
An Improved HILIC HPLC-MS/MS Method for the Determination of β-ODAP and Its α Isomer in Lathyrus sativus

 , , , and
, , , and
Abstract
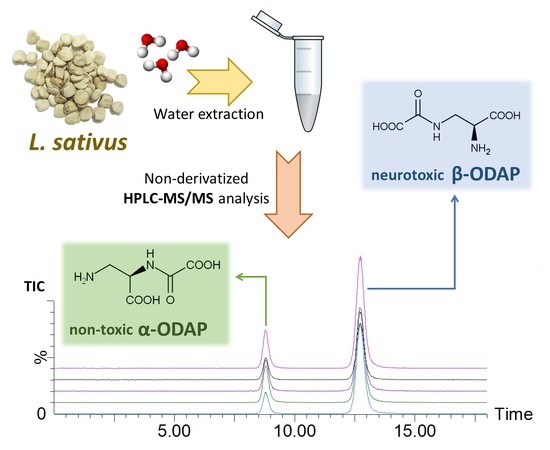
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the Extraction Procedure
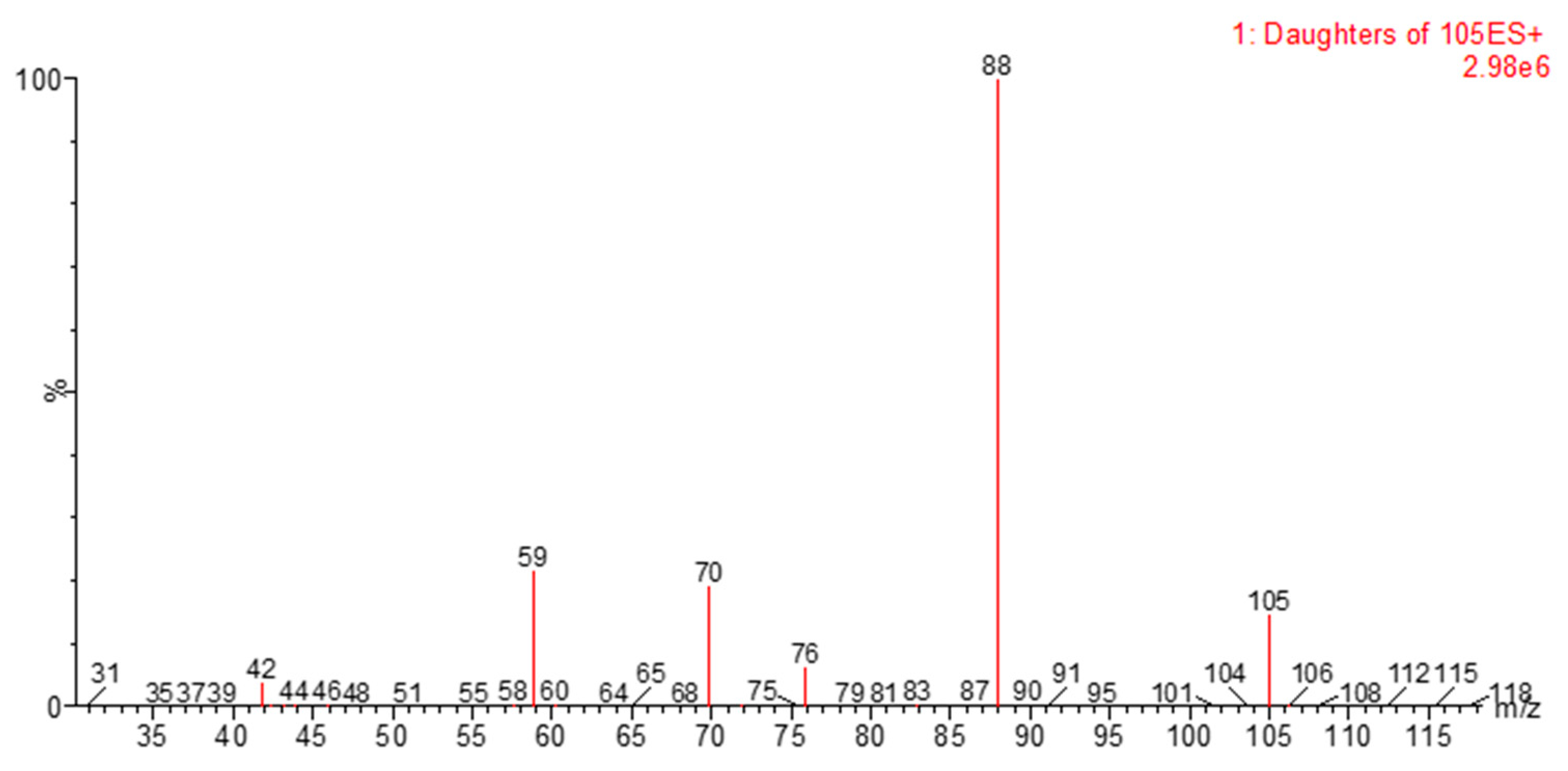
2.2. HPLC-MS/MS Method Development
2.3. Method Validation
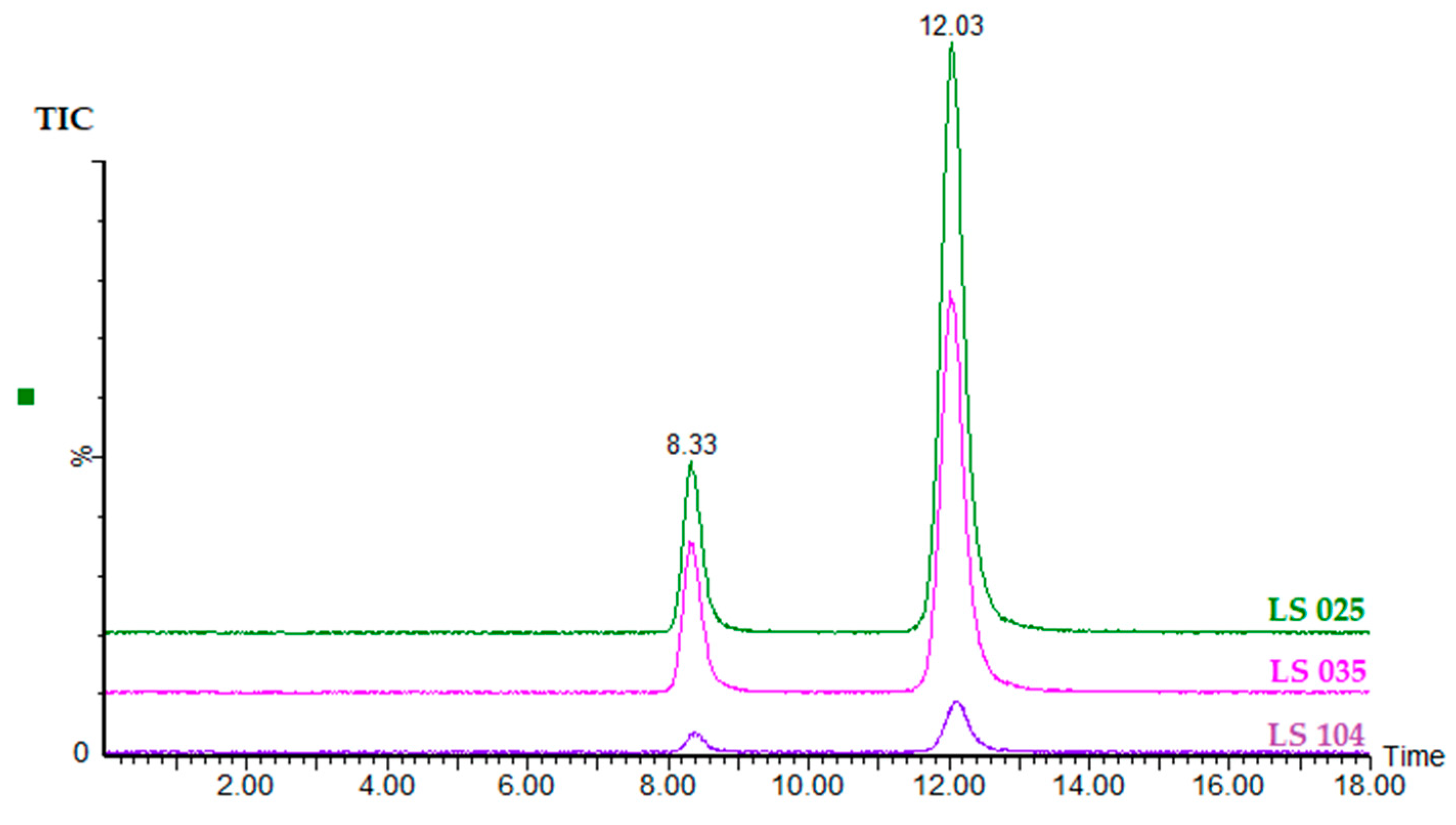
2.3.1. Specificity and Selectivity
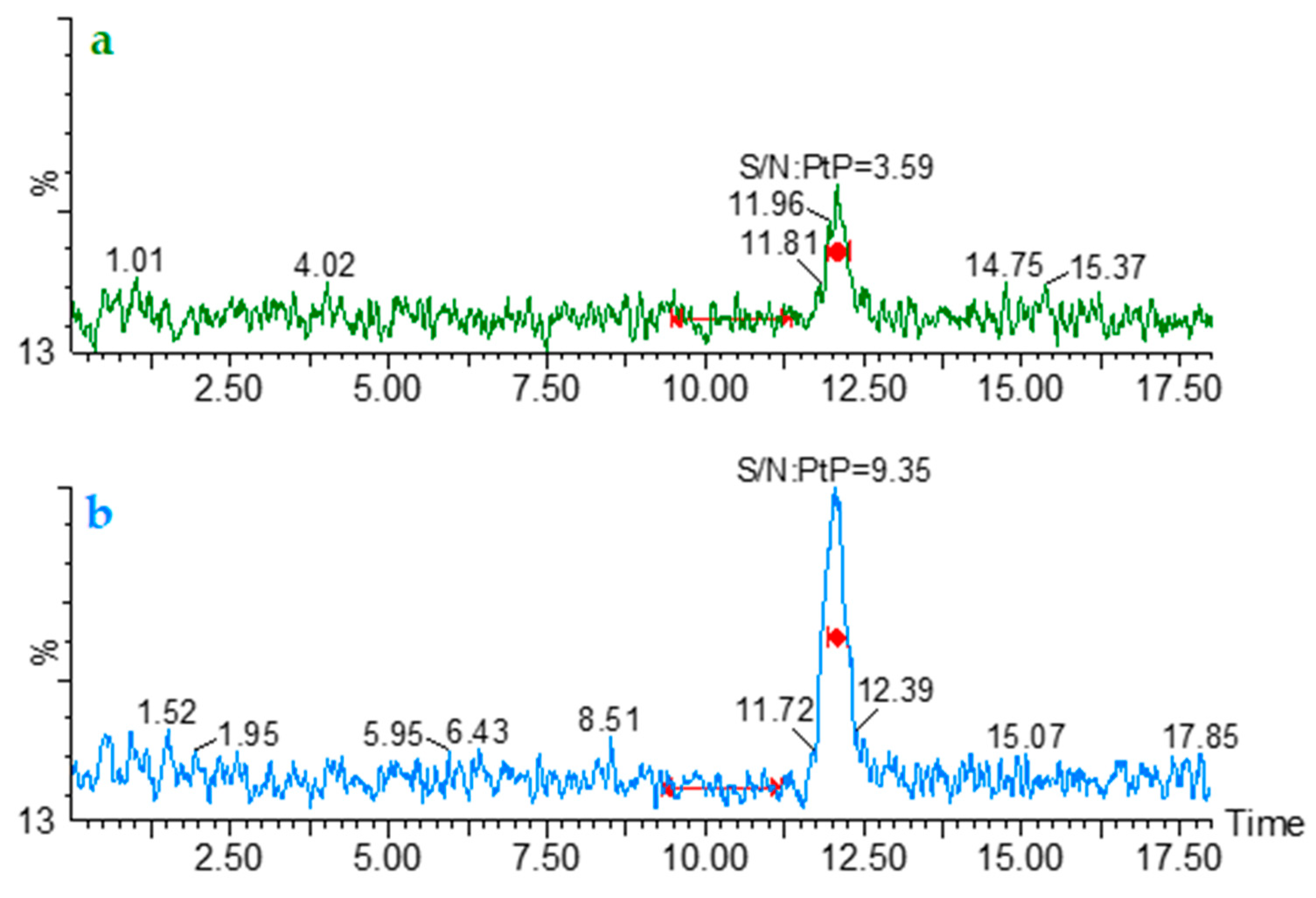
2.3.2. Limit of Detection (LOD) and Lower Limit of Quantification (LLOQ)
2.3.3. Linearity and Linear Range
2.3.4. Precision and Accuracy
2.3.5. Matrix Effect
2.3.6. Dilution Integrity
2.3.7. Method Recovery
2.3.8. Stability Assays
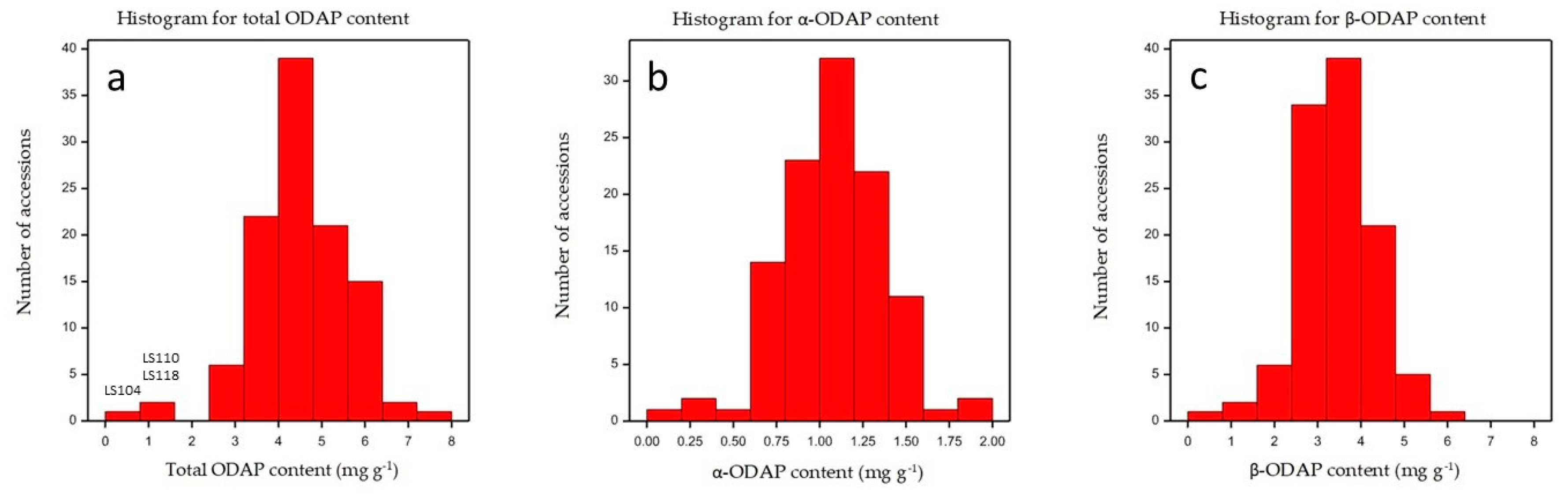
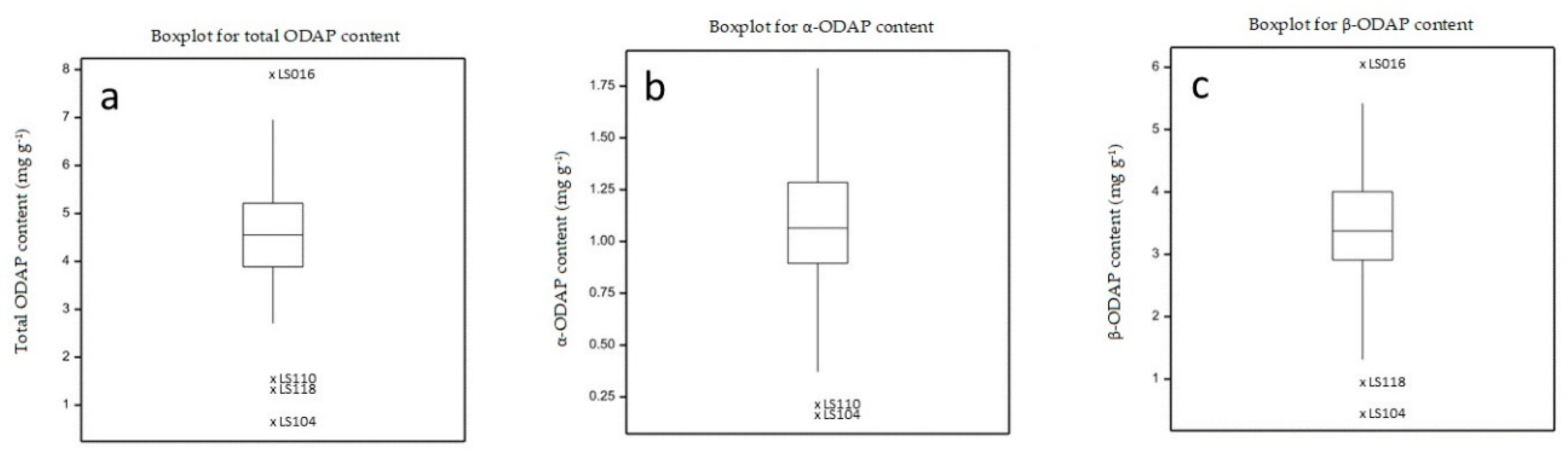
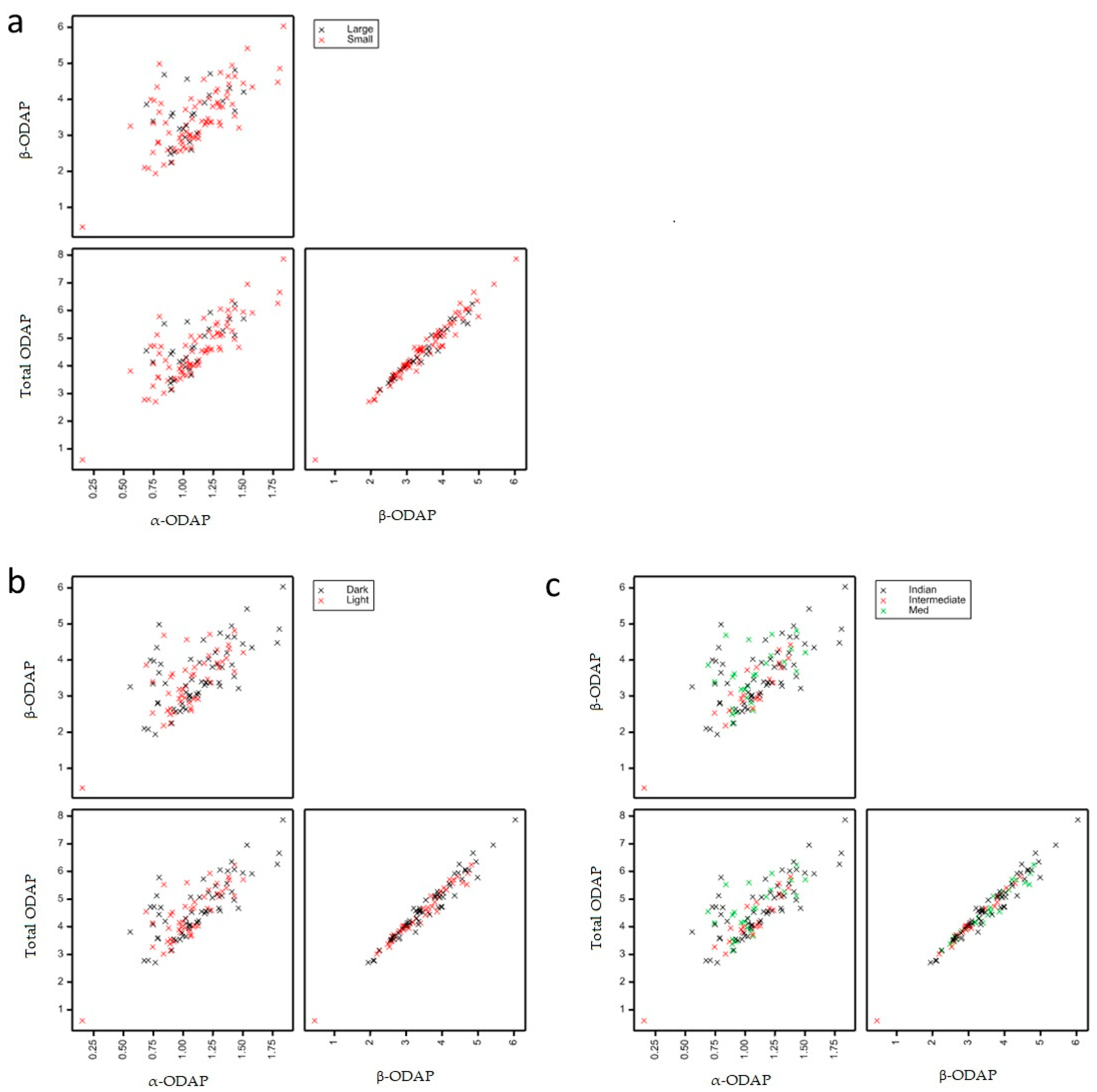
2.4. Determination of ODAP Content in Lathyrus sativus Accessions
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Samples
3.3. Optimization of the Extraction Procedure
3.4. HPLC-MS/MS Method Development
3.5. Method Validation
3.5.1. Specificity
3.5.2. Limit of Detection (LOD) and Lower Limit of Quantification (LLOQ)
3.5.3. Linearity and Linear Range
3.5.4. Precision and Accuracy
3.5.5. Matrix Effect
3.5.6. Dilution Integrity
3.5.7. Method Recovery
3.5.8. Stability Assays
3.6. Application of the Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Vaz Patto, M.; Rubiales, D. Lathyrus diversity: Available resources with relevance to crop improvement—L. sativus and L. cicera as case studies. Ann. Bot. 2014, 113, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.A. Nonprotein amino acids of plants: Significance in medicine, nutrition, and agriculture. J. Agric. Food Chem. 2003, 51, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Fratianni, F.; Cardinale, F.; Cozzolino, A.; Granese, T.; Albanese, D.; Di Matteo, M.; Zaccardelli, M.; Coppola, R.; Nazzaro, F. Polyphenol composition and antioxidant activity of different grass pea (Lathyrus sativus), lentils (Lens culinaris), and chickpea (Cicer arietinum) ecotypes of the Campania region (Southern Italy). J. Funct. Foods 2014, 7, 551–557. [Google Scholar] [CrossRef]
- Enneking, D. The nutritive value of grasspea (Lathyrus sativus) and allied species, their toxicity to animals and the role of malnutrition in neurolathyrism. Food Chem. Toxicol. 2011, 49, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Lambein, F.; Kuo, Y.-H.; Kusama-Eguchi, K.; Ikegami, F. 3-N-oxalyl-L-2, 3-diaminopropanoic acid, a multifunctional plant metabolite of toxic reputation. Arkivoc 2007, 45–52. [Google Scholar]
- Van Moorhem, M.; Lambein, F.; Leybaert, L. Unraveling the mechanism of β-N-oxalyl-α, β-diaminopropionic acid (β-ODAP) induced excitotoxicity and oxidative stress, relevance for neurolathyrism prevention. Food Chem. Toxicol. 2011, 49, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Rao, S. A look at the brighter facets of β-N-oxalyl-l-α, β-diaminopropionic acid, homoarginine and the grass pea. Food Chem. Toxicol. 2011, 49, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.L.; Lau, A.J.; Chan, E.C.Y. Hydrophilic interaction liquid chromatography with tandem mass spectrometry for the determination of underivatized dencichine (β-N-oxalyl-L-α, β-diaminopropionic acid) in Panax medicinal plant species. Rapid Commun. Mass Spectrom. 2005, 19, 1237–1244. [Google Scholar] [CrossRef]
- Xie, G.-X.; Qiu, Y.-P.; Qiu, M.-F.; Gao, X.-F.; Liu, Y.-M.; Jia, W. Analysis of dencichine in Panax notoginseng by gas chromatography–mass spectrometry with ethyl chloroformate derivatization. J. Pharm. Biomed. Anal. 2007, 43, 920–925. [Google Scholar] [CrossRef]
- Padmajaprasad, V.; Kaladhar, M.; Bhat, R.V. Thermal isomerisation of β-N-oxalyl-L-α, β-diaminopropionic acid, the neurotoxin in Lathyrus sativus, during cooking. Food Chem. 1997, 59, 77–80. [Google Scholar] [CrossRef]
- Abegaz, B.M.; Nunn, P.B.; De Bruyn, A.; Lambein, F. Thermal isomerization of N-oxalyl derivatives of diamino acids. Phytochemistry 1993, 33, 1121–1123. [Google Scholar] [CrossRef]
- Rao, S. A sensitive and specific colorimetric method for the determination of α, β-diaminopropionic acid and the Lathyrus sativus neurotoxin. Anal. Biochem. 1978, 86, 386–395. [Google Scholar] [CrossRef]
- Ghosh, B.; Mitra, J.; Chakraborty, S.; Bhattacharyya, J.; Chakraborty, A.; Sen, S.K.; Neerathilingam, M. Simple Detection Methods for Antinutritive Factor β-ODAP Present in Lathyrus sativus L. by High Pressure Liquid Chromatography and Thin Layer Chromatography. Plos ONE 2015, 10, e0140649. [Google Scholar] [CrossRef] [PubMed]
- Gresta, F.; Rocco, C.; Lombardo, G.M.; Avola, G.; Ruberto, G. Agronomic characterization and α-and β-ODAP determination through the adoption of new analytical strategies (HPLC-ELSD and NMR) of ten Sicilian accessions of grass pea. J. Agric. Food Chem. 2014, 62, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Piergiovanni, A.R.; Damascelli, A. L-homoarginine accumulation in grass pea (Lathyrus sativus L.) dry seeds. A preliminary survey. Food Nutr. Sci. 2011, 2, 207. [Google Scholar] [CrossRef]
- Onar, A.N.; Erdoğan, B.Y.; Ayan, I.; Acar, Z. Homoarginine, β-ODAP, and asparagine contents of grass pea landraces cultivated in Turkey. Food Chem. 2014, 143, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Jiao, C.J.; Li, F.; Liang, Y.; Li, Z. Analysis of Toxin β-N-Oxalyl-α, β-diaminopropionic Acid (β-ODAP), Its Isomer α-ODAP and Other Free Amino Acids in Lathyrus sativus. Chin. Chem. Lett. 2005, 16, 627–630. [Google Scholar]
- Megías, C.; Cortés-Giraldo, I.; Alaiz, M.; Girón-Calle, J.; Vioque, J.; Santana-Méridas, O.; Herraiz-Peñalver, D.; Sánchez-Vioque, R. Determination of the Neurotoxin 3-N-Oxalyl-2, 3-Diaminopropionic Acid and Other Free Amino Acids in Lathyrus cicera and L. sativus Seeds by Reversed-Phase High-Performance Liquid Chromatography. Food Anal. Methods 2015, 8, 1953–1961. [Google Scholar]
- Xiong, J.-L.; Xiong, Y.-C.; Bai, X.; Kong, H.-Y.; Tan, R.-Y.; Zhu, H.; Siddique, K.H.; Wang, J.-Y.; Turner, N.C. Genotypic variation in the concentration of β-N-Oxalyl-L-α, β-diaminopropionic acid (β-ODAP) in Grass Pea (Lathyrus sativus L.) seeds is associated with an accumulation of leaf and pod β-ODAP during vegetative and reproductive stages at three levels of water stress. J. Agric. Food Chem. 2015, 63, 6133–6141. [Google Scholar]
- Qian, C.; Yuan, Y.; He, X.; Liu, J.; Shao, Q.; Wu, H.; Qiao, H. A Sensitive HPLC-MS/MS Analysis of Dencichine in Rat Plasma and Its Application to Pharmacokinetics. Am. J. Anal. Chem. 2012, 3, 596. [Google Scholar] [CrossRef]
- Arslan, M.; Oten, M.; Erkaymaz, T.; Tongur, T.; Kilic, M.; Elmasulu, S.; Cinar, A. β-N-oxalyl-L-2, 3-diaminopropionic acid, L-homoarginine, and asparagine contents in the seeds of different genotypes Lathyrus sativus L. as determined by UHPLC-MS/MS. Int. J. Food Prop. 2017, 20, S108–S118. [Google Scholar] [CrossRef]
- Wang, F.; Chen, X.; Chen, Q.; Qin, X.; Li, Z. Determination of neurotoxin 3-N-oxalyl-2, 3-diaminopropionic acid and non-protein amino acids in Lathyrus sativus by precolumn derivatization with 1-fluoro-2, 4-dinitrobenzene. J. Chromatogr. A 2000, 883, 113–118. [Google Scholar] [CrossRef]
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Xiao, B.; Weng, N. Impact of column temperature and mobile phase components on selectivity of hydrophilic interaction chromatography (HILIC). J. Sep. Sci. 2008, 31, 1449–1464. [Google Scholar] [CrossRef]
- Xiong, X.; Liu, Y. Chromatographic behavior of 12 polar pteridines in hydrophilic interaction chromatography using five different HILIC columns coupled with tandem mass spectrometry. Talanta 2016, 150, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Chirita, R.-I.; West, C.; Finaru, A.-L.; Elfakir, C. Approach to hydrophilic interaction chromatography column selection: Application to neurotransmitters analysis. J. Chromatogr. A 2010, 1217, 3091–3104. [Google Scholar] [CrossRef] [PubMed]
- SANTE/11813/2017. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed; European Commission: Geneva, Switzerland, 2017. [Google Scholar]
- McKeown, A.P. A Simple, Generally Applicable HILIC Method Development Platform Based Upon Selectivity. Chromatography Today, December 2015. [Google Scholar]
- Sacristán, M.; Varela, A.; Pedrosa, M.M.; Burbano, C.; Cuadrado, C.; Legaz, M.E.; Muzquiz, M. Determination of β-N-oxalyl-l-α, β-diaminopropionic acid and homoarginine in Lathyrus sativus and Lathyrus cicera by capillary zone electrophoresis. J. Sci. Food Agric. 2015, 95, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- European Commission. European Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Commun. 2002, L221. [Google Scholar]
- ICH Harmonised Tripartite Guideline. Validation of analytical procedures: Text and methodology Q2 (R1). In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, 2005. [Google Scholar]
- European Medicines Agency. Guideline on bioanalytical method validation. Committee for Medicinal Products for Human Use (EMEA/CHMP/EWP/192217/2009), 2011. [Google Scholar]
- U.S. Department of Health and Human Services. Bioanalytical Method Validation Guidance for Industry; Food and Drug Administration: Silver Spring, MD, USA, 2018.
- El-Moneim, A.M.A.; Van Dorrestein, B.; Baum, M.; Bejiga, J.R.G. Role of ICARDA in improving the nutritional quality and yield potential of grasspea (Lathyrus sativus L.), for subsistence farmers in dry areas. Lathyrus Lathyrism Newsl. 2001, 4, 55–58. [Google Scholar]
- Jiao, C.-J.; Jiang, J.-L.; Ke, L.-M.; Cheng, W.; Li, F.-M.; Li, Z.-X.; Wang, C.-Y. Factors affecting β-ODAP content in Lathyrus sativus and their possible physiological mechanisms. Food Chem. Toxicol. 2011, 49, 543–549. [Google Scholar] [CrossRef]
- Fikre, A.; Negwo, T.; Kuo, Y.-H.; Lambein, F.; Ahmed, S. Climatic, edaphic and altitudinal factors affecting yield and toxicity of Lathyrus sativus grown at five locations in Ethiopia. Food Chem. Toxicol. 2011, 49, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Lambein, F.; Travella, S.; Kuo, Y.-H.; Van Montagu, M.; Heijde, M. Grass pea (Lathyrus sativus L.): Orphan crop, nutraceutical or just plain food? Planta 2019, 250, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, X.; Li, G.; Chen, X.; Hu, Z. Kinetics studies on thermal isomerization of β-N-oxalyl-L-α, β-diaminopropionic acid by capillary zone electrophoresis. Phys. Chem. Chem. Phys. 1999, 1, 3771–3773. [Google Scholar] [CrossRef]
- Hanbury, C.; Siddique, K.; Galwey, N.; Cocks, P. Genotype-environment interaction for seed yield and ODAP concentration of Lathyrus sativus L. and L. cicera L. in Mediterranean-type environments. Euphytica 1999, 110, 45–60. [Google Scholar] [CrossRef]
- Mata, A.; Ferreira, J.; Oliveira, B.; Batoréu, M.; Crespo, M.B.; Pereira, V.; Bronze, M. Bottled water: Analysis of mycotoxins by LC–MS/MS. Food Chem. 2015, 176, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; Alamoudi, J.A.; Alnouti, Y. Quantitative analysis of endogenous compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (ng mL−1) | Intra-Day Precision | Inter-Day Precision | Intra-Day Accuracy | Inter-Day Accuracy | |
|---|---|---|---|---|---|
| Injection Repeatability | Experimental Repeatability | ||||
| 25 | 5.3 | 4.5 | 4.4 | 0.7 | 1.1 |
| 75 | 3.3 | 5.8 | 6.5 | 3.5 | 0.4 |
| 1300 | 0.6 | 2.0 | 5.5 | 2.2 | 1.3 |
| 2500 | 1.0 | 0.6 | 5.1 | 0.9 | 0.1 |
| Solvent | LS 043 | LS 067 | LS 076 | LS 104 | LS 114 | LS 118 | Average (Matrix) | |
|---|---|---|---|---|---|---|---|---|
| Slope | 45.32 | 40.26 | 41.19 | 45.20 | 39.16 | 46.79 | 44.40 | 43.16 ± 2.94 |
| MF (%) | 100 | 86 | 103 | 98 | 100 | 95 | 89 | 95 ± 6.49 |
| LS 103 | LS 104 | LS 114 | LS 076 | LS 067 | LS 043 |
|---|---|---|---|---|---|
| 3 | 13 | 12 | 6 | 3 | 2 |
| Concentration (ng mL−1) | LS 059 | LS 075 | LS 104 | LS 114 | Average |
|---|---|---|---|---|---|
| 75 | 94 | 107 | 99 | 114 | 104 ± 9 |
| 1300 | 94 | 100 | 86 | 107 | 97 ± 9 |
| 2500 | 90 | 92 | 86 | 95 | 91 ± 4 |
| Theoretical Concentration (ng mL−1) | 75 | 1300 | 2500 | ||
|---|---|---|---|---|---|
| Freshly prepared solutions | Solution 1 | 0 | 1 | 1 | |
| Solution 2 | 7 | 1 | 3 | ||
| Room temperature (23 °C) | 1 week | W 1 | 10 | 1 | 1 |
| W 2 | 0 | 3 | 5 | ||
| 1 week | S 1 | 3 | 10 | 9 | |
| S 2 | 41 | 9 | 9 | ||
| 2 months | S 1 | 16 | 21 | 22 | |
| S 2 | 19 | 20 | 19 | ||
| Autosampler (10 °C) | 3 days | W 1 | 6 | 8 | 7 |
| W 2 | 2 | 6 | 5 | ||
| 1 week | W 1 | 11 | 1 | 2 | |
| W 2 | 3 | 1 | 1 | ||
| Freeze-thaw (−20 °C) | Cycle 1 | S 1 | 3 | 6 | 5 |
| S 2 | 5 | 5 | 6 | ||
| Cycle 2 | S 1 | 10 | 11 | 13 | |
| S 2 | 8 | 9 | 11 | ||
| Cycle 3 | S 1 | 3 | 3 | 3 | |
| S 2 | 3 | 5 | 7 | ||
| Freezer (−20 °C) | 3 months | S 1 | 8 | 5 | 4 |
| S 2 | 6 | 4 | 4 | ||
| Samples | LS 104 | LS 035 | LS 025 | |||||
|---|---|---|---|---|---|---|---|---|
| α and β-ODAP | α | β | α | β | α | β | ||
| Room temperature (23 °C) | 24 h | W 1 | 2 | 2 | 1 | 5 | 2 | 0 |
| W 2 | 2 | 1 | 5 | 11 | 6 | 6 | ||
| W 3 | 6 | 1 | 8 | 12 | 4 | 10 | ||
| A:W 1 | 3 | 2 | 0 | 2 | 2 | 3 | ||
| A:W 2 | 2 | 0 | 3 | 8 | 6 | 8 | ||
| A:W 3 | 0 | 1 | 5 | 10 | 8 | 12 | ||
| 1 week | W 1 | 10 | 66 | 7 | 3 | 97 | 22 | |
| W 2 | 23 | 6 | 4 | 4 | 80 | 18 | ||
| W 3 | 22 | 5 | 5 | 2 | 48 | 22 | ||
| A:W 1 | 21 | 10 | 16 | 7 | 25 | 17 | ||
| A:W 2 | 25 | 9 | 14 | 1 | 21 | 9 | ||
| A:W 3 | 25 | 11 | 15 | 10 | 35 | 27 | ||
| Autosampler (10 °C) | 1 week | A:W 1 | 1 | 4 | 5 | 8 | 2 | 2 |
| A:W 2 | 3 | 5 | 8 | 14 | 4 | 7 | ||
| A:W 3 | 7 | 2 | 8 | 12 | 4 | 5 | ||
| Fridge (4°C) | 1 week | W 1 | 3 | 1 | 6 | 8 | 2 | 1 |
| W 2 | 6 | 1 | 7 | 12 | 7 | 6 | ||
| W 3 | 4 | 1 | 9 | 13 | 7 | 12 | ||
| A:W 1 | 20 | 19 | 4 | 5 | 13 | 11 | ||
| A:W 2 | 22 | 16 | 2 | 3 | 7 | 4 | ||
| A:W 3 | 37 | 30 | 1 | 1 | 2 | 2 | ||
| Freeze-thaw (−20 °C) | Cycle 1 | W 1 | 2 | 3 | 1 | 1 | 1 | 1 |
| W 2 | 0 | 2 | 2 | 8 | 5 | 6 | ||
| W 3 | 6 | 3 | 5 | 9 | 9 | 11 | ||
| A:W 1 | 6 | 12 | 1 | 2 | 3 | 3 | ||
| A:W 2 | 16 | 9 | 0 | 5 | 6 | 5 | ||
| A:W 3 | 17 | 7 | 0 | 2 | 1 | 2 | ||
| Cycle 2 | W 1 | 14 | 5 | 3 | 0 | 5 | 5 | |
| W 2 | 12 | 2 | 0 | 6 | 1 | 0 | ||
| W 3 | 11 | 8 | 3 | 7 | 2 | 5 | ||
| A:W 1 | 15 | 9 | 6 | 3 | 5 | 2 | ||
| A:W 2 | 13 | 14 | 3 | 4 | 8 | 5 | ||
| A:W 3 | 28 | 21 | 3 | 3 | 6 | 0 | ||
| Cycle 3 | W 1 | 7 | 2 | 1 | 0 | 11 | 10 | |
| W 2 | 12 | 5 | 0 | 6 | 10 | 9 | ||
| W 3 | 13 | 11 | 6 | 3 | 7 | 4 | ||
| A:W 1 | 11 | 5 | 3 | 4 | 1 | 3 | ||
| A:W 2 | 10 | 2 | 6 | 11 | 1 | 1 | ||
| A:W 3 | 33 | 18 | 5 | 8 | 2 | 6 | ||
| Freezer (−20 °C) | 3 months | W 1 | 7 | 4 | 1 | 3 | 4 | 3 |
| W 2 | 8 | 7 | 0 | 7 | 1 | 2 | ||
| W 3 | 13 | 6 | 2 | 6 | 3 | 6 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bento-Silva, A.; Gonçalves, L.; Mecha, E.; Pereira, F.; Vaz Patto, M.C.; Bronze, M.d.R. An Improved HILIC HPLC-MS/MS Method for the Determination of β-ODAP and Its α Isomer in Lathyrus sativus. Molecules 2019, 24, 3043. https://doi.org/10.3390/molecules24173043
Bento-Silva A, Gonçalves L, Mecha E, Pereira F, Vaz Patto MC, Bronze MdR. An Improved HILIC HPLC-MS/MS Method for the Determination of β-ODAP and Its α Isomer in Lathyrus sativus. Molecules. 2019; 24(17):3043. https://doi.org/10.3390/molecules24173043
Chicago/Turabian StyleBento-Silva, Andreia, Letice Gonçalves, Elsa Mecha, Filipe Pereira, Maria Carlota Vaz Patto, and Maria do Rosário Bronze. 2019. "An Improved HILIC HPLC-MS/MS Method for the Determination of β-ODAP and Its α Isomer in Lathyrus sativus" Molecules 24, no. 17: 3043. https://doi.org/10.3390/molecules24173043
APA StyleBento-Silva, A., Gonçalves, L., Mecha, E., Pereira, F., Vaz Patto, M. C., & Bronze, M. d. R. (2019). An Improved HILIC HPLC-MS/MS Method for the Determination of β-ODAP and Its α Isomer in Lathyrus sativus. Molecules, 24(17), 3043. https://doi.org/10.3390/molecules24173043