Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy

Abstract
1. Introduction
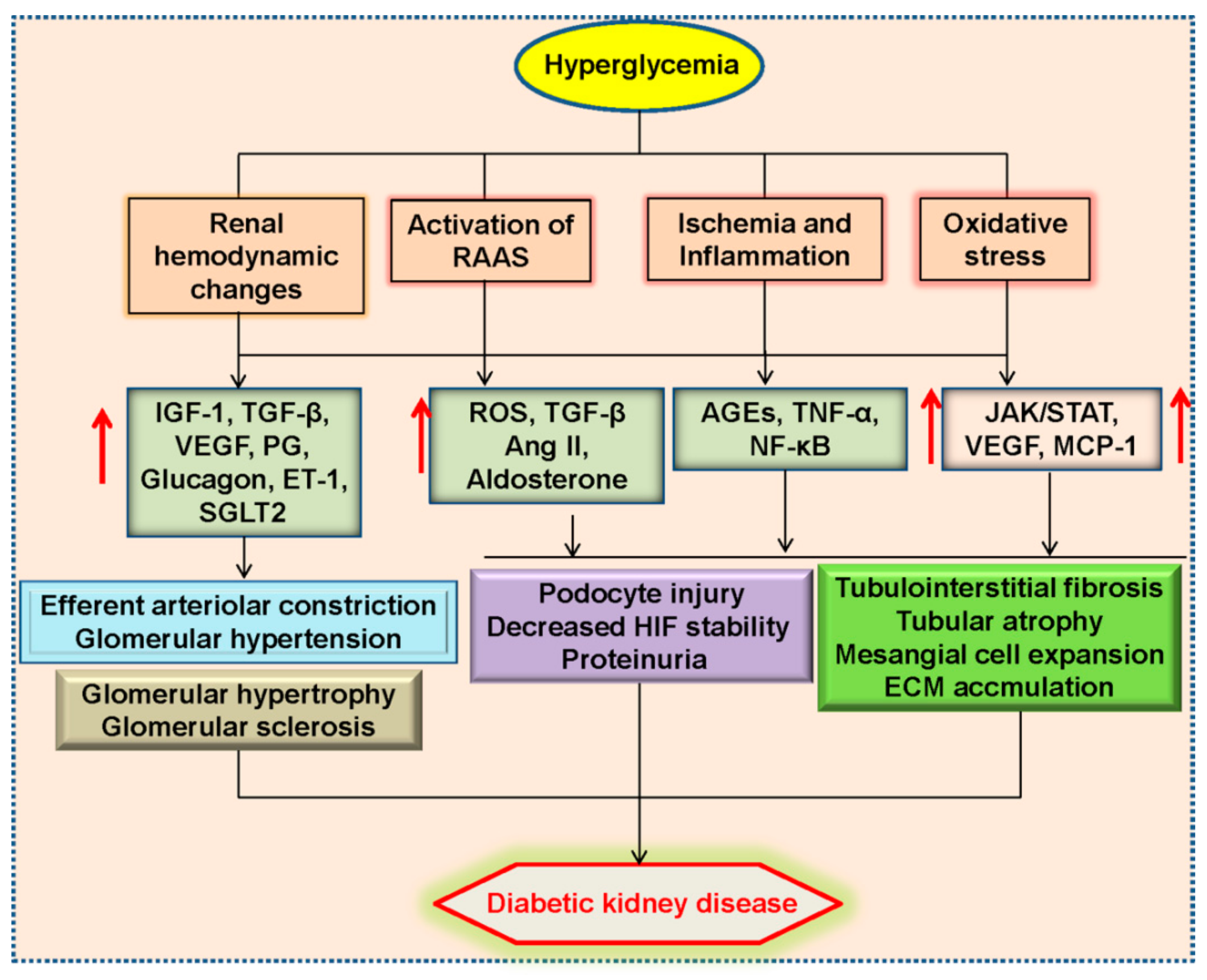
2. Pathophysiology of Diabetic Kidney Disease
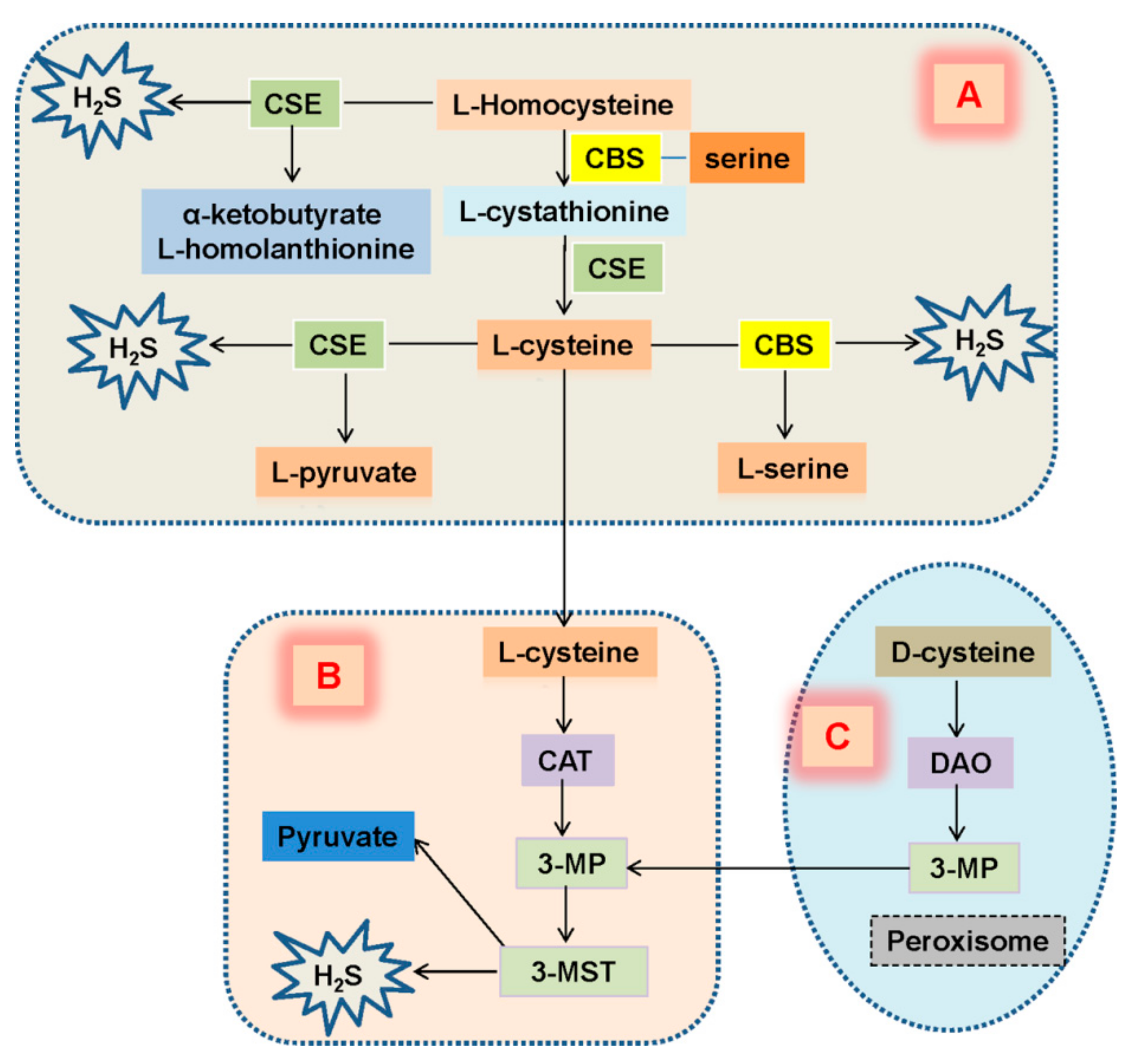
3. Expression of H2S in Diabetic Kidney Disease
4. H2S Regulation of Renal Function
4.1. H2S and Renal Excretory Function
4.2. H2S and Oxygen Sensing
5. Role of H2S in Diabetic Kidney Disease
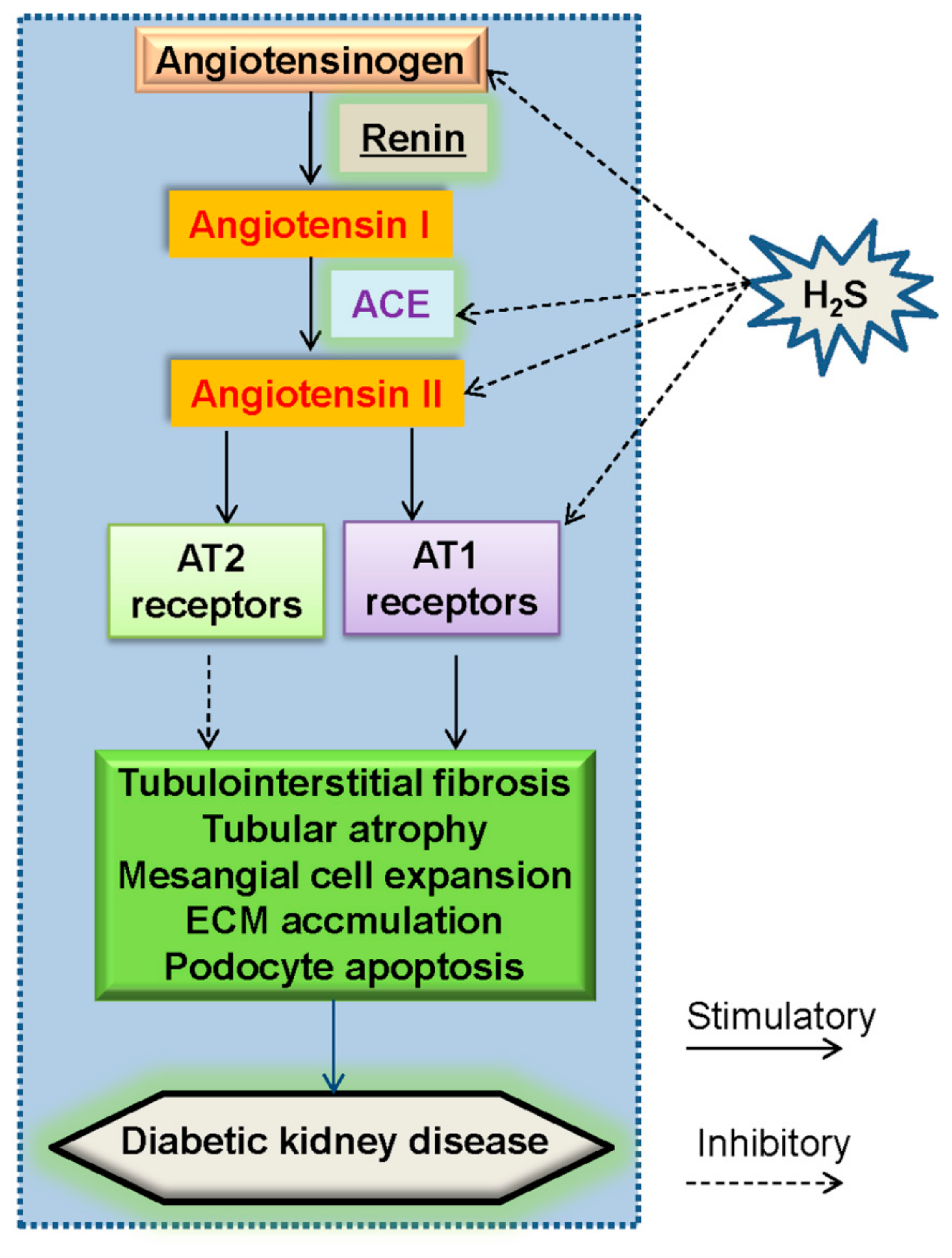
5.1. Renin–Angiotensin System (RAS) and H2S in Diabetic Nephropathy
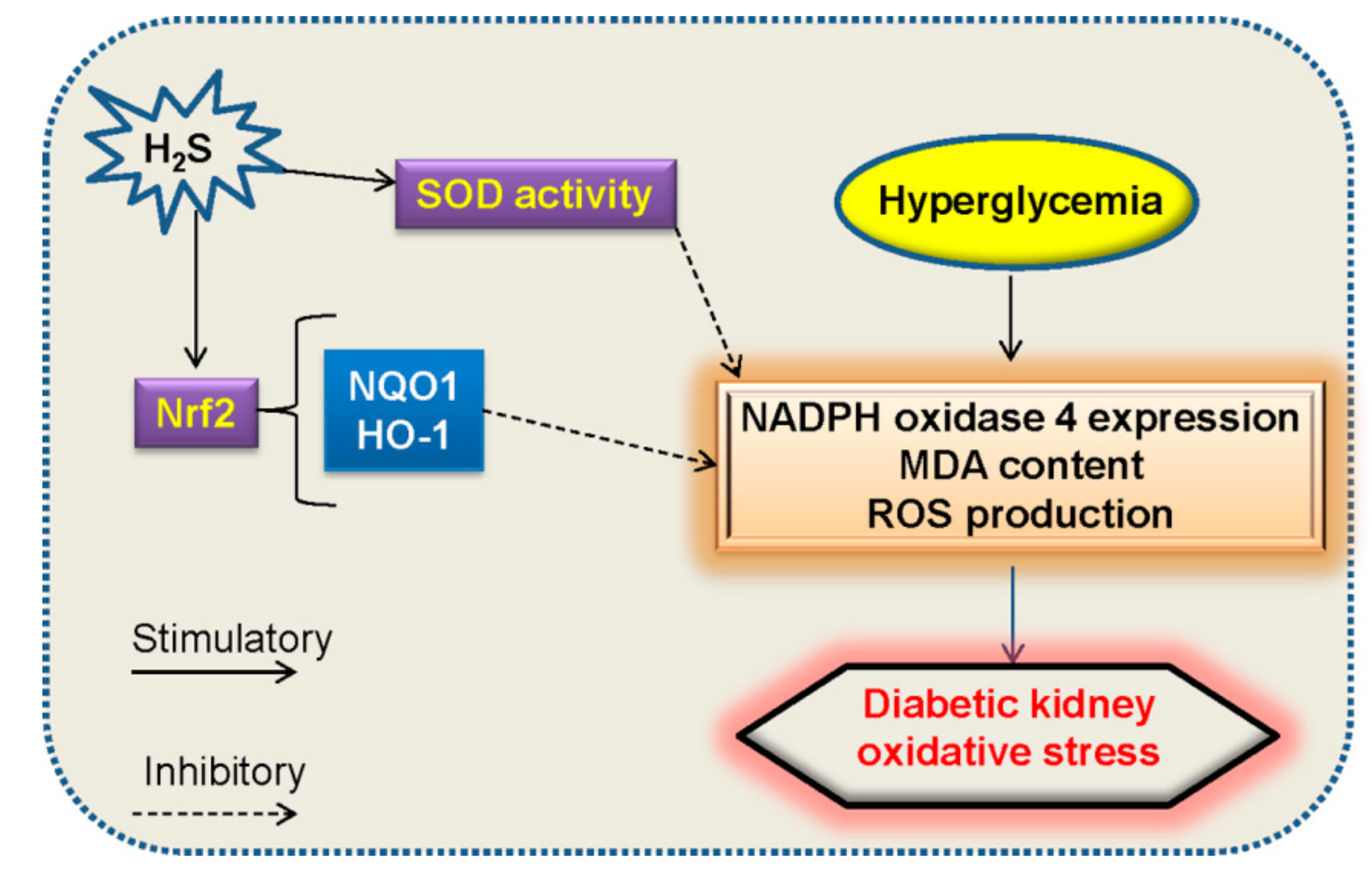
5.2. Oxidative Stress and H2S in Diabetic Nephropathy
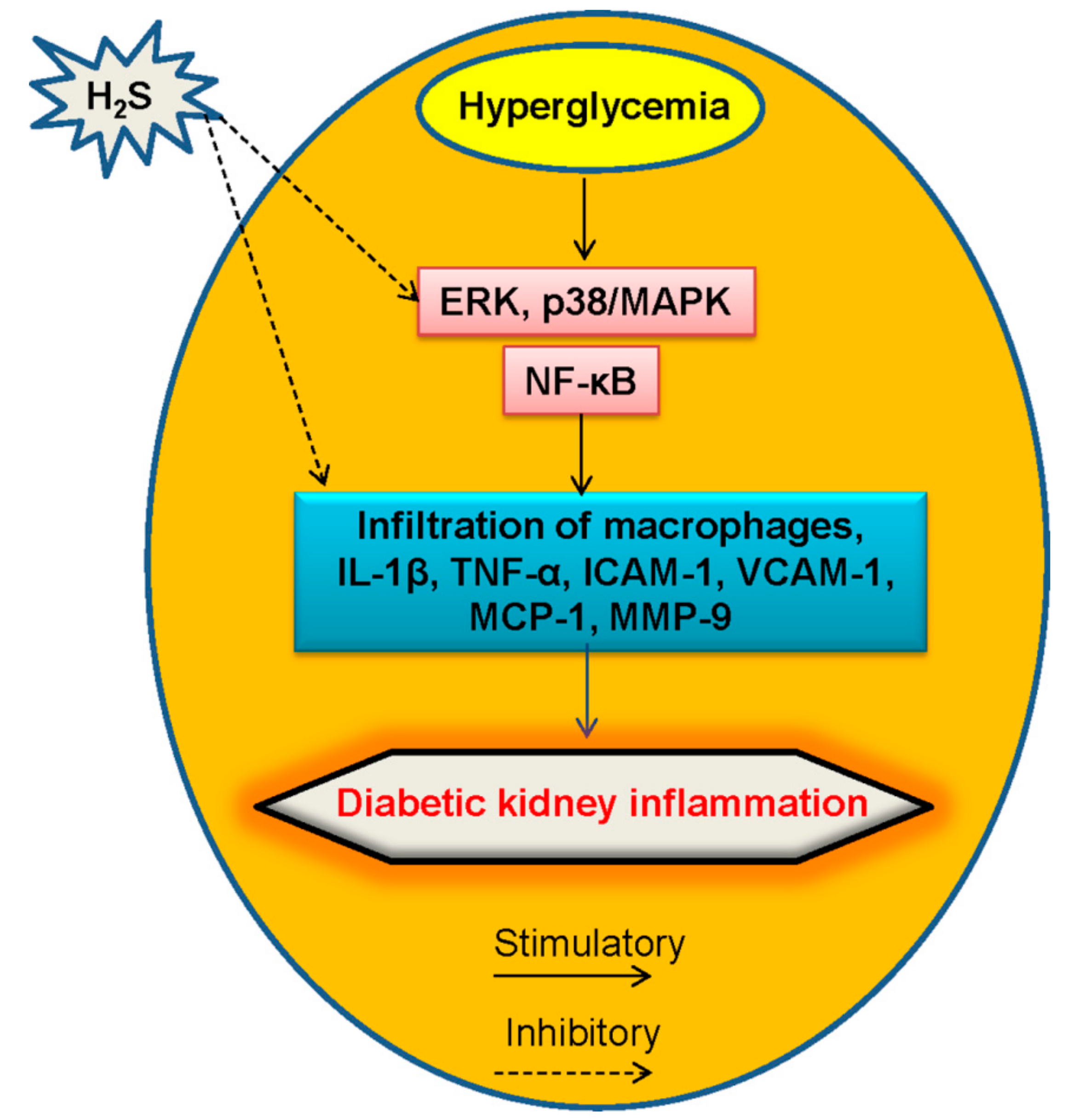
5.3. Inflammation and H2S in Diabetic Kidney Disease
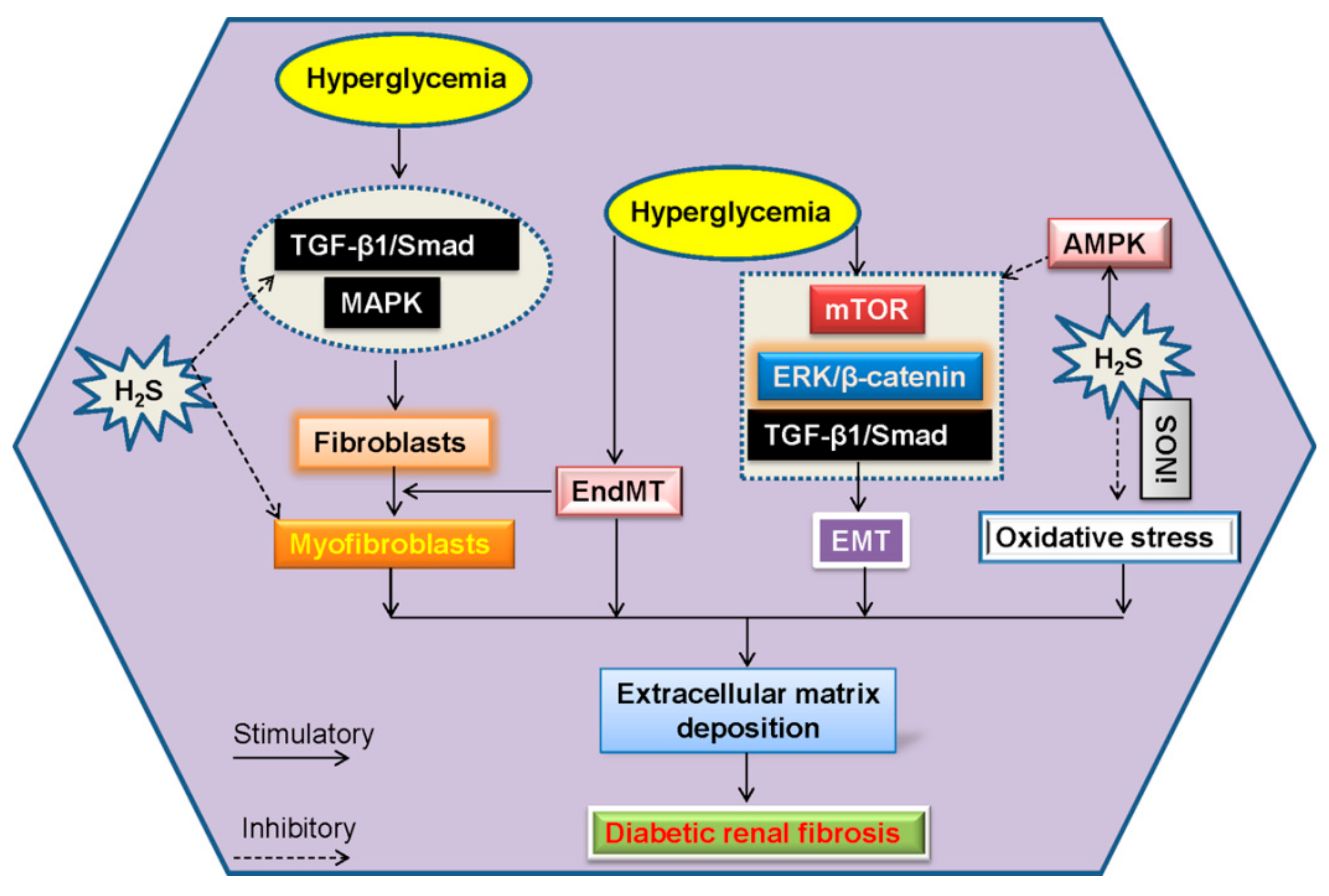
6. Renal Fibrosis and H2S in Diabetic Kidney Disease
6.1. EndMT and H2S in Diabetic Renal Fibrosis
6.2. EMT and H2S in Diabetic Renal Fibrosis
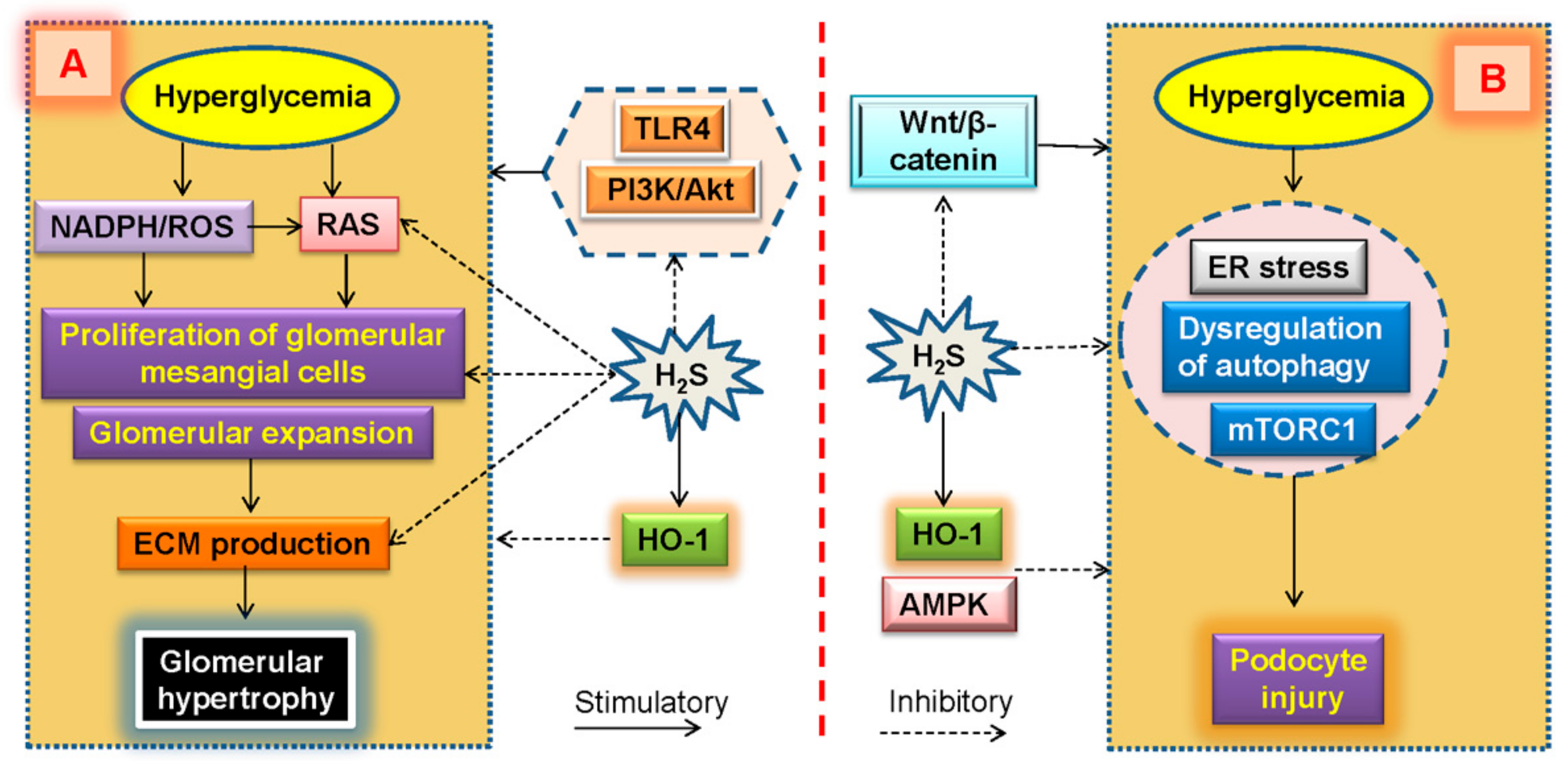
7. Glomerular Expansion and H2S in Diabetic Kidney Disease
8. Podocyte Injury and H2S in Diabetic Kidney Disease
9. Phytopharmaceuticals/Agents-Mediated H2S Induction in Diabetic Kidney Disease
10. Current Molecular Mechanisms of H2S in Diabetic Kidney Disease
11. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Duran-Salgado, M.B.; Rubio-Guerra, A.F. Diabetic nephropathy and inflammation. World J. Diabetes 2014, 5, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.H.; Kang, K.S.; Kwak, M.K. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules 2014, 19, 12727–12759. [Google Scholar] [CrossRef] [PubMed]
- Feliers, D.; Lee, H.J.; Kasinath, B.S. Hydrogen sulfide in renal physiology and disease. Antioxid. Redox Signal. 2016, 25, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Afkarian, M.; Sachs, M.C.; Kestenbaum, B.; Hirsch, I.B.; Tuttle, K.R.; Himmelfarb, J.; de Boer, I.H. Kidney disease and increased mortality risk in type 2 diabetes. J. Am. Soc. Nephrol. JASN 2013, 24, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Gruden, G.; Perin, P.C.; Camussi, G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Curr. Diabetes Rev. 2005, 1, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Dugbartey, G.J. Diabetic nephropathy: A potential savior with ‘rotten-egg’ smell. Pharmacol. Rep. PR 2017, 69, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kasinath, B.S.; Feliers, D.; Lee, H.J. Hydrogen sulfide as a regulatory factor in kidney health and disease. Biochem. Pharmacol. 2018, 149, 29–41. [Google Scholar] [CrossRef]
- Cao, X.; Bian, J.S. The role of hydrogen sulfide in renal system. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef]
- Kashfi, K. The role of hydrogen sulfide in health and disease. Biochem. Pharmacol. 2018, 149, 1–4. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, W.; Moore, P.K.; Bian, J. Protective smell of hydrogen sulfide and polysulfide in cisplatin-induced nephrotoxicity. Int. J. Mol. Sci. 2019, 20, 313. [Google Scholar] [CrossRef]
- Koning, A.M.; Frenay, A.R.; Leuvenink, H.G.; van Goor, H. Hydrogen sulfide in renal physiology, disease and transplantation—The smell of renal protection. Nitric Oxide Biol. Chem. 2015, 46, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Lobb, I.; Sonke, E.; Aboalsamh, G.; Sener, A. Hydrogen sulphide and the kidney: Important roles in renal physiology and pathogenesis and treatment of kidney injury and disease. Nitric Oxide Biol. Chem. 2015, 46, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic kidney disease: Challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. CJASN 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, Y.H.; Yang, S.Y.; Wu, K.D.; Chu, T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med Assoc. Taiwan Yi Zhi 2018, 117, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R. Back to the future: Glomerular hyperfiltration and the diabetic kidney. Diabetes 2017, 66, 14–16. [Google Scholar] [CrossRef]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- De Zeeuw, D.; Coll, B.; Andress, D.; Brennan, J.J.; Tang, H.; Houser, M.; Correa-Rotter, R.; Kohan, D.; Lambers Heerspink, H.J.; Makino, H.; et al. The endothelin antagonist atrasentan lowers residual albuminuria in patients with type 2 diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2014, 25, 1083–1093. [Google Scholar] [CrossRef]
- Prabhakar, S.S. Role of nitric oxide in diabetic nephropathy. Semin. Nephrol. 2004, 24, 333–344. [Google Scholar] [CrossRef]
- Bernhardt, W.M.; Schmitt, R.; Rosenberger, C.; Munchenhagen, P.M.; Grone, H.J.; Frei, U.; Warnecke, C.; Bachmann, S.; Wiesener, M.S.; Willam, C.; et al. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006, 69, 114–122. [Google Scholar] [CrossRef]
- Giacchetti, G.; Sechi, L.A.; Rilli, S.; Carey, R.M. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol. Metab. TEM 2005, 16, 120–126. [Google Scholar] [CrossRef]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Gurley, S.B.; Coffman, T.M. The renin-angiotensin system and diabetic nephropathy. Semin. Nephrol. 2007, 27, 144–152. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Tomaschitz, A. Aldosterone, a vasculotoxic agent—Novel functions for an old hormone. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2009, 24, 2302–2305. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ziyadeh, F.N.; Wolf, G. Pathogenesis of the podocytopathy and proteinuria in diabetic glomerulopathy. Curr. Diabetes Rev. 2008, 4, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Brosius, F.C.; Tuttle, K.R.; Kretzler, M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia 2016, 59, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1792–1798. [Google Scholar] [CrossRef]
- Li, L.; Hsu, A.; Moore, P.K. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation–A tale of three gases! Pharmacol. Ther. 2009, 123, 386–400. [Google Scholar] [CrossRef]
- Mathai, J.C.; Missner, A.; Kugler, P.; Saparov, S.M.; Zeidel, M.L.; Lee, J.K.; Pohl, P. No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. USA 2009, 106, 16633–16638. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. H(2)S signalling through protein sulfhydration and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [Google Scholar] [CrossRef]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide and polysulfides as biological mediators. Molecules 2014, 19, 16146–16157. [Google Scholar] [CrossRef] [PubMed]
- Modis, K.; Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 601–611. [Google Scholar] [CrossRef]
- Tanizawa, K. Production of H2S by 3-mercaptopyruvate sulphurtransferase. J. Biochem. 2011, 149, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Lu, M.; Hu, L.F.; Wong, P.T.; Webb, G.D.; Bian, J.S. Hydrogen sulfide in the mammalian cardiovascular system. Antioxid. Redox Signal. 2012, 17, 141–185. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, P. Endogenous production of hydrogen sulfide in mammals. Amino Acids 2004, 26, 243–254. [Google Scholar] [CrossRef]
- Song, K.; Wang, F.; Li, Q.; Shi, Y.B.; Zheng, H.F.; Peng, H.; Shen, H.Y.; Liu, C.F.; Hu, L.F. Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 2014, 85, 1318–1329. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, C.; Zhou, F.; Yuan, Z.; Wang, H.; Cui, W.; Zhang, G. Hydrogen sulfide as a potential therapeutic target in fibrosis. Oxid. Med. Cell. Longev. 2015, 2015, 593407. [Google Scholar] [CrossRef]
- Bos, E.M.; Wang, R.; Snijder, P.M.; Boersema, M.; Damman, J.; Fu, M.; Moser, J.; Hillebrands, J.L.; Ploeg, R.J.; Yang, G.; et al. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J. Am. Soc. Nephrol. JASN 2013, 24, 759–770. [Google Scholar] [CrossRef]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.L.; Li, N. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Kabil, O.; Vitvitsky, V.; Xie, P.; Banerjee, R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal. 2011, 15, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Hao, D.D.; Sun, J.P.; Li, W.W.; Zhao, M.M.; Li, X.H.; Chen, Y.; Zhu, J.H.; Ding, Y.J.; Liu, J.; et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid. Redox Signal. 2013, 19, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Xue, H.; Zhou, L.; Qu, L.; Li, C.; Wang, Z.; Ni, J.; Yu, C.; Yao, T.; Huang, Y.; et al. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2011, 26, 2119–2126. [Google Scholar] [CrossRef]
- Jain, S.K.; Bull, R.; Rains, J.L.; Bass, P.F.; Levine, S.N.; Reddy, S.; McVie, R.; Bocchini, J.A. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid. Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Gooding, K.M.; Whatmore, J.L.; Ball, C.I.; Mawson, D.; Skinner, K.; Tooke, J.E.; Shore, A.C. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 2010, 53, 1722–1726. [Google Scholar] [CrossRef]
- Li, H.; Feng, S.J.; Zhang, G.Z.; Wang, S.X. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif. 2014, 38, 188–194. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, D.Y.; Mariappan, M.M.; Feliers, D.; Ghosh-Choudhury, G.; Abboud, H.E.; Gorin, Y.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J. Biol. Chem. 2017, 292, 5665–5675. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, Y.; Zhan, Z.; Chen, J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J. Biol. Chem. 2014, 289, 28827–28834. [Google Scholar] [CrossRef]
- Ge, S.N.; Zhao, M.M.; Wu, D.D.; Chen, Y.; Wang, Y.; Zhu, J.H.; Cai, W.J.; Zhu, Y.Z.; Zhu, Y.C. Hydrogen sulfide targets EGFR Cys797/Cys798 residues to induce Na (+)/K (+)-ATPase endocytosis and inhibition in renal tubular epithelial cells and increase sodium excretion in chronic salt-loaded rats. Antioxid. Redox Signal. 2014, 21, 2061–2082. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Liu, H.; Zhang, B.; Zhao, Y.; Ma, K.; Zhao, D.; Wang, Q.; Ma, H.; Zhang, Z. Hydrogen sulfide prevents hydrogen peroxide-induced activation of epithelial sodium channel through a PTEN/PI (3,4,5)P3 dependent pathway. PLoS ONE 2013, 8, e64304. [Google Scholar] [CrossRef]
- Agne, A.M.; Baldin, J.P.; Benjamin, A.R.; Orogo-Wenn, M.C.; Wichmann, L.; Olson, K.R.; Walters, D.V.; Althaus, M. Hydrogen sulfide decreases beta-adrenergic agonist-stimulated lung liquid clearance by inhibiting ENaC-mediated transepithelial sodium absorption. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R636–R649. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.H.; Bian, J.S. Bicarbonate-dependent effect of hydrogen sulfide on vascular contractility in rat aortic rings. Am. J. Physiol. Cell Physiol. 2010, 299, C866–C872. [Google Scholar] [CrossRef][Green Version]
- Lee, E.J.; Hyun, S.H.; Chun, J.; Kang, S.S. Human NIMA-related kinase 6 is one of the Fe65 WW domain binding proteins. Biochem. Biophys. Res. Commun. 2007, 358, 783–788. [Google Scholar] [CrossRef]
- Olson, K.R. Hydrogen sulfide as an oxygen sensor. Antioxid. Redox Signal. 2015, 22, 377–397. [Google Scholar] [CrossRef]
- Kumar, P.; Prabhakar, N.R. Peripheral chemoreceptors: Function and plasticity of the carotid body. Compr. Physiol. 2012, 2, 141–219. [Google Scholar]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J. Exp. Biol. 2006, 209, 4011–4023. [Google Scholar] [CrossRef]
- Olson, K.R.; Whitfield, N.L. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid. Redox Signal. 2010, 12, 1219–1234. [Google Scholar] [CrossRef]
- Hu, H.; Shi, Y.; Chen, Q.; Yang, W.; Zhou, H.; Chen, L.; Tang, Y.; Zheng, Y. Endogenous hydrogen sulfide is involved in regulation of respiration in medullary slice of neonatal rats. Neuroscience 2008, 156, 1074–1082. [Google Scholar] [CrossRef]
- Dombkowski, R.A.; Naylor, M.G.; Shoemaker, E.; Smith, M.; DeLeon, E.R.; Stoy, G.F.; Gao, Y.; Olson, K.R. Hydrogen sulfide (H2S) and hypoxia inhibit salmonid gastrointestinal motility: Evidence for H2S as an oxygen sensor. J. Exp. Biol. 2011, 214, 4030–4040. [Google Scholar] [CrossRef][Green Version]
- Hirakawa, Y.; Tanaka, T.; Nangaku, M. Renal hypoxia in CKD; Pathophysiology and detecting methods. Front. Physiol. 2017, 8, 99. [Google Scholar] [CrossRef]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar] [CrossRef]
- Beltowski, J. Hypoxia in the renal medulla: Implications for hydrogen sulfide signaling. J. Pharmacol. Exp. Ther. 2010, 334, 358–363. [Google Scholar] [CrossRef]
- Kurtz, A. Endocrine functions of the renal interstitium. Pflug. Arch. Eur. J. Physiol. 2017, 469, 869–876. [Google Scholar] [CrossRef]
- Zeisberg, M.; Kalluri, R. Physiology of the renal interstitium. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1831–1840. [Google Scholar] [CrossRef]
- Tong, L.; Adler, S.G. Diabetic kidney disease. Clin. J. Am. Soc. Nephrol. CJASN 2018, 13, 335–338. [Google Scholar] [CrossRef]
- Hostetter, T.H.; Troy, J.L.; Brenner, B.M. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int. 1981, 19, 410–415. [Google Scholar] [CrossRef]
- Anderson, S.; Meyer, T.W.; Rennke, H.G.; Brenner, B.M. Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J. Clin. Investig. 1985, 76, 612–619. [Google Scholar] [CrossRef]
- Zatz, R.; Dunn, B.R.; Meyer, T.W.; Anderson, S.; Rennke, H.G.; Brenner, B.M. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J. Clin. Investig. 1986, 77, 1925–1930. [Google Scholar] [CrossRef]
- Bermejo, S.; Garcia, C.O.; Rodriguez, E.; Barrios, C.; Otero, S.; Mojal, S.; Pascual, J.; Soler, M.J. The renin-angiotensin-aldosterone system blockade in patients with advanced diabetic kidney disease. Nefrologia 2018, 38, 197–206. [Google Scholar] [CrossRef]
- Hsueh, W.A. Treatment of type 2 diabetic nephropathy by blockade of the renin-angiotensin system: A comparison of angiotensin-converting-enzyme inhibitors and angiotensin receptor antagonists. Curr. Opin. Pharmacol. 2002, 2, 182–188. [Google Scholar] [CrossRef]
- Jacobsen, P.K. Preventing end-stage renal disease in diabetic patients—Dual blockade of the renin-angiotensin system (Part II). J. Renin Angiotensin Aldosterone Syst. JRAAS 2005, 6, 55–68. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Stafylas, P.C.; Kanaki, A.I.; Lasaridis, A.N. Effects of renin-angiotensin system blockers on renal outcomes and all-cause mortality in patients with diabetic nephropathy: An updated meta-analysis. Am. J. Hypertens. 2008, 21, 922–929. [Google Scholar] [CrossRef]
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A. Renin release. Physiology 2007, 22, 310–319. [Google Scholar] [CrossRef]
- Lim, J.J.; Liu, Y.H.; Khin, E.S.; Bian, J.S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1261–C1270. [Google Scholar] [CrossRef]
- Yong, Q.C.; Pan, T.T.; Hu, L.F.; Bian, J.S. Negative regulation of beta-adrenergic function by hydrogen sulphide in the rat hearts. J. Mol. Cell. Cardiol. 2008, 44, 701–710. [Google Scholar] [CrossRef]
- Lu, M.; Liu, Y.H.; Goh, H.S.; Wang, J.J.; Yong, Q.C.; Wang, R.; Bian, J.S. Hydrogen sulfide inhibits plasma renin activity. J. Am. Soc. Nephrol. JASN 2010, 21, 993–1002. [Google Scholar] [CrossRef]
- Liu, Y.H.; Lu, M.; Xie, Z.Z.; Hua, F.; Xie, L.; Gao, J.H.; Koh, Y.H.; Bian, J.S. Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in isoproterenol-treated rats. Antioxid. Redox Signal. 2014, 20, 759–769. [Google Scholar] [CrossRef]
- Li, Z.; Organ, C.L.; Kang, J.; Polhemus, D.J.; Trivedi, R.K.; Sharp, T.E., 3rd; Jenkins, J.S.; Tao, Y.X.; Xian, M.; Lefer, D.J. Hydrogen sulfide attenuates renin angiotensin and aldosterone pathological signaling to preserve kidney function and improve exercise tolerance in heart failure. JACC Basic Transl. Sci. 2018, 3, 796–809. [Google Scholar] [CrossRef]
- Kennefick, T.M.; Anderson, S. Role of angiotensin II in diabetic nephropathy. Semin. Nephrol. 1997, 17, 441–447. [Google Scholar]
- Wolf, G.; Butzmann, U.; Wenzel, U.O. The renin-angiotensin system and progression of renal disease: From hemodynamics to cell biology. Nephron. Physiol. 2003, 93, P3–P13. [Google Scholar] [CrossRef]
- Zhuo, J.L.; Li, X.C. New insights and perspectives on intrarenal renin-angiotensin system: Focus on intracrine/intracellular angiotensin II. Peptides 2011, 32, 1551–1565. [Google Scholar] [CrossRef]
- Sonkodi, S.; Mogyorosi, A. Treatment of diabetic nephropathy with angiotensin II blockers. Nephrol. Dial. Transplant. 2003, 18 (Suppl. 5), v21–v23. [Google Scholar] [CrossRef]
- Xue, H.; Yuan, P.; Ni, J.; Li, C.; Shao, D.; Liu, J.; Shen, Y.; Wang, Z.; Zhou, L.; Zhang, W.; et al. H2S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation. PLoS ONE 2013, 8, e74366. [Google Scholar] [CrossRef]
- Kashihara, N.; Haruna, Y.; Kondeti, V.K.; Kanwar, Y.S. Oxidative stress in diabetic nephropathy. Curr. Med. Chem. 2010, 17, 4256–4269. [Google Scholar] [CrossRef]
- LeBaron, T.W.; Kura, B.; Kalocayova, B.; Tribulova, N.; Slezak, J. A new approach for the prevention and treatment of cardiovascular disorders. Molecular hydrogen significantly reduces the effects of oxidative stress. Molecules 2019, 24, 2076. [Google Scholar] [CrossRef]
- Lee, H.B.; Yu, M.R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2003, 14 (Suppl. 3), S241–S245. [Google Scholar] [CrossRef]
- Ha, H.; Yu, M.R.; Choi, Y.J.; Kitamura, M.; Lee, H.B. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J. Am. Soc. Nephrol. JASN 2002, 13, 894–902. [Google Scholar]
- Ha, H.; Lee, H.B. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int. Suppl. 2000, 77, S19–S25. [Google Scholar] [CrossRef]
- Iglesias-De La Cruz, M.C.; Ruiz-Torres, P.; Alcami, J.; Diez-Marques, L.; Ortega-Velazquez, R.; Chen, S.; Rodriguez-Puyol, M.; Ziyadeh, F.N.; Rodriguez-Puyol, D. Hydrogen peroxide increases extracellular matrix mRNA through TGF-beta in human mesangial cells. Kidney Int. 2001, 59, 87–95. [Google Scholar] [CrossRef]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.T.; Lee, H.B. Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. JASN 2005, 16, 667–675. [Google Scholar] [CrossRef]
- Arora, M.K.; Singh, U.K. Oxidative stress: Meeting multiple targets in pathogenesis of diabetic nephropathy. Curr. Drug Targets 2014, 15, 531–538. [Google Scholar] [CrossRef]
- Yang, R.; Liu, X.F.; Ma, S.F.; Gao, Q.; Li, Z.H.; Jia, Q. Protective effect of hydrogen sulfide on kidneys of type 1 diabetic rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi Zhongguo Yingyong Shenglixue Zazhi Chin. J. Appl. Physiol. 2016, 32, 181–184. [Google Scholar]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef]
- Itoh, K.; Mimura, J.; Yamamoto, M. Discovery of the negative regulator of Nrf2, Keap1: A historical overview. Antioxid. Redox Signal. 2010, 13, 1665–1678. [Google Scholar] [CrossRef]
- Keum, Y.S.; Choi, B.Y. Molecular and chemical regulation of the Keap1-Nrf2 signaling pathway. Molecules 2014, 19, 10074–10089. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells Devoted Mol. Cell. Mech. 2001, 6, 857–868. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Katoh, Y.; Iida, K.; Kang, M.I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.D.; Itoh, K.; Yamamoto, M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 2005, 433, 342–350. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of three major cysteine sensors of Keap1 in stress response. Mol. Cell. Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef]
- Ruiz, S.; Pergola, P.E.; Zager, R.A.; Vaziri, N.D. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013, 83, 1029–1041. [Google Scholar] [CrossRef]
- Cui, W.; Bai, Y.; Luo, P.; Miao, L.; Cai, L. Preventive and therapeutic effects of MG132 by activating Nrf2-ARE signaling pathway on oxidative stress-induced cardiovascular and renal injury. Oxid. Med. Cell. Longev. 2013, 2013, 306073. [Google Scholar] [CrossRef]
- Tan, S.M.; de Haan, J.B. Combating oxidative stress in diabetic complications with Nrf2 activators: How much is too much? Redox Rep. Commun. Free Radic. Res. 2014, 19, 107–117. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 system to prevent kidney disease progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef]
- Cui, W.; Min, X.; Xu, X.; Du, B. Role of nuclear factor erythroid 2-related factor 2 in diabetic nephropathy. J. Diabetes Res. 2017, 2017, 3797802. [Google Scholar] [CrossRef]
- Zhou, X.; An, G.; Lu, X. Hydrogen sulfide attenuates the development of diabetic cardiomyopathy. Clin. Sci. 2015, 128, 325–335. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Yang, H.; Mao, Y.; Tan, B.; Luo, S.; Zhu, Y. The protective effects of endogenous hydrogen sulfide modulator, S-propargyl-cysteine, on high glucose-induced apoptosis in cardiomyocytes: A novel mechanism mediated by the activation of Nrf2. Eur. J. Pharmacol. 2015, 761, 135–143. [Google Scholar] [CrossRef]
- Cao, X.; Nie, X.; Xiong, S.; Cao, L.; Wu, Z.; Moore, P.K.; Bian, J.S. Renal protective effect of polysulfide in cisplatin-induced nephrotoxicity. Redox Biol. 2018, 15, 513–521. [Google Scholar] [CrossRef]
- Perez-Morales, R.E.; Del Pino, M.D.; Valdivielso, J.M.; Ortiz, A.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammation in diabetic kidney disease. Nephron 2018, 10, 1–5. [Google Scholar] [CrossRef]
- Donath, M.Y. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia 2016, 59, 679–682. [Google Scholar] [CrossRef]
- Pickup, J.C.; Crook, M.A. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998, 41, 1241–1248. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Johnson, E.J.; Tuttle, K.R. Inflammatory mechanisms as new biomarkers and therapeutic targets for diabetic kidney disease. Adv. Chronic Kidney Dis. 2018, 25, 181–191. [Google Scholar] [CrossRef]
- Garcia-Garcia, P.M.; Getino-Melian, M.A.; Dominguez-Pimentel, V.; Navarro-Gonzalez, J.F. Inflammation in diabetic kidney disease. World J. Diabetes 2014, 5, 431–443. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora, C. Role of inflammation in diabetic complications. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc. Eur. Ren. Assoc. 2005, 20, 2601–2604. [Google Scholar] [CrossRef]
- Mora, C.; Navarro, J.F. Inflammation and diabetic nephropathy. Curr. Diabetes Rep. 2006, 6, 463–468. [Google Scholar] [CrossRef]
- Wang, M.; Tang, W.; Zhu, Y.Z. An update on AMPK in hydrogen sulfide pharmacology. Front. Pharmacol. 2017, 8, 810. [Google Scholar] [CrossRef]
- Sen, U.; Basu, P.; Abe, O.A.; Givvimani, S.; Tyagi, N.; Metreveli, N.; Shah, K.S.; Passmore, J.C.; Tyagi, S.C. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. Am. J. Physiol. Ren. Physiol. 2009, 297, F410–F419. [Google Scholar] [CrossRef]
- Toba, H.; Lindsey, M.L. Extracellular matrix roles in cardiorenal fibrosis: Potential therapeutic targets for CVD and CKD in the elderly. Pharmacol. Ther. 2019, 193, 99–120. [Google Scholar] [CrossRef]
- Hayashi, T.; Takai, S.; Yamashita, C. Impact of the renin-angiotensin-aldosterone-system on cardiovascular and renal complications in diabetes mellitus. Curr. Vasc. Pharmacol. 2010, 8, 189–197. [Google Scholar] [CrossRef]
- Kundu, S.; Pushpakumar, S.B.; Tyagi, A.; Coley, D.; Sen, U. Hydrogen sulfide deficiency and diabetic renal remodeling: Role of matrix metalloproteinase-9. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1365–E1378. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Koya, D.; Kanasaki, K. MicroRNAs in kidney fibrosis and diabetic nephropathy: Roles on EMT and EndMT. BioMed Res. Int. 2013, 2013, 125469. [Google Scholar] [CrossRef]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic modification mechanisms involved in inflammation and fibrosis in renal pathology. Mediators Inflamm 2018, 2018, 2901049. [Google Scholar] [CrossRef]
- Kanasaki, K.; Shi, S.; Kanasaki, M.; He, J.; Nagai, T.; Nakamura, Y.; Ishigaki, Y.; Kitada, M.; Srivastava, S.P.; Koya, D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 2014, 63, 2120–2131. [Google Scholar] [CrossRef]
- Harris, R.C.; Neilson, E.G. Toward a unified theory of renal progression. Annu. Rev. Med. 2006, 57, 365–380. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. JASN 2008, 19, 2282–2287. [Google Scholar] [CrossRef]
- Villeneuve, L.M.; Reddy, M.A.; Natarajan, R. Epigenetics: Deciphering its role in diabetes and its chronic complications. Clin. Exp. Pharmacol. Physiol. 2011, 38, 451–459. [Google Scholar] [CrossRef]
- Kanasaki, K.; Nagai, T.; Nitta, K.; Kitada, M.; Koya, D. N-acetyl-seryl-aspartyl-lysyl-proline: A valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front. Pharmacol. 2014, 5, 70. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Wang, X.; Chen, Z.; Peng, K.; Lu, X.; Meng, L.; Liu, G.; Guan, G.; Wang, F. Serum response factor induces endothelial-mesenchymal transition in glomerular endothelial cells to aggravate proteinuria in diabetic nephropathy. Physiol. Genom. 2016, 48, 711–718. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Li, J.; Bertram, J.F. Review: Endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrology 2010, 15, 507–512. [Google Scholar] [CrossRef]
- Medici, D.; Kalluri, R. Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 2012, 22, 379–384. [Google Scholar] [CrossRef]
- Ying, R.; Wang, X.Q.; Yang, Y.; Gu, Z.J.; Mai, J.T.; Qiu, Q.; Chen, Y.X.; Wang, J.F. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016, 144, 208–217. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Zeisberg, M.; Bottiglio, C.; Kumar, N.; Maeshima, Y.; Strutz, F.; Muller, G.A.; Kalluri, R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003, 285, F1060–F1067. [Google Scholar] [CrossRef]
- Yeung, K.T.; Yang, J. Epithelial-mesenchymal transition in tumor metastasis. Mol. Oncol. 2017, 11, 28–39. [Google Scholar] [CrossRef]
- Carew, R.M.; Wang, B.; Kantharidis, P. The role of EMT in renal fibrosis. Cell Tissue Res. 2012, 347, 103–116. [Google Scholar] [CrossRef]
- Loeffler, I.; Wolf, G. Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef]
- Fragiadaki, M.; Mason, R.M. Epithelial-mesenchymal transition in renal fibrosis—Evidence for and against. Int. J. Exp. Pathol. 2011, 92, 143–150. [Google Scholar] [CrossRef]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef]
- Hills, C.E.; Squires, P.E. The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011, 22, 131–139. [Google Scholar]
- Lin, S.; Visram, F.; Liu, W.; Haig, A.; Jiang, J.; Mok, A.; Lian, D.; Wood, M.E.; Torregrossa, R.; Whiteman, M.; et al. GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J. Urol. 2016, 196, 1778–1787. [Google Scholar] [CrossRef]
- Lin, S.; Lian, D.; Liu, W.; Haig, A.; Lobb, I.; Hijazi, A.; Razvi, H.; Burton, J.; Whiteman, M.; Sener, A. Daily therapy with a slow-releasing H2S donor GYY4137 enables early functional recovery and ameliorates renal injury associated with urinary obstruction. Nitric Oxide Biol. Chem. 2018, 76, 16–28. [Google Scholar] [CrossRef]
- Lin, S.; Juriasingani, S.; Sener, A. Is hydrogen sulfide a potential novel therapy to prevent renal damage during ureteral obstruction? Nitric Oxide Biol. Chem. 2018, 73, 15–21. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, Z.; Huang, Y.; Mao, Z.; Yang, X.; Nakamura, Y.; Sawada, N.; Mitsui, T.; Takeda, M.; Yao, J. Induction of inactive TGF-beta1 monomer formation by hydrogen sulfide contributes to its suppressive effects on Ang II- and TGF-beta1-induced EMT in renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2018, 501, 534–540. [Google Scholar] [CrossRef]
- Guo, L.; Peng, W.; Tao, J.; Lan, Z.; Hei, H.; Tian, L.; Pan, W.; Wang, L.; Zhang, X. Hydrogen sulfide inhibits transforming growth Factor-Beta1-induced EMT via Wnt/Catenin pathway. PLoS ONE 2016, 11, e0147018. [Google Scholar]
- Lee, H.J.; Mariappan, M.M.; Feliers, D.; Cavaglieri, R.C.; Sataranatarajan, K.; Abboud, H.E.; Choudhury, G.G.; Kasinath, B.S. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J. Biol. Chem. 2012, 287, 4451–4461. [Google Scholar] [CrossRef]
- Pfeilschifter, J. Does nitric oxide, an inflammatory mediator of glomerular mesangial cells, have a role in diabetic nephropathy? Kidney Int. Suppl. 1995, 51, S50–S60. [Google Scholar]
- Ha, H.; Kim, K.H. Pathogenesis of diabetic nephropathy: The role of oxidative stress and protein kinase C. Diabetes Res. Clin. Pract. 1999, 45, 147–151. [Google Scholar] [CrossRef]
- Makino, H.; Sugiyama, H.; Kashihara, N. Apoptosis and extracellular matrix-cell interactions in kidney disease. Kidney Int. Suppl. 2000, 77, S67–S75. [Google Scholar] [CrossRef]
- Sugiyama, H.; Kashihara, N.; Maeshima, Y.; Okamoto, K.; Kanao, K.; Sekikawa, T.; Makino, H. Regulation of survival and death of mesangial cells by extracellular matrix. Kidney Int. 1998, 54, 1188–1196. [Google Scholar] [CrossRef]
- Guan, Y.; Breyer, M.D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2001, 60, 14–30. [Google Scholar] [CrossRef]
- Kobori, H.; Nangaku, M.; Navar, L.G.; Nishiyama, A. The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol. Rev. 2007, 59, 251–287. [Google Scholar] [CrossRef]
- Malek, V.; Sharma, N.; Sankrityayan, H.; Gaikwad, A.B. Concurrent neprilysin inhibition and renin-angiotensin system modulations prevented diabetic nephropathy. Life Sci. 2019, 221, 159–167. [Google Scholar] [CrossRef]
- De Morais, R.B.; do Couto Muniz, V.P.; Nunes Costa, E.; Filho, S.R.F.; Nakamura Hiraki, K.R.; Bispo-da-Silva, L.B.; Coelho Balbi, A.P. Mast cell population in the development of diabetic nephropathy: Effects of renin angiotensin system inhibition. Biomed. Pharmacother. 2018, 107, 1115–1118. [Google Scholar] [CrossRef]
- Brewster, U.C.; Perazella, M.A. The renin-angiotensin-aldosterone system and the kidney: Effects on kidney disease. Am. J. Med. 2004, 116, 263–272. [Google Scholar] [CrossRef]
- Ohashi, N.; Urushihara, M.; Satou, R.; Kobori, H. Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen species—ERK/JNK pathways. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2010, 33, 1174–1181. [Google Scholar] [CrossRef]
- Catherwood, M.A.; Powell, L.A.; Anderson, P.; McMaster, D.; Sharpe, P.C.; Trimble, E.R. Glucose-induced oxidative stress in mesangial cells. Kidney Int. 2002, 61, 599–608. [Google Scholar] [CrossRef]
- D’Araio, E.; Shaw, N.; Millward, A.; Demaine, A.; Whiteman, M.; Hodgkinson, A. Hydrogen sulfide induces heme oxygenase-1 in human kidney cells. Acta Diabetol. 2014, 51, 155–157. [Google Scholar] [CrossRef]
- Ding, T.; Chen, W.; Li, J.; Ding, J.; Mei, X.; Hu, H. High glucose induces mouse mesangial cell overproliferation via inhibition of hydrogen sulfide synthesis in a TLR-4-dependent manner. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 1035–1043. [Google Scholar] [CrossRef]
- Reddy, G.R.; Kotlyarevska, K.; Ransom, R.F.; Menon, R.K. The podocyte and diabetes mellitus: Is the podocyte the key to the origins of diabetic nephropathy? Curr. Opin. Nephrol. Hypertens. 2008, 17, 32–36. [Google Scholar] [CrossRef]
- Wolf, G.; Chen, S.; Ziyadeh, F.N. From the periphery of the glomerular capillary wall toward the center of disease: Podocyte injury comes of age in diabetic nephropathy. Diabetes 2005, 54, 1626–1634. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Shankland, S.J.; Pichler, R.H. Proteinuria in diabetic kidney disease: A mechanistic viewpoint. Kidney Int. 2008, 74, 22–36. [Google Scholar] [CrossRef]
- Hishikawa, A.; Hayashi, K.; Itoh, H. Transcription factors as therapeutic targets in chronic kidney disease. Molecules 2018, 23, 1123. [Google Scholar] [CrossRef]
- Turkmen, K. Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: The four horsemen of the apocalypse. Int. Urol. Nephrol. 2017, 49, 837–844. [Google Scholar] [CrossRef]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef]
- Stitt-Cavanagh, E.; MacLeod, L.; Kennedy, C. The podocyte in diabetic kidney disease. Sci. World J. 2009, 9, 1127–1139. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.P.; Xu, K.F.; Mao, X.D.; Lu, Y.B.; Fang, L.; Yang, J.W.; Liu, C. Effect of taurine-conjugated ursodeoxycholic acid on endoplasmic reticulum stress and apoptosis induced by advanced glycation end products in cultured mouse podocytes. Am. J. Nephrol. 2008, 28, 1014–1022. [Google Scholar] [CrossRef]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Ruegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]
- Godel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mor, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, H.; Qiang, Y.; Qian, G.; Lu, S.; Chen, J.; Wang, X.; Guan, Q.; Liu, Y.; Fu, Y. Effects of hydrogen sulfide on high glucose-induced glomerular podocyte injury in mice. Int. J. Clin. Exp. Pathol. 2015, 8, 6814–6820. [Google Scholar]
- Qian, X.; Li, X.; Ma, F.; Luo, S.; Ge, R.; Zhu, Y. Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine, prevents STZ-induced diabetic nephropathy. Biochem. Biophys. Res. Commun. 2016, 473, 931–938. [Google Scholar] [CrossRef]
- Lee, H.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Choudhury, G.G.; Gorin, Y.; Kasinath, B.S. Tadalafil integrates nitric oxide-hydrogen sulfide signaling to inhibit high glucose-induced matrix protein synthesis in podocytes. J. Biol. Chem. 2015, 290, 12014–12026. [Google Scholar] [CrossRef]
- Nasri, H.; Rafieian-Kopaei, M. Metformin and diabetic kidney disease: A mini-review on recent findings. Iran. J. Pediatr. 2014, 24, 565–568. [Google Scholar]
- Wilinski, B.; Wilinski, J.; Somogyi, E.; Piotrowska, J.; Opoka, W. Metformin raises hydrogen sulfide tissue concentrations in various mouse organs. Pharmacol. Rep. PR 2013, 65, 737–742. [Google Scholar] [CrossRef]
- John, A.; Kundu, S.; Pushpakumar, S.; Fordham, M.; Weber, G.; Mukhopadhyay, M.; Sen, U. GYY4137, a hydrogen sulfide donor modulates miR194-dependent collagen realignment in diabetic kidney. Sci. Rep. 2017, 7, 10924. [Google Scholar] [CrossRef]
- Qian, L.L.; Liu, X.Y.; Chai, Q.; Wang, R.X. Hydrogen sulfide in diabetic complications: Focus on molecular mechanisms. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 470–476. [Google Scholar] [CrossRef]
- Kanwar, Y.S.; Sun, L.; Xie, P.; Liu, F.Y.; Chen, S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 2011, 6, 395–423. [Google Scholar] [CrossRef]
- Sharma, K. Obesity, oxidative stress, and fibrosis in chronic kidney disease. Kidney Int. Suppl. 2014, 4, 113–117. [Google Scholar] [CrossRef]
- Dimas, G.G.; Didangelos, T.P.; Grekas, D.M. Matrix gelatinases in atherosclerosis and diabetic nephropathy: Progress and challenges. Curr. Vasc. Pharmacol. 2017, 15, 557–565. [Google Scholar] [CrossRef]
- Lloberas, N.; Cruzado, J.M.; Franquesa, M.; Herrero-Fresneda, I.; Torras, J.; Alperovich, G.; Rama, I.; Vidal, A.; Grinyo, J.M. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J. Am. Soc. Nephrol. JASN 2006, 17, 1395–1404. [Google Scholar] [CrossRef]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef]
- Eid, A.A.; Ford, B.M.; Block, K.; Kasinath, B.S.; Gorin, Y.; Ghosh-Choudhury, G.; Barnes, J.L.; Abboud, H.E. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. J. Biol. Chem. 2010, 285, 37503–37512. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N (G)-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636.e1–636.e72. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2S Donors | Cell Type | Main Findings | Ref. |
|---|---|---|---|
| NaHS | Renal fibroblasts | H2S inhibits the proliferation of renal fibroblasts. Furthermore, the differentiation of quiescent renal fibroblasts to myofibroblasts is prevented by H2S, which involves the inhibition of TGF-β1-Smad and MAPK signaling pathways. | [38] |
| NaHS | Glomerular mesangial cells | NaHS inhibits the ROS generation and cell proliferation, and downregulates the expressions of TGF-β1 and collagen IV in high glucose-incubated cells. | [45] |
| NaHS | Renal tubular epithelial cells. | The activation of AMPK by H2S prevents high glucose-induced NOX4 expression in epithelial cells. NaHS augments the expression of iNOS, this effect is involved in the protective effect of H2S against high glucose-induced NOX4 expression, ROS generation, and matrix laminin expression. | [49] |
| NaHS | Glomerular mesangial cells | H2S activates the Nrf2 signaling pathway to restrain high glucose-induced oxidative stress. H2S exerts anti-inflammatory effects by blocking NF-κB signaling. Additionally, the cell proliferation induced by high glucose is mediated by MAPK signaling pathways, which is impeded by H2S. | [50] |
| NaHS | Glomerular mesangial cells | Supplementation of H2S represses the cell proliferation, inhibits TGF-β1 and collagen IV expressions, and attenuates the elevation of ROS in high glucose-treated cells. Meanwhile, AGT, ACE and AT1 receptor mRNA levels and Ang II concentration are upregulated in high glucose-challenged cells, which are diminished by H2S. | [86] |
| NaHS | Renal tubular epithelial cells | Blockade of ERK- and β-catenin-dependent pathways may be involved in the suppressive effect of H2S on TGF-β1-induced renal EMT in renal tubular epithelial cells, as evidenced by upregulated levels of E-cadherin, along with downregulated expressions of α-SMA and fibronectin. | [156] |
| NaHS | Renal tubular epithelial cells | The activation of mTORC1 and inactivation of AMPK are involved in global matrix protein synthesis, and these events are all reversed by NaHS. Importantly, NaHS stimulates AMPK phosphorylation and restores AMPK phosphorylation induced by high glucose. | [157] |
| AP39, AP106, AP72, AP67, GYY4134 | Glomerular mesangial cells, podocytes | H2S upregulates the expression of HO-1 in both mesangial and podocyte cells. H2S might have the ability to upregulate this antioxidant enzyme, which may be a potential mechanism by which H2S exerts its protective effects. | [169] |
| NaHS | Glomerular mesangial cells | Exogenous H2S treatment mitigates the proliferation of mesangial cells. Furthermore, H2S supplementation remarkably inhibits TLR4 expression and curbs the mesangial cell proliferation. | [170] |
| NaHS | Mouse podocytes | High glucose stimulation significantly reduces nephrin, ZO-2, and CSE expression levels, and elevates β-catenin production in mouse podocytes. Supplementation of NaHS rectifies these changes. Exogenous H2S may alleviate high glucose-induced podocyte injury possibly through ZO-2 upregulation and the subsequent suppression of Wnt/β-catenin pathway. | [182] |
| GYY4137 | Glomerular endothelial cells | GYY4137 upregulates miR-194 level to mitigate ROS production under high glucose condition. | [187] |
| H2S Donors | Animal Models | Main Findings | Ref. |
|---|---|---|---|
| NaHS | STZ-induced diabetic rats | Administration of NaHS reverses the increases in TGF-β1 and collagen IV in diabetic rats. | [45] |
| NaHS | STZ-induced diabetic rats | H2S attenuates glomerular basement membrane thickening, mesangial matrix deposition, and renal interstitial fibrosis, thereby improving improve renal function in diabetic rats. The protein expressions of ACE and AT1 receptors as well as Ang II are significantly up-regulated in diabetic kidneys and down-regulated after treatment with H2S. | [50] |
| NaHS | STZ-induced diabetic rats | In STZ-induced diabetic rats, the changes in RAS are reversed by H2S supplementation without affecting blood glucose concentration. | [86] |
| NaHS | STZ-induced diabetic rats | The increased 24 h urinary protein, fasting blood glucose (FBG), blood urea nitrogen (BUN), serum creatinine (Scr) and renal index, as well as the elevated amount of glomerular mesangial matrix in diabetic rats are all ameliorated by H2S treatment. In addition, the diabetic kidney shows the increased MDA content, caspase-3 activity and Bax expression, but decreased SOD activity and BCl−2 expression, which are normalized by administration of H2S. | [95] |
| NaHS | Mice with type 1 diabetes or type 2 diabetes | Renal cortical contents of CBS and CSE are significantly reduced, alongside with renal hypertrophy and matrix accumulation in mice with type 1 diabetes or type 2 diabetes. | [157] |
| GYY4137 | Diabetic Akita mice | GYY4137 prevents collagen deposition and realignment and renal fibrosis in mice. The increased expressions of MMP-9, MMP-13 and MMP-14, and reduced vascular density in diabetic kidney are reversed by GYY4137. | [187] |
| H2S-Releasing Compounds | Animal Models/Cell Type | Main Finding | Ref. |
|---|---|---|---|
| S-propargyl-cysteine | STZ-induced diabetic rat/mesangial cells | S-propargyl-cysteine, a H2S-releasing compound, reduces the level of creatinine, kidney to body weight ratio and 24-h urine microalbuminuria excretion in STZ-induced diabetic kidney injury. The renal fibrosis, inflammation, and hypertrophy are suppressed by this compound. The renal protective effects of this compound may be mediated by inhibition of TGF-β1/Smad3 pathway and blockade of MAPK signaling pathway. | [183] |
| Tadalafil | Podocytes | Tadalafil, increases the expression and activity of the H2S-generating enzyme CSE by accelerating its translation. It can effectively abrogate high glucose-induced global protein synthesis in podocytes. Tadalafil activates AMPK by stimulating calcium-calmodulin kinase β, thus attenuating the activation of mTOR induced by high glucose. Furthermore, in tadalafil-treated podocytes, the iNOS expression is rapidly upregulated. Knockdown or inhibition of iNOS abolished the effect of tadalafil on CSE expression and AMPK phosphorylation in podocytes. | [184] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.-J.; Wu, Z.-Y.; Cao, L.; Zhu, M.-Y.; Liu, T.-T.; Guo, L.; Lin, Y.; Nie, X.-W.; Bian, J.-S. Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy. Molecules 2019, 24, 2857. https://doi.org/10.3390/molecules24152857
Sun H-J, Wu Z-Y, Cao L, Zhu M-Y, Liu T-T, Guo L, Lin Y, Nie X-W, Bian J-S. Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy. Molecules. 2019; 24(15):2857. https://doi.org/10.3390/molecules24152857
Chicago/Turabian StyleSun, Hai-Jian, Zhi-Yuan Wu, Lei Cao, Meng-Yuan Zhu, Teng-Teng Liu, Lei Guo, Ye Lin, Xiao-Wei Nie, and Jin-Song Bian. 2019. "Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy" Molecules 24, no. 15: 2857. https://doi.org/10.3390/molecules24152857
APA StyleSun, H.-J., Wu, Z.-Y., Cao, L., Zhu, M.-Y., Liu, T.-T., Guo, L., Lin, Y., Nie, X.-W., & Bian, J.-S. (2019). Hydrogen Sulfide: Recent Progression and Perspectives for the Treatment of Diabetic Nephropathy. Molecules, 24(15), 2857. https://doi.org/10.3390/molecules24152857