
Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease

 , , ,
, , ,  ,
,  and
and
Abstract
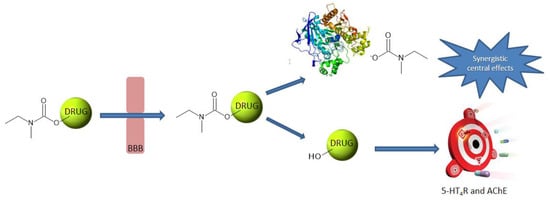
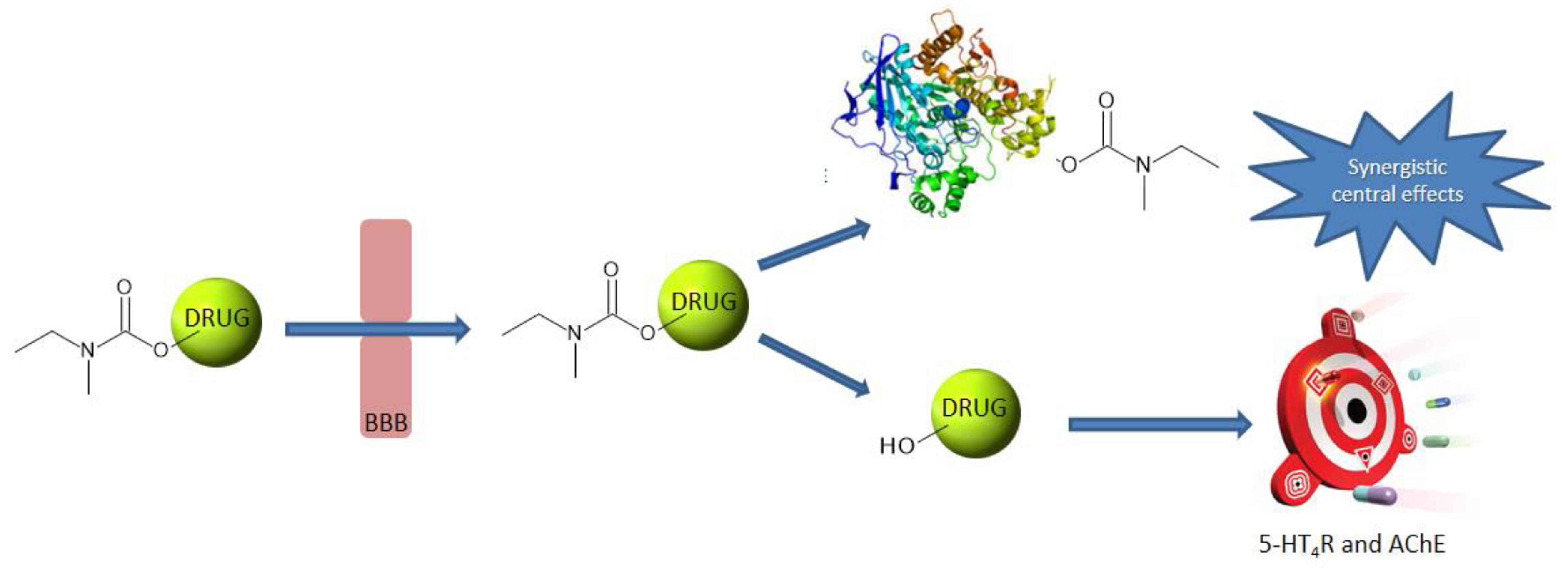
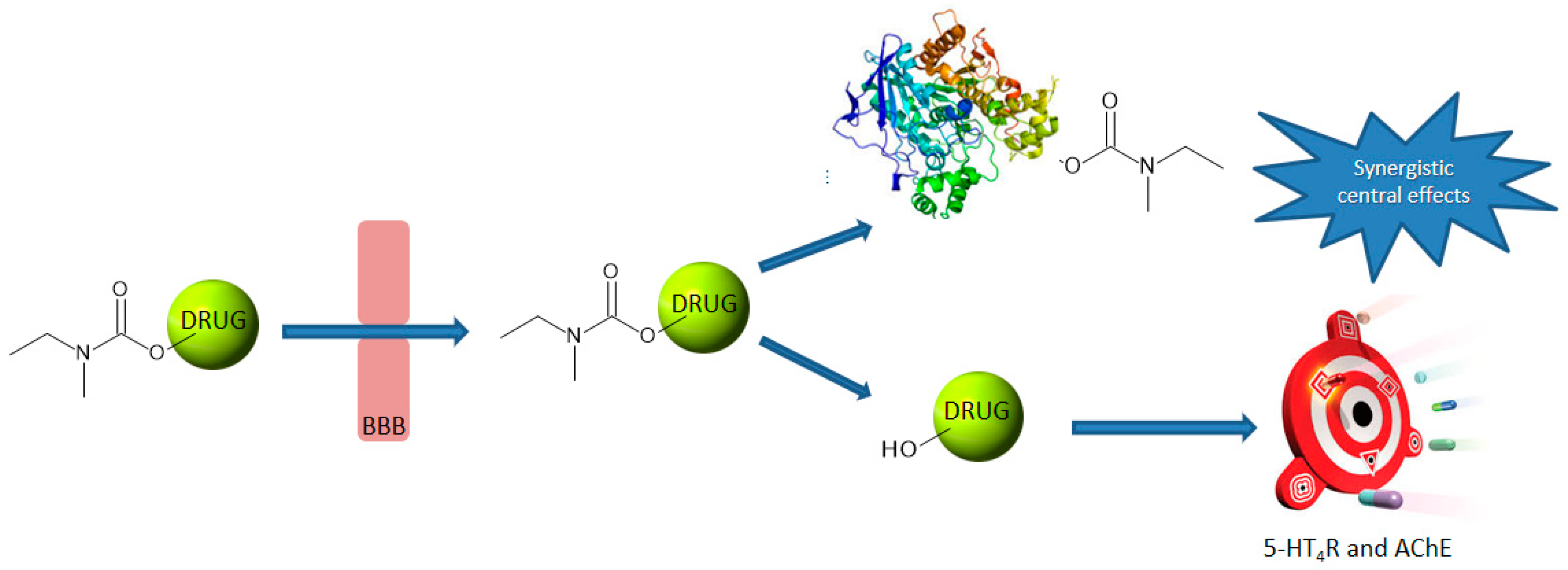
1. Introduction
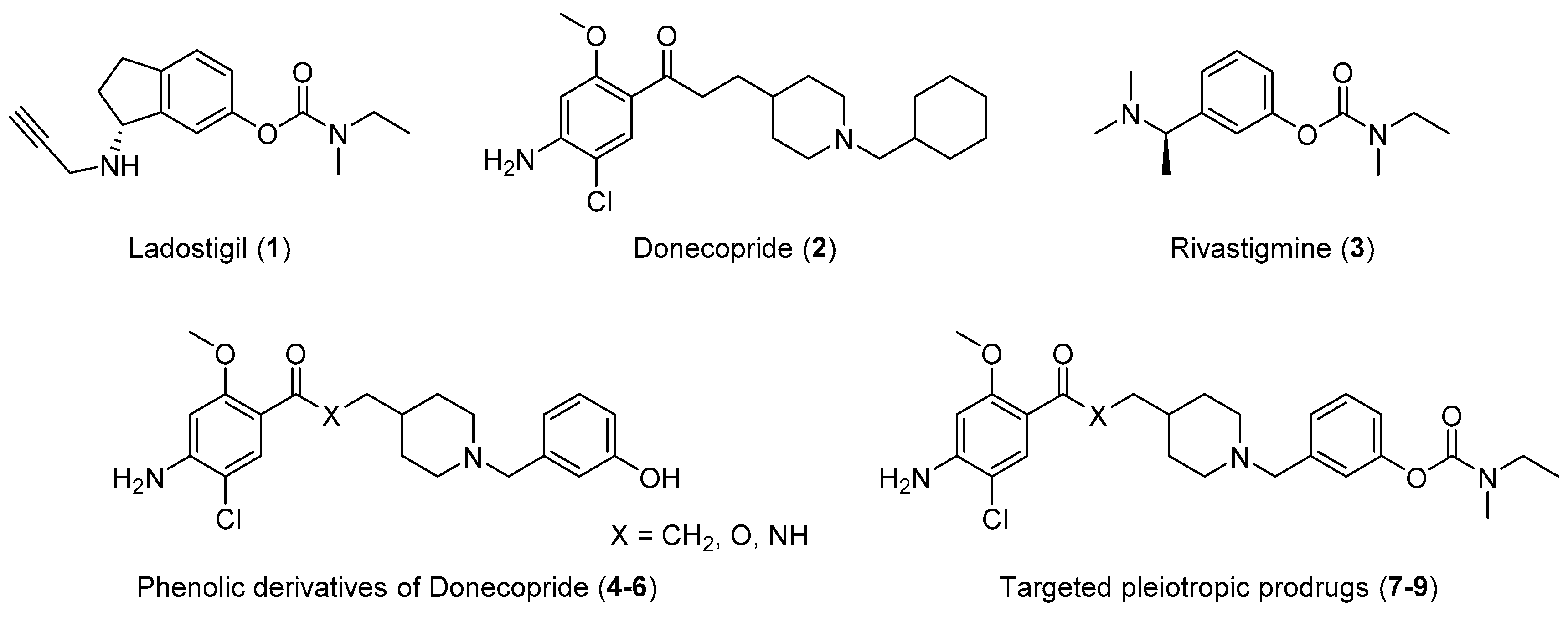
2. Results
2.1. Chemistry
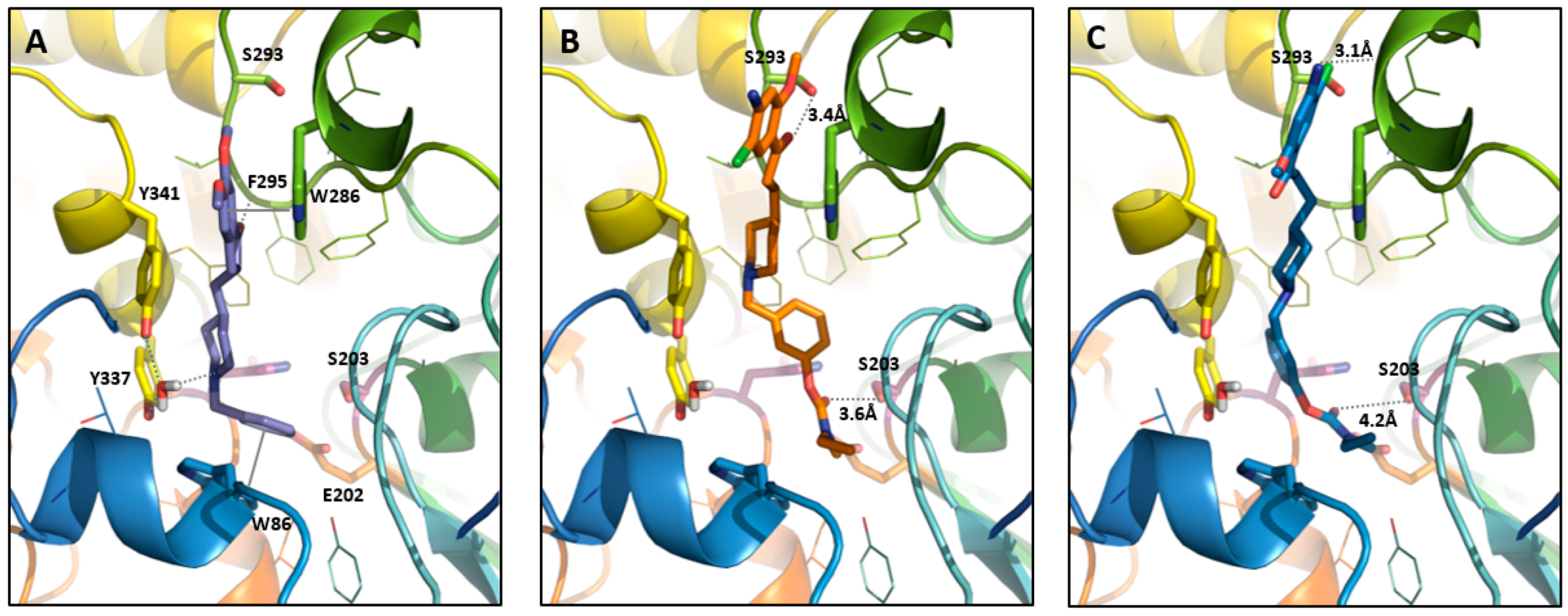
2.2. In Silico Results
2.3. In Vitro Results
2.3.1. AChE Inhibition and 5-HT4R Binding
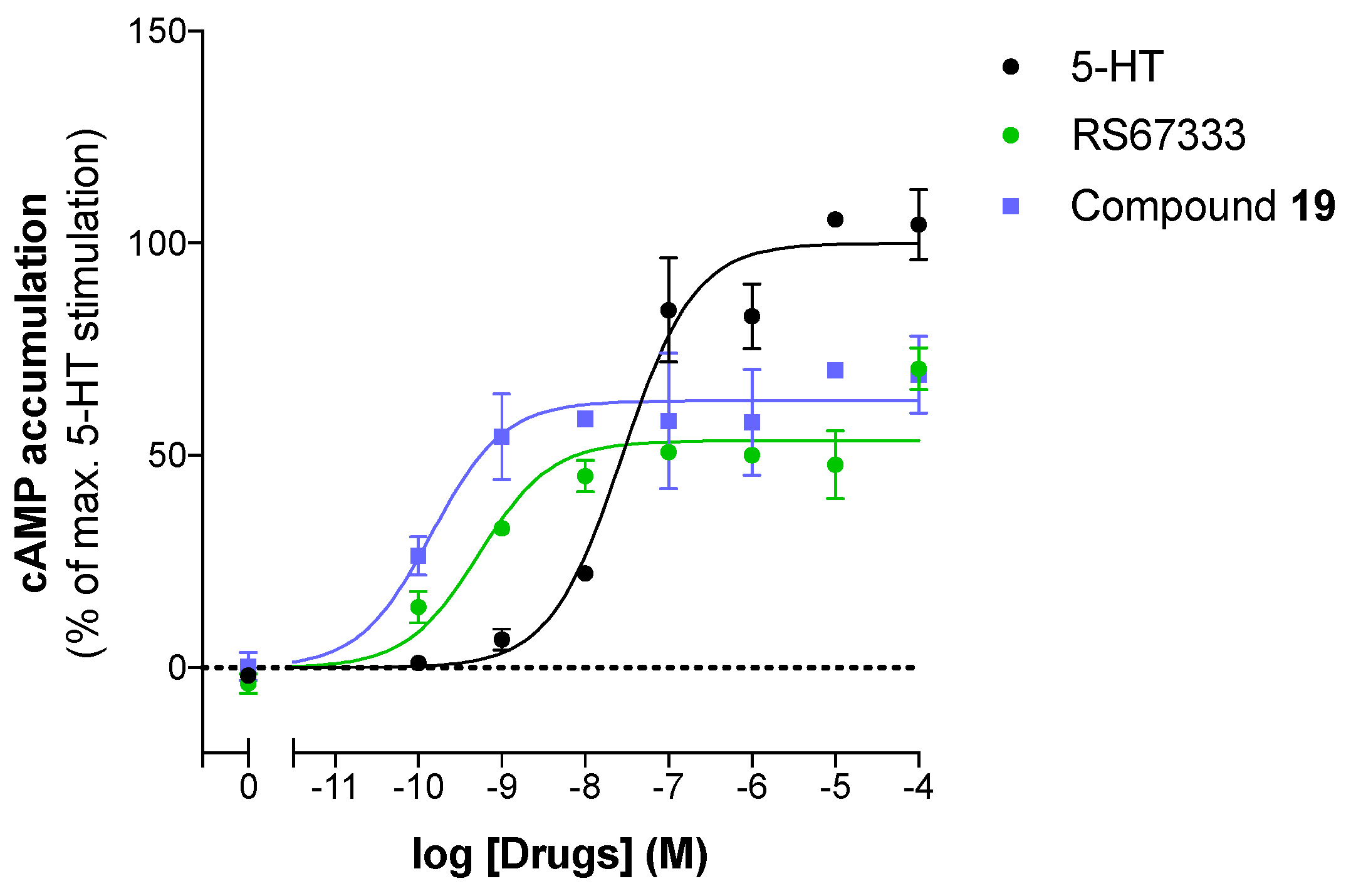
2.3.2. Pharmacological Profile Results
2.3.3. Brain Penetration
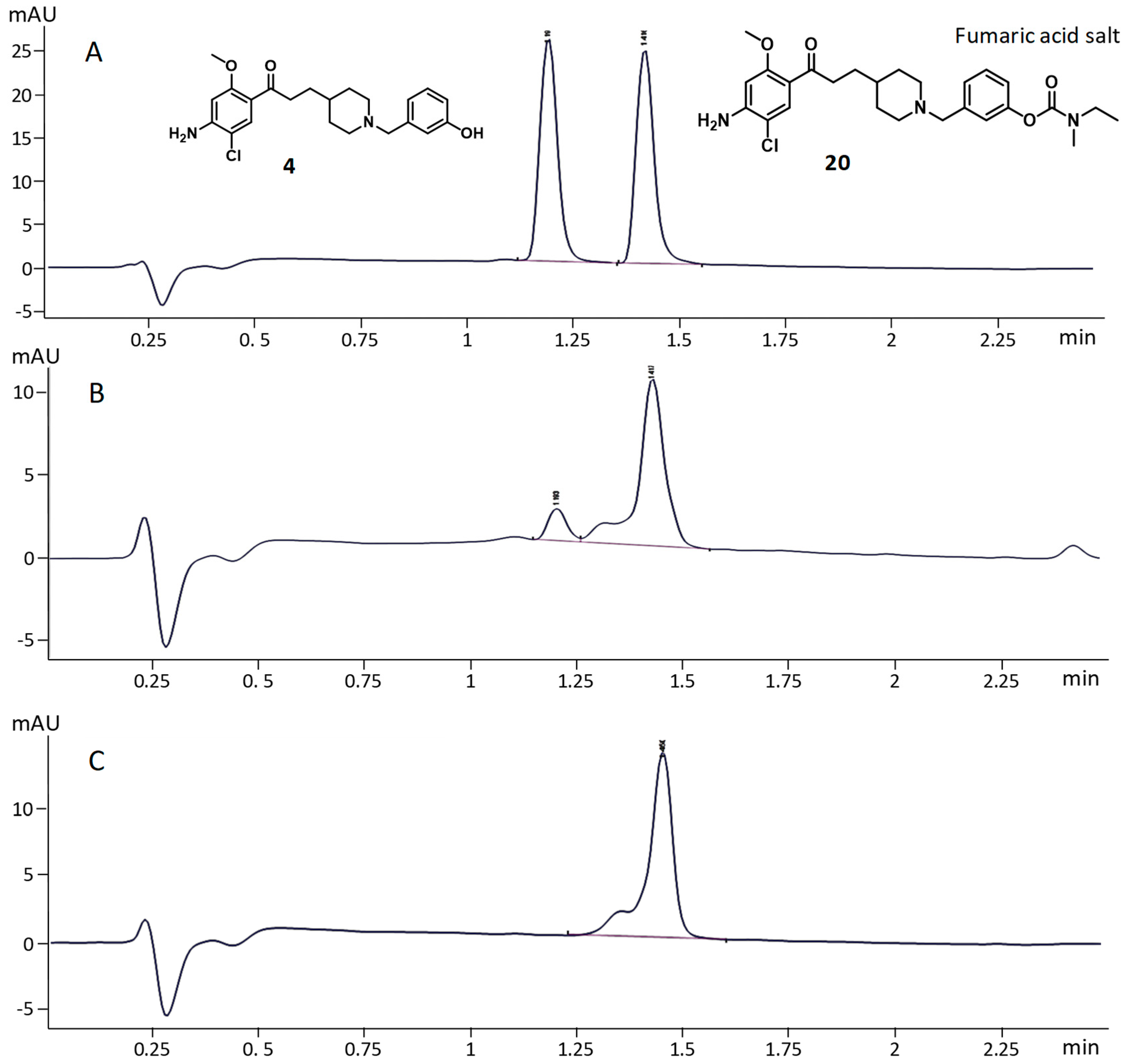
2.3.4. AChE-Dependent Decarbamoylation
2.4. In Vivo Results
2.4.1. Pharmacological Screening
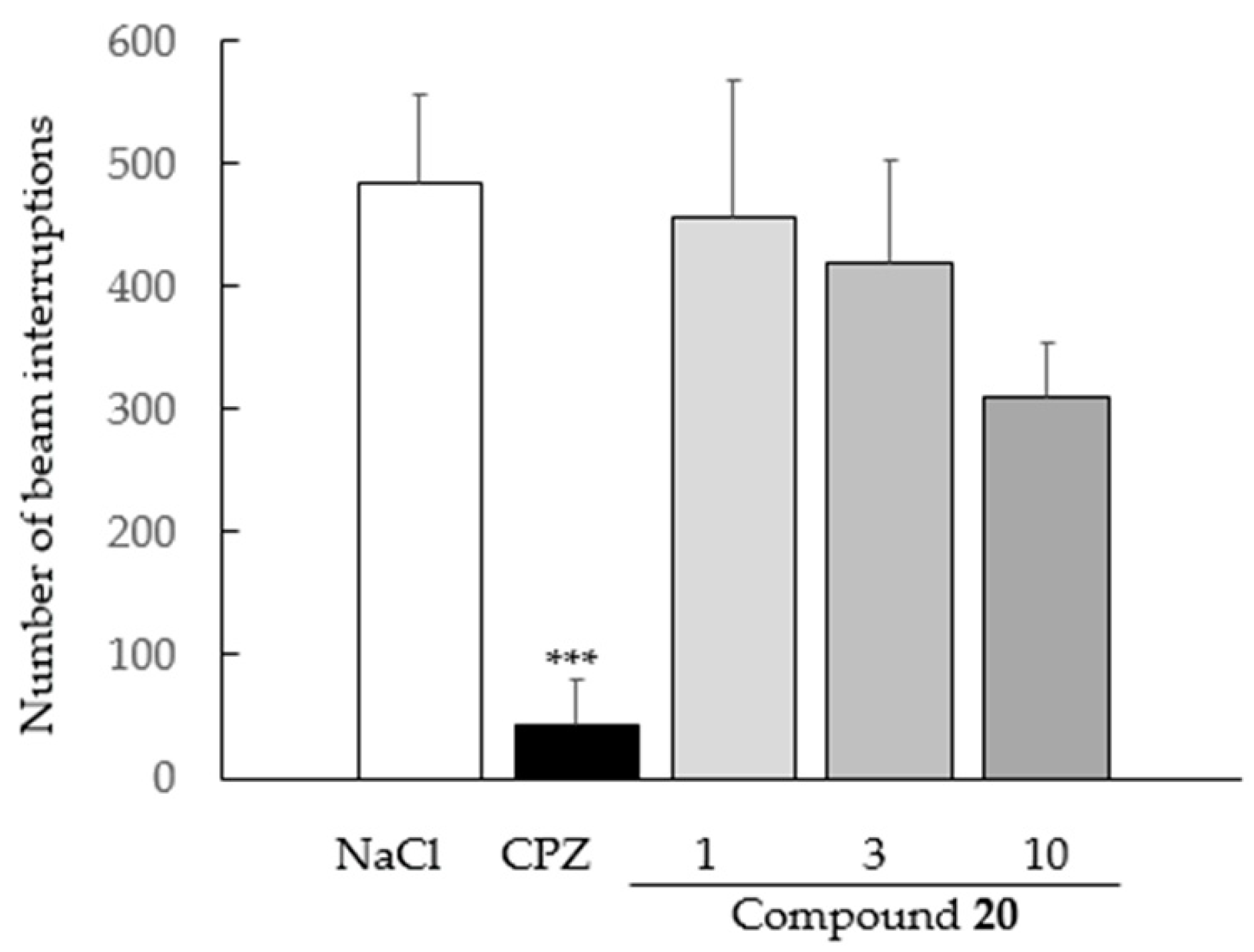
2.4.2. Spontaneous Locomotor Activity
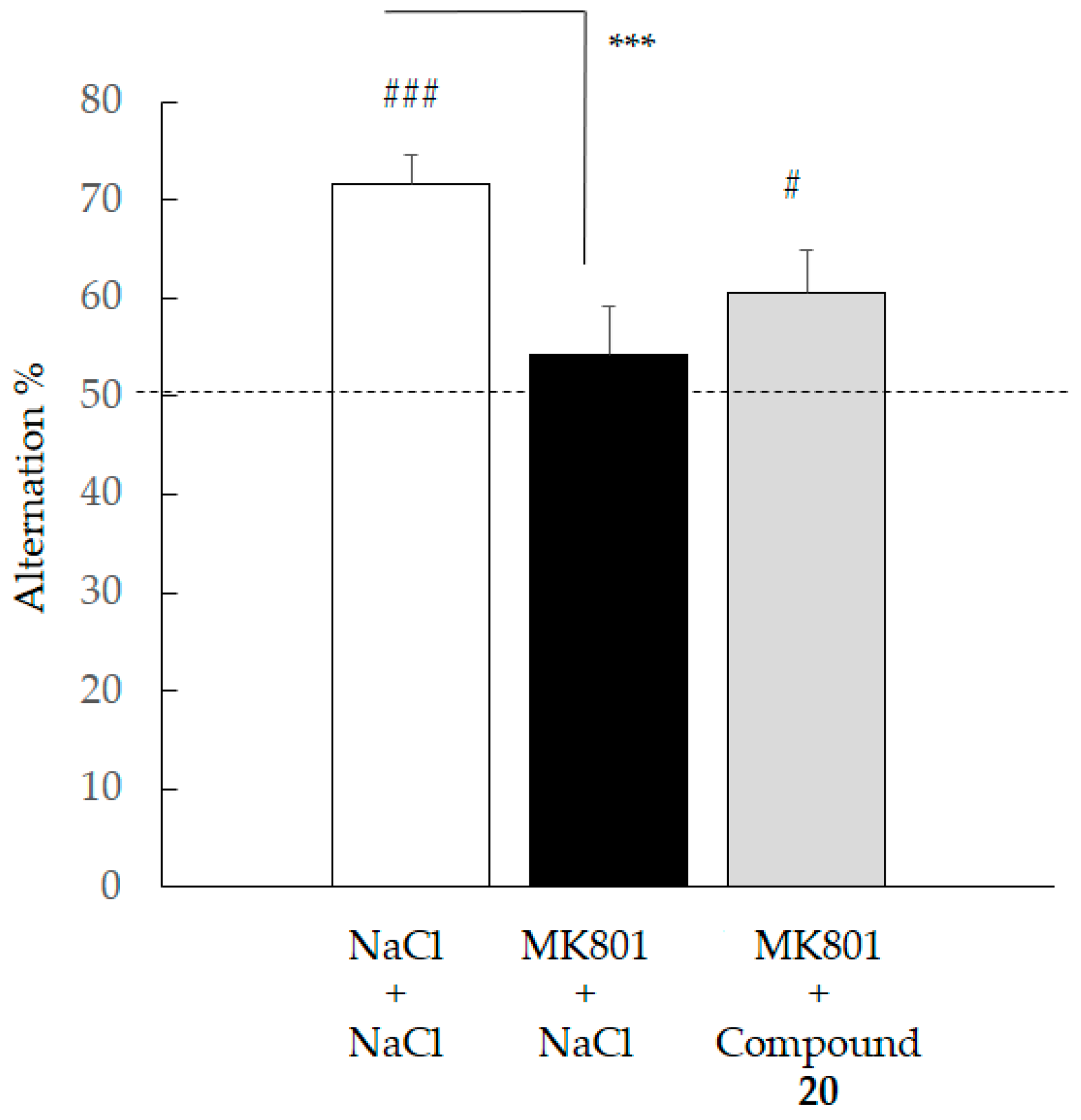
2.4.3. Spontaneous Alternation Deficit
3. Discussion
4. Materials and Methods
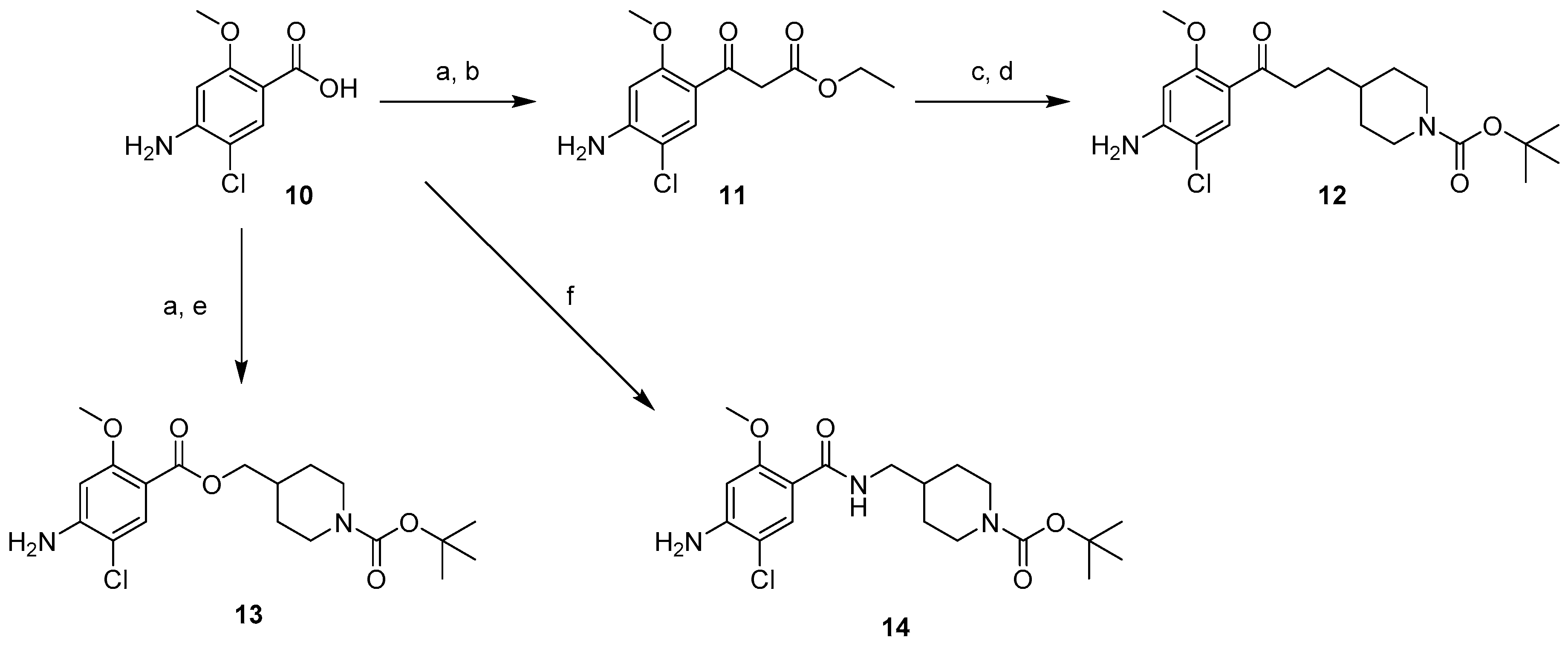
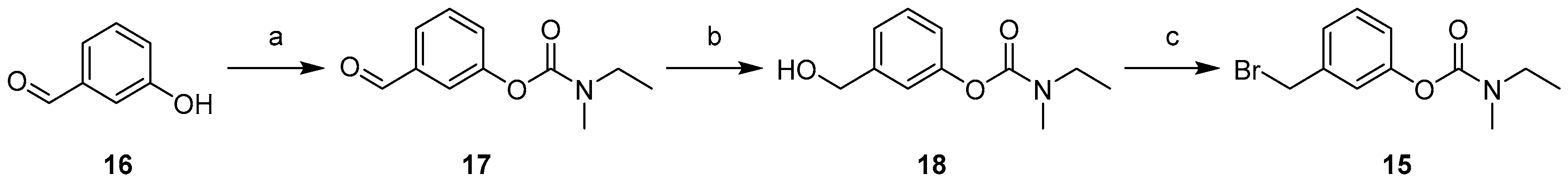
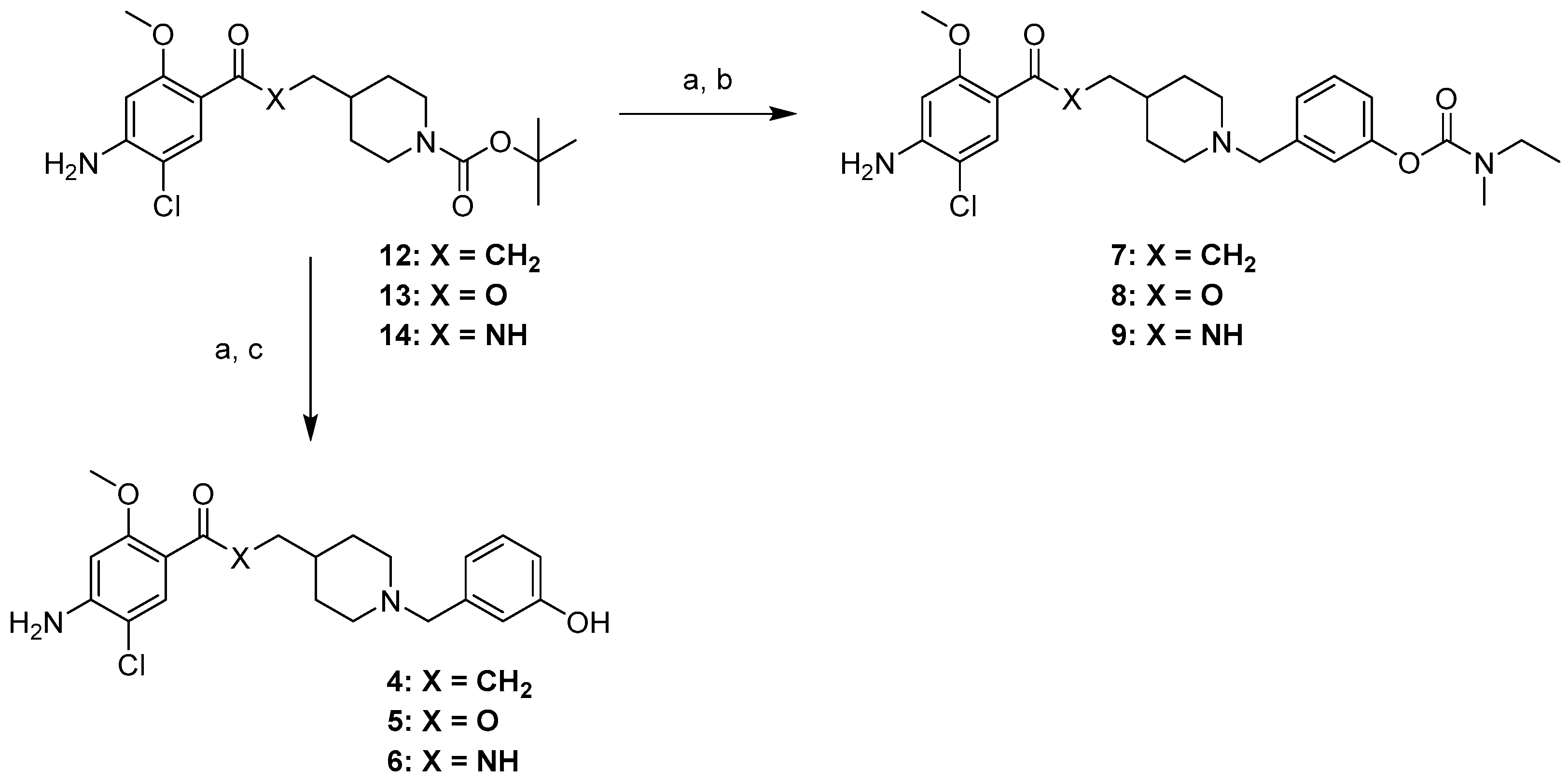
4.1. Chemistry
4.1.1. General Methods
4.1.2. Synthesis of Compounds (11–18)
4.1.3. General Procedure for the Synthesis of Compounds (4–6)
4.1.4. General Procedure for the Synthesis of Compounds (7–9)
4.1.5. General Procedure for the Preparation of Fumarate Salts (19–20)
4.2. In Silico Study
4.3. Biological Evaluation
4.3.1. In Vitro Tests of AChE Inhibitory Activity
4.3.2. Kinetic Study for AChE Inhibition
4.3.3. Pharmacological Characterization of Drugs on Human 5-HT4R
4.3.4. Determination of cAMP Production
4.3.5. Parallel Artificial Membrane Permeability Assay
4.3.6. AChE-Dependent Decarbamoylation
4.3.7. In Vivo Biological Studies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 8 July 2019).
- Wang, T.; Liu, X.; Guan, J.; Ge, S.; Wu, M.-B.; Lin, J.; Yang, L. Advancement of multi-target drug discoveries and promising applications in the field of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 169, 200–223. [Google Scholar] [CrossRef] [PubMed]
- Lecoutey, C.; Hedou, D.; Freret, T.; Giannoni, P.; Gaven, F.; Since, M.; Bouet, V.; Ballandonne, C.; Corvaisier, S.; Malzert Freon, A.; et al. Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc. Natl. Acad. Sci. USA 2014, 111, E3825–E3830. [Google Scholar] [CrossRef]
- Rochais, C.; Lecoutey, C.; Gaven, F.; Giannoni, P.; Hamidouche, K.; Hedou, D.; Dubost, E.; Genest, D.; Yahiaoui, S.; Freret, T.; et al. Novel multitarget-directed ligands (MTDLs) with acetylcholinesterase (AChE) inhibitory and serotonergic subtype 4 receptor (5-HT4R) agonist activities as potential agents against Alzheimer’s disease: The design of donecopride. J. Med. Chem. 2015, 58, 3172–3187. [Google Scholar] [CrossRef]
- Yogev-Falach, M.; Bar-Am, O.; Amit, T.; Weinreb, O.; Youdim, M.B.H. A multifunctional, neuroprotective drug, ladostigil (TV3326), regulates holo-APP translation and processing. Faseb J. 2006, 20, 2177–2179. [Google Scholar] [CrossRef]
- Shoham, S.; Bejar, C.; Kovalev, E.; Schorer-Apelbaum, D.; Weinstock, M. Ladostigil prevents gliosis, oxidative–nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology 2007, 52, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H. Reconsideration of anticholinesterase therapeutic strategies against Alzheimer’s disease. Acs Chem. Neurosci. 2019, 10, 852–862. [Google Scholar] [CrossRef]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Pérez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-target-directed ligands for Alzheimer’s disease: Discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef]
- Nagai, T.; Sakurai, S.; Natori, N.; Hataoka, M.; Kinoshita, T.; Inoue, H.; Hanaya, K.; Shoji, M.; Sugai, T. Synthesis of enantiomerically enriched drug precursors and an insect pheromone via reduction of ketones using commercially available carbonyl reductase screening kit “Chiralscreen® OH.”. Bioorg. Med. Chem. 2018, 26, 1304–1313. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. An expedient and highly selective iodination of alcohols using a KI/BF3.Et2O system. Tetrahedron Lett. 2001, 42, 951–953. [Google Scholar] [CrossRef]
- Bacalhau, P.; San Juan, A.A.; Goth, A.; Caldeira, A.T.; Martins, R.; Burke, A.J. Insights into (S)-rivastigmine inhibition of butyrylcholinesterase (BuChE): Molecular docking and saturation transfer difference NMR (STD-NMR). Bioorg. Chem. 2016, 67, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharm. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Grossman, C.J.; Kilpatrick, G.J.; Bunce, K.T. Development of a radioligand binding assay for 5-HT4 receptors in guinea-pig and rat brain. Br. J. Pharm. 1993, 109, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.-S.; Zhu, X.; Holloway, H.W.; Whittaker, N.F.; Brossi, A.; Greig, N.H. Anticholinesterase activity of compounds related to geneserine tautomers. N -oxides and 1,2-oxazines ‖. J. Med. Chem. 2002, 45, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Irving, H.R.; Tochon-Danguy, N.; Chinkwo, K.A.; Li, J.G.; Grabbe, C.; Shapiro, M.; Pouton, C.W.; Coupar, I.M. Investigations into the binding affinities of different human 5-HT4 receptor splice variants. Pharmacology 2010, 85, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-Y.; Kang, Y.-S. The inhibitory effect of rivastigmine and galantamine on choline transport in brain capillary endothelial cells. Biomol. 2010, 18, 65–70. [Google Scholar] [CrossRef]
- Alix, F.; Gembus, V.; Coquet, L.; Hubert-Roux, M.; Chan, P.; Truong, L.; Sebban, M.; Coadou, G.; Oulyadi, H.; Papamicaël, C.; et al. Dihydroquinoline carbamate DQS1-02 as a prodrug of a potent acetylcholinesterase inhibitor for Alzheimer’s disease therapy: Multigram-scale synthesis, mechanism investigations, in vitro safety pharmacology, and preliminary in vivo toxicology profile. Acs Omega 2018, 3, 18387–18397. [Google Scholar] [CrossRef]
- Deiana, S.; Harrington, C.R.; Wischik, C.M.; Riedel, G. Methylthioninium chloride reverses cognitive deficits induced by scopolamine: Comparison with rivastigmine. Psychopharmacology 2009, 202, 53–65. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, D.; Sheng, R.; Wu, H.; Hu, Y.; Wang, F.; Cai, T.; Yang, B.; He, Q. BZYX. A novel acetylcholinesterase inhibitor, significantly improved chemicals-induced learning and memory impairments on rodents and protected PC12 cells from apoptosis induced by hydrogen peroxide. Eur. J. Pharm. 2009, 613, 1–9. [Google Scholar] [CrossRef]
- Robinson, L.; Goonawardena, A.V.; Pertwee, R.; Hampson, R.E.; Platt, B.; Riedel, G. WIN55,212-2 induced deficits in spatial learning are mediated by cholinergic hypofunction. Behav. Brain Res. 2010, 208, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Lecoutey, C.; Rochais, C.; Genest, D.; Butt-Gueulle, S.; Ballandonne, C.; Corvaisier, S.; Dulin, F.; Lepailleur, A.; Sopkova-de Oliveira Santos, J.; Dallemagne, P. Synthesis of dual AChE/5-HT4 receptor multi-target directed ligands. Medchemcomm 2012, 3, 627. [Google Scholar] [CrossRef]
- Freret, T.; Bouet, V.; Quiedeville, A.; Nee, G.; Dallemagne, P.; Rochais, C.; Boulouard, M. Synergistic effect of acetylcholinesterase inhibition (donepezil) and 5-HT4 receptor activation (RS67333) on object recognition in mice. Behav. Brain Res. 2012, 230, 304–308. [Google Scholar] [CrossRef]
- Hooper, N.; Fraser, C.; Stone, T.W. Effects of purine analogues on spontaneous alternation in mice. Psychopharmacology 1996, 123, 250–257. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all the synthesized compounds are available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | X | R | (h)AChE IC50 (nM) or % Inhibition at 10−6 M | (h)5-HT4R Ki (nM) or % Inhibition at 10−8 M |
|---|---|---|---|---|
| Donepezil | - | - | 7.0 ± 1.5 | - |
| Rivastigmine | - | - | 4.150 ± 160 1 | - |
| RS67333 | - | - | - | 5.1 ± 0.5 2 |
| 4 | CH2 | OH | 148.6 ± 34.9 | 5.1 ± 1.1 |
| 7 | CH2 | OCON(Me)Et | 4151 ± 794 | 3% |
| 5 | O | OH | 11% | 0.6 ± 0.4 |
| 8 | O | OCON(Me)Et | 16,290 ± 240 | 23% |
| 6 | NH | OH | 4% | 5.8 ± 1.2 |
| 9 | NH | OCON(Me)Et | 10,077 ± 2,988 | 13% |
| 19 | CH2 | OH, fumaric salt | 72.0 ± 1.4 | 6.9 ± 1.2 |
| 20 | CH2 | OCON(Me)Et, fumaric salt | 6070 ± 404 | 16% |
| Compound | Log(EC50) 1 | % Control Agonist Response 2 | Profile |
|---|---|---|---|
| RS67333 | −8.8 ± 0.2 | 48.7 ± 5.2 | Partial agonist |
| 19 | −9.90 ± 0.0 | 59.0 ± 3.9 | Partial agonist |
| Compound | LogPe |
|---|---|
| Corticosterone | −4.84 ± 0.02 |
| Theophylline | −6.52 ± 0.05 |
| 20 | −4.39 ± 0.12 |
| Compound | Dose (mg/kg) | LD50 (mg/kg) | Symptoms (Subtoxic Doses) |
|---|---|---|---|
| 20 | 1–10–100 | >100 | 1–10, no symptoms |
| 100, tremors, hypoactivity full recovery after 24h | |||
| Amphetamine 1 | 2 | - | Hyperactivity, exophtalmy, irritability |
| Chlorpromazine 1 | 10 | - | Hypoactivity, ataxia, sleep |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toublet, F.-X.; Lecoutey, C.; Lalut, J.; Hatat, B.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Sopkova de Oliveira Santos, J.; Claeysen, S.; et al. Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease. Molecules 2019, 24, 2786. https://doi.org/10.3390/molecules24152786
Toublet F-X, Lecoutey C, Lalut J, Hatat B, Davis A, Since M, Corvaisier S, Freret T, Sopkova de Oliveira Santos J, Claeysen S, et al. Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease. Molecules. 2019; 24(15):2786. https://doi.org/10.3390/molecules24152786
Chicago/Turabian StyleToublet, François-Xavier, Cédric Lecoutey, Julien Lalut, Bérénice Hatat, Audrey Davis, Marc Since, Sophie Corvaisier, Thomas Freret, Jana Sopkova de Oliveira Santos, Sylvie Claeysen, and et al. 2019. "Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease" Molecules 24, no. 15: 2786. https://doi.org/10.3390/molecules24152786
APA StyleToublet, F.-X., Lecoutey, C., Lalut, J., Hatat, B., Davis, A., Since, M., Corvaisier, S., Freret, T., Sopkova de Oliveira Santos, J., Claeysen, S., Boulouard, M., Dallemagne, P., & Rochais, C. (2019). Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease. Molecules, 24(15), 2786. https://doi.org/10.3390/molecules24152786