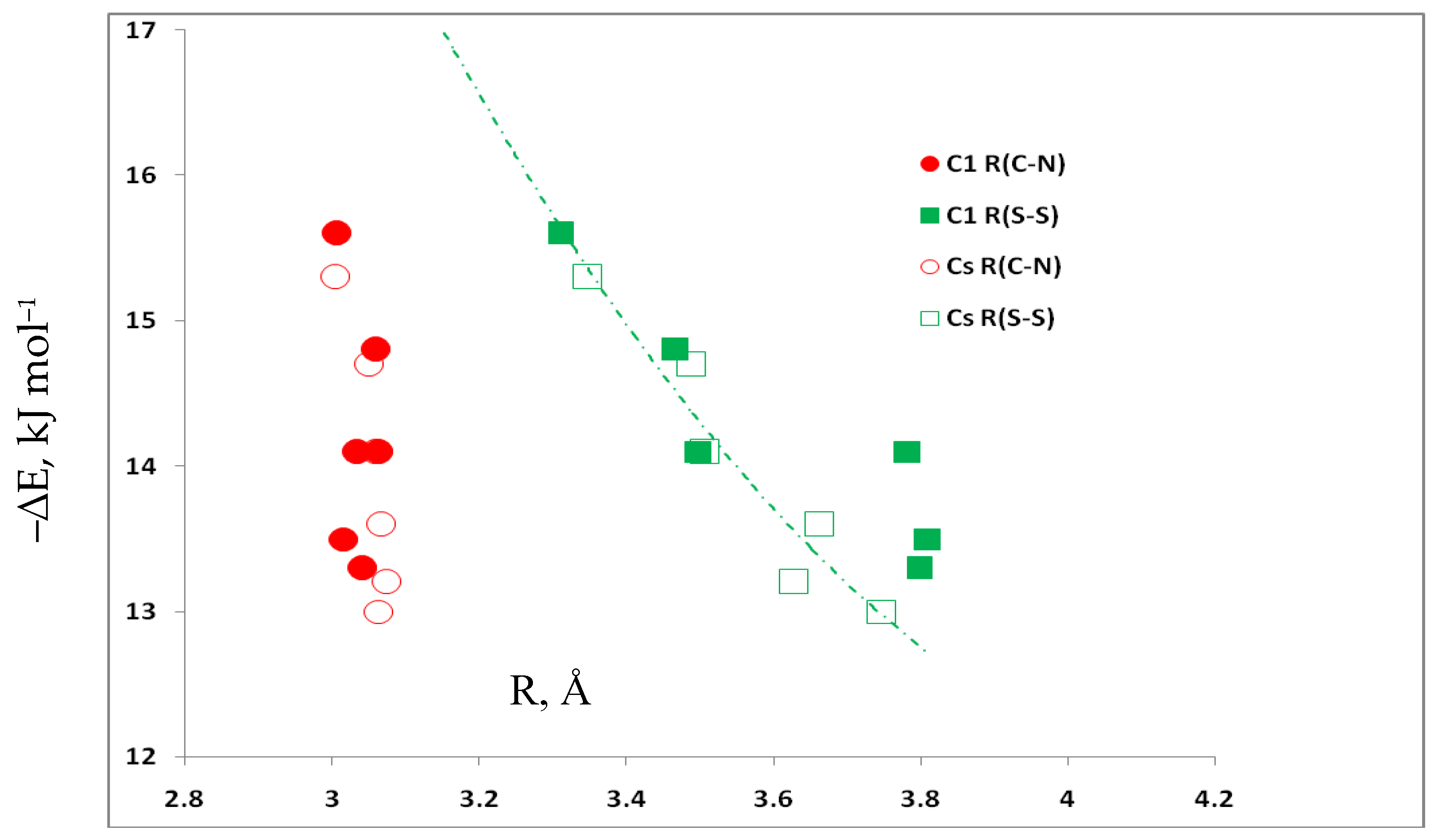
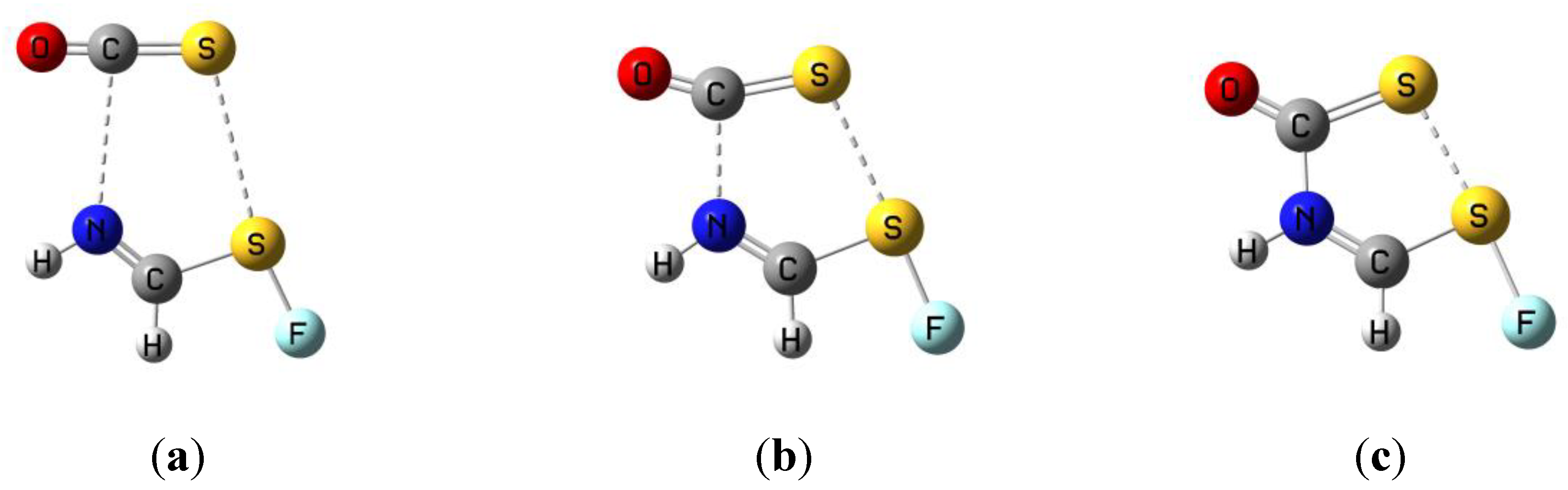
3.5. Comparison of Cs Complexes, Molecules, and Transition Structures HN(CH)SX:SCO and HN(CH)SX:OCS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman and Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Metrangolo, P.; Resnati, G. Halogen Bonding. Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2008; p. 126. [Google Scholar]
- Metrangolo, P.; Resnati, G. Halogen Bonding I. Impact on Materials Chemistry and Life Sciences; Springer: Berlin/Heidelberg, Germany, 2015; p. 358. [Google Scholar]
- Halogen Bonding in Supramolecular and Solid State Chemistry—Faraday Discussion; 10–12 July 2017, Ottawa, Canada; The Royal Society of Chemistry: Cambridge, England, 2017.
- Minyaev, R.M.; Minkin, V.I. Theoretical Study of O → X (S, Se, Te) Coordination in Organic Compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- Sanz, P.; Yáñez, M.; Mó, O. Resonance-Assisted Intramolecular Chalcogen–Chalcogen Interactions? Chem. Eur. J. 2003, 9, 4548–4555. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical Investigations on Chalcogen−Chalcogen Interactions: What Makes These Nonbonded Interactions Bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Intermolecular Weak Interactions in HTeXH Dimers (X=O, S, Se, Te): Hydrogen Bonds, Chalcogen–Chalcogen Contacts and Chiral Discrimination. ChemPhysChem 2012, 13, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S.A. New Noncovalent Force: Comparison of P···N Interaction with Hydrogen and Halogen Bonds. J. Chem. Phys. 2011, 134, 094315. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin−Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces; Challenges and Advances in Computational Chemistry and Physics; Scheiner, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 19, pp. 191–263. [Google Scholar]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel Bond-σ-Hole Bond as a Preliminary Stage of the SN2 Reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding and Other σ-Hole Interactions: A Perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbenes as Electron-Pair Donors to CO2 for C···C Tetrel Bonds and C−C Covalent Bonds. J. Phys. Chem. A 2017, 121, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Azines as Electron-Pair Donors to CO2 for N···C Tetrel Bonds. J. Phys. Chem. A 2017, 121, 8017–8025. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbon−Carbon Bonding between Nitrogen Heterocyclic Carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Complexes of CO2 with the Azoles: Tetrel Bonds, Hydrogen Bonds and Other Secondary Interactions. Molecules 2018, 23, 906. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Potential Energy Surfaces of HN(CH)SX:CO2 for X=F, Cl, NC, CN, CCH, and H: N···C Tetrel Bonds and O···S Chalcogen Bonds. J. Phys. Chem. A 2019, 123, 7270–7277. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Complexes of O=C=S with Nitrogen Bases: Chalcogen Bonds, Tetrel Bonds, and Other Secondary Interactions. ChemPhysChem 2018, 19, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Exploring N···C Tetrel and O···S Chalcogen Bonds in HN(CH)SX:OCS Systems, for X=F, NC, Cl, CN, CCH, and H. Chem. Phys. Lett. 2019, 730, 466–471. [Google Scholar] [CrossRef]
- Elguero, J.; Katritzky, A.R.; Marzin, C.; Linda, P. The Tautomerism of Heterocycles; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Salina, E.; Ryabova, O.; Kaprelyants, A.; Makarov, V. New 2-Thiopyridines as Potential Candidates for Killing both Actively Growing and Dormant Mycobacterium tuberculosis Cells. Antimicrob. Agents Chemother. 2014, 58, 55–60. [Google Scholar] [CrossRef]
- Pople, J.A.; Binkley, J.S.; Seeger, R. Theoretical Models Incorporating Electron Correlation. Int. J. Quantum Chem. 1976, 10, 1–19. [Google Scholar] [CrossRef]
- Krishnan, R.; Pople, J.A. Approximate Fourth-Order Perturbation Theory of the Electron Correlation Energy. Int. J. Quantum Chem. 1978, 14, 91–100. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Silver, D.M. Many-Body Perturbation Theory Applied to Electron Pair Correlation Energies. I. Closed-Shell First Row Diatomic Hydrides. J. Chem. Phys. 1975, 62, 3258–3268. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Purvis, G.D. Many-Body Perturbation Theory, Coupled-Pair Many-Electron Theory, and the Importance of Quadruple Excitations for the Correlation Problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Del Bene, J.E. Proton Affinities of Ammonia, Water, and Hydrogen Fluoride and Their Anions: A Quest for the Basis-Set Limit Using the Dunning Augmented Correlation-Consistent Basis Sets. J. Phys. Chem. 1993, 97, 107–110. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; et al. Gaussian 16, Revision A. 03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor–Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Prentice Hall: Harlow, UK, 2000. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Keith, T.A. AIMAll (Version 17.11.14B); TK Gristmill Software: Overland Park, KS, USA, 2017. [Google Scholar]
- Perera, S.A.; Nooijen, M.; Bartlett, R.J. Electron Correlation Effects on the Theoretical Calculation of NuclearMagnetic Resonance Spin-Spin Coupling Constants. J. Chem. Phys. 1996, 104, 3290–3305. [Google Scholar] [CrossRef]
- Perera, S.A.; Sekino, H.; Bartlett, R.J. Coupled-Cluster Calculations of Indirect Nuclear Coupling Constants: The Importance of Non-Fermi Contact Contributions. J. Chem. Phys. 1994, 101, 2186–2196. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Watts, J.D.; Nooijen, J.M.; Oliphant, N.; Perera, S.A.; Szalay, P.G.; Lauderdale, W.J.; Gwaltney, S.R.; Beck, S.; et al. ACES II; University of Florida: Gainesville, FL, USA, 1991. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X = | –ΔE | R(N4–C1) | R(S3–S6) | <N4–C1–S3 | <S3–S6–A7 a | dih<S3–C1–N4–C5 |
|---|---|---|---|---|---|---|
| F | 15.6 | 3.006 | 3.310 | 93 | 169 | 46 |
| Cl | 14.8 | 3.059 | 3.467 | 95 | 166 | 49 |
| NC | 14.1 | 3.064 | 3.498 | 96 | 166 | 20 |
| CCH | 14.1 | 3.034 | 3.782 | 97 | 123 | 43 |
| H | 13.5 | 3.016 | 3.810 | 97 | 131 | 39 |
| CN | 13.3 | 3.041 | 3.798 | 98 | 124 | 44 |
| X = | S3lp1 → σ*S6–A | S3lp2 → σ*S6–A | Nlp → π*C–O | Nlp → π*C–S3 |
|---|---|---|---|---|
| F | 12.2 (12.8) | 1.9 (1.7) | 5.9 (6.6) | 0.3 (0.3) |
| Cl | 8.0 (9.5) | 1.5 (0.8) | 4.9 (5.4) | 0.3 (0.3) |
| NC | 5.9 (7.5) | 1.6 (1.0) | 4.9 (5.0) | 0.3 (0.3) |
| CCH | 0.5 (4.7) | ---b (0.5) | 2.6 (4.8) | ---b (0.3) |
| H | 0.9 (4.0) | ---b (0.4) | 3.6 (5.1) | 0.3 (0.3) |
| CN | 0.5 (3.5) | ---b (0.5) | 2.5 (5.2) | ---b (0.4) |
| X = | –ΔE | R(N4–C1) | R(S3–S6) | <N4–C1–S3 | <S3–S6–A7 b | 1cJ(S–S) |
|---|---|---|---|---|---|---|
| F | 15.3 | 3.004 | 3.348 | 95 | 169 | 5.7 |
| Cl | 14.7 | 3.051 | 3.489 | 96 | 166 | 3.8 |
| NC | 14.1 | 3.060 | 3.508 | 97 | 168 | 3.6 |
| CCH | 13.6 | 3.067 | 3.663 | 97 | 169 | 2.1 |
| H | 13.0 | 3.064 | 3.747 | 97 | 174 | 1.4 |
| CN | 13.2 | 3.075 | 3.628 | 97 | 169 | 2.3 |
| X = | –ΔE | R(N4–C1) | R(S3–S6) | <N4–C1–S3 | <S3–S6–A7 a |
|---|---|---|---|---|---|
| F | 48.1 | 1.436 | 2.292 | 131 | 175 |
| Cl | 24.3 | 1.438 | 2.314 | 131 | 177 |
| NC | –9.6 | 1.443 | 2.294 | 131 | 174 |
| CCH | –65.1 | 1.536 | 2.803 | 137 | 175 |
| H | –64.0 | 1.548 | 2.885 | 137 | 170 |
| CN | –69.9 | 1.523 | 2.672 | 136 | 174 |
| X = F | –ΔE | R(N4–C1) | R(S3–S6) | R(S6–F7) | <N4–C1–S3 | <S3–S6–F7 | <S3–C1–O2 |
| complex | 15.3 | 3.004 | 3.348 | 1.636 | 95 | 167 | 179 |
| TS | –18.8 | 2.036 | 2.614 | 1.683 | 100 | 178 | 154 |
| molecule | 48.1 | 1.436 | 2.292 | 1.798 | 109 | 175 | 131 |
| X = Cl | –ΔE | R(N4–C1) | R(S3–S6) | R(S6–Cl7) | <N4–C1–S3 | <S3–S6–Cl7 | <S3–C1–O2 |
| complex | 14.7 | 3.051 | 3.489 | 2.037 | 96 | 166 | 179 |
| TS | –37.1 | 1.967 | 2.676 | 2.113 | 102 | 180 | 152 |
| molecule | 24.3 | 1.438 | 2.314 | 2.311 | 109 | 177 | 131 |
| X = | Complex 1tJ(N1–C4) a | TS 1tJ(N1–C4) | Molecule 1J(N–C) | Complex 1cJ(S–S) | TS 1cJ(S–S) | Molecule 1cJ(S–S) |
|---|---|---|---|---|---|---|
| F | –0.3 | 17.9 | –7.8 | 5.7 | 22.7 | 13.1 |
| Cl | –0.3 | 21.0 | –7.6 | 3.8 | 21.5 | 12.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Bene, J.E.; Alkorta, I.; Elguero, J. N…C and S…S Interactions in Complexes, Molecules, and Transition Structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN. Molecules 2019, 24, 3232. https://doi.org/10.3390/molecules24183232
Del Bene JE, Alkorta I, Elguero J. N…C and S…S Interactions in Complexes, Molecules, and Transition Structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN. Molecules. 2019; 24(18):3232. https://doi.org/10.3390/molecules24183232
Chicago/Turabian StyleDel Bene, Janet E., Ibon Alkorta, and José Elguero. 2019. "N…C and S…S Interactions in Complexes, Molecules, and Transition Structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN" Molecules 24, no. 18: 3232. https://doi.org/10.3390/molecules24183232
APA StyleDel Bene, J. E., Alkorta, I., & Elguero, J. (2019). N…C and S…S Interactions in Complexes, Molecules, and Transition Structures HN(CH)SX:SCO, for X = F, Cl, NC, CCH, H, and CN. Molecules, 24(18), 3232. https://doi.org/10.3390/molecules24183232