
Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands


 , ,
, ,  ,
, 
 and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
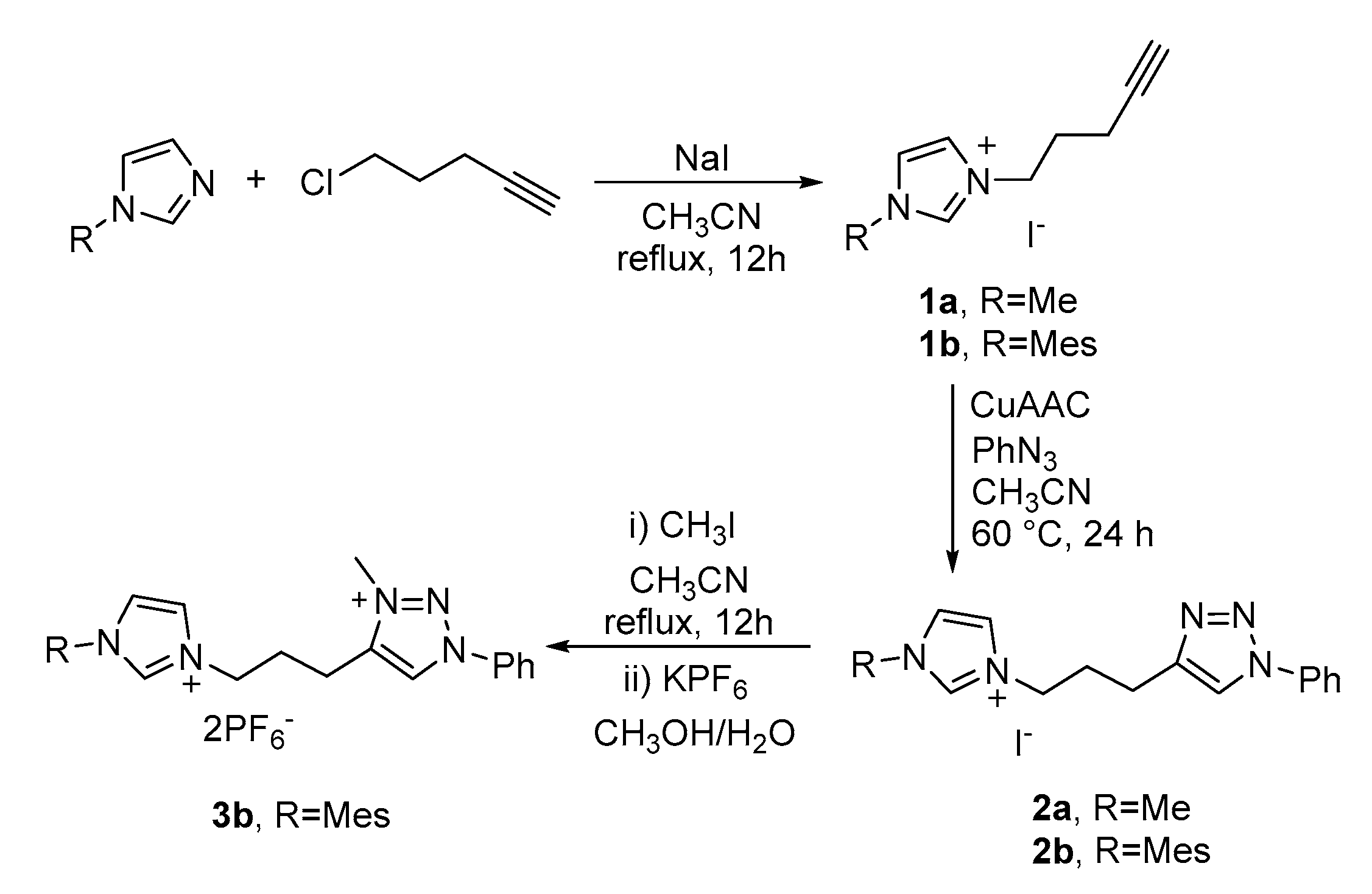
2.1. Synthesis of the Proligands
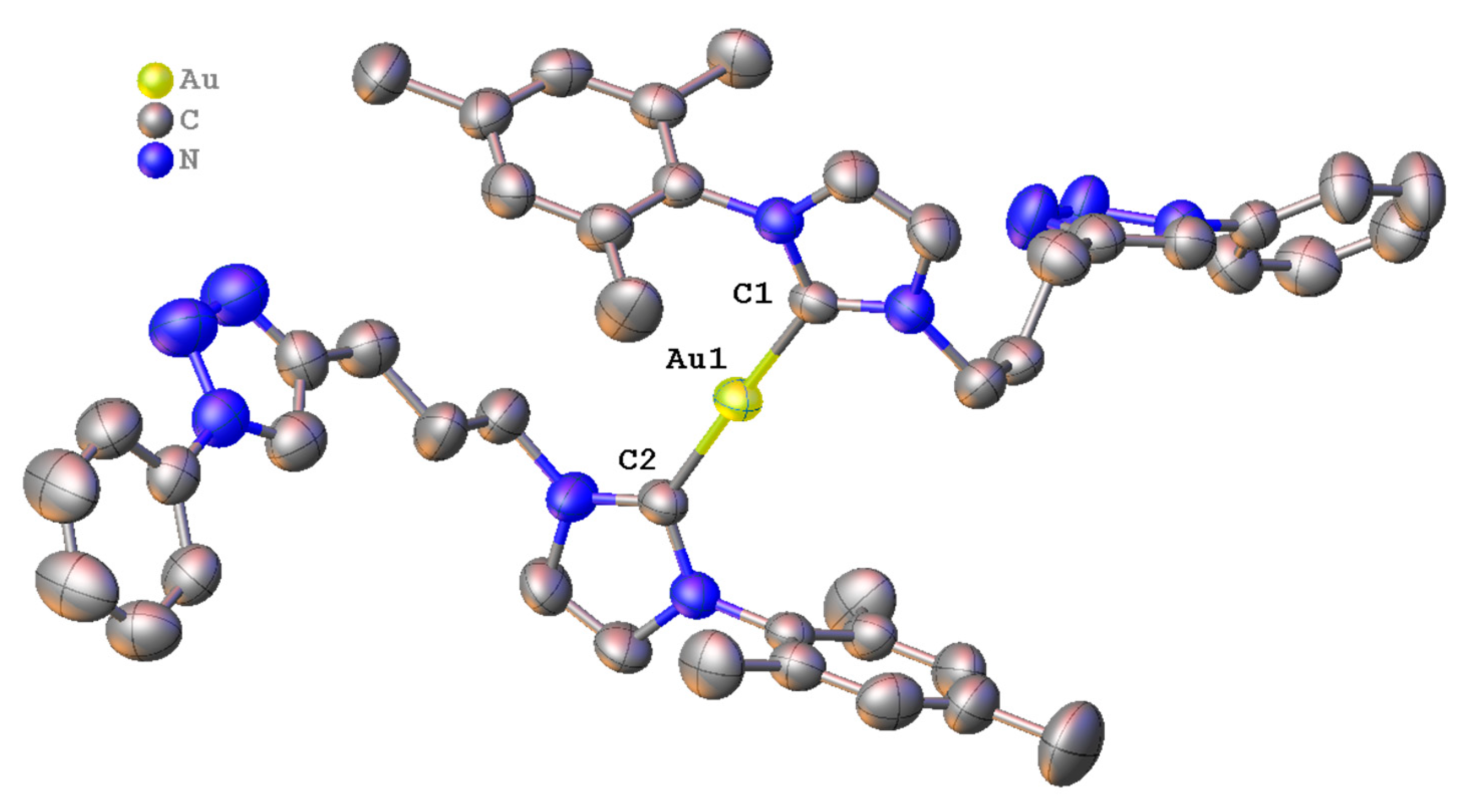
2.2. Synthesis and Properties of Mononuclear Gold(I) Complexes
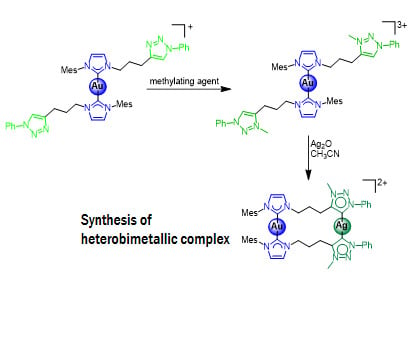
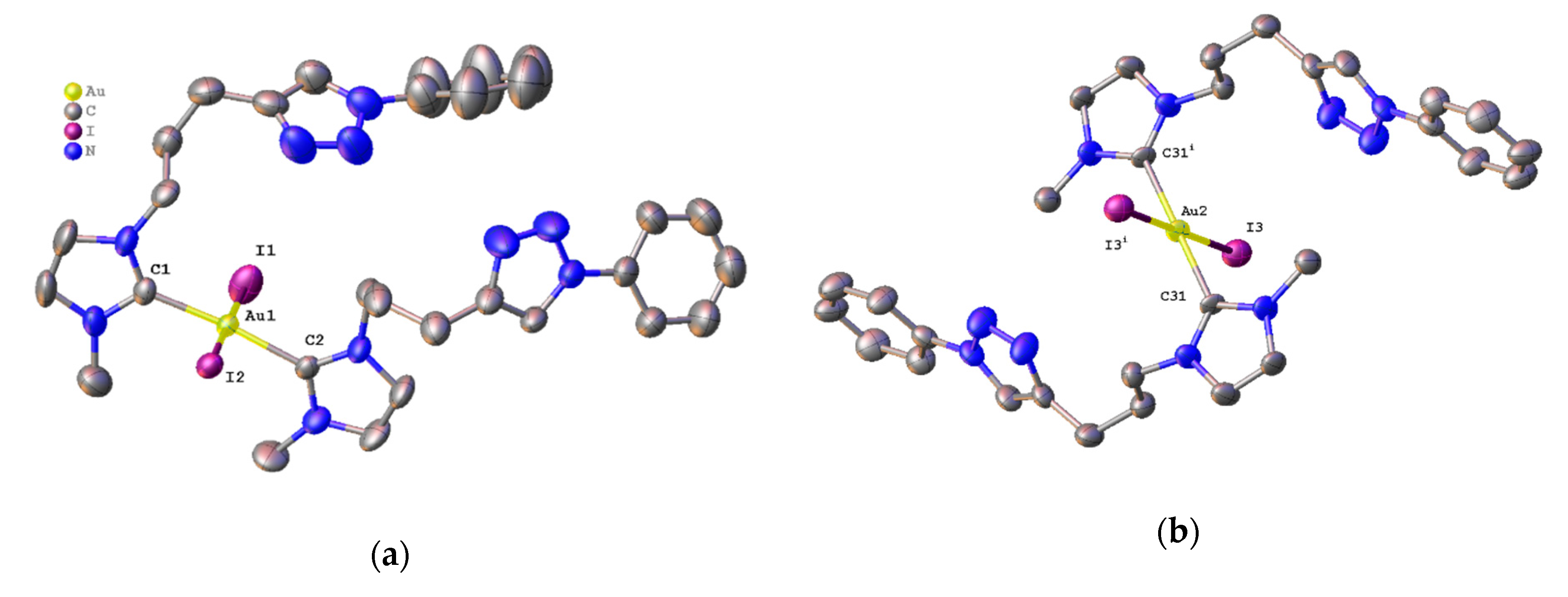
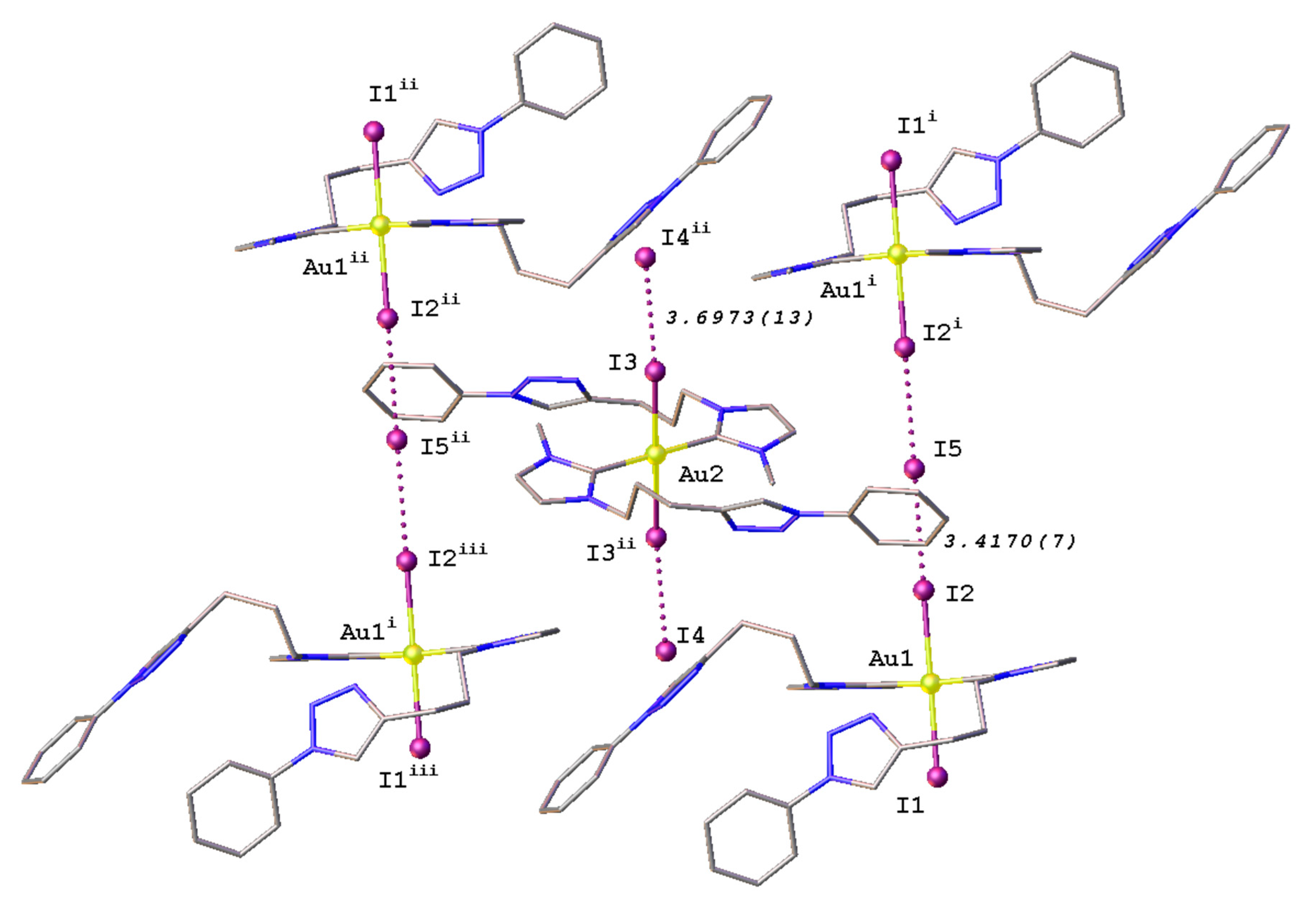
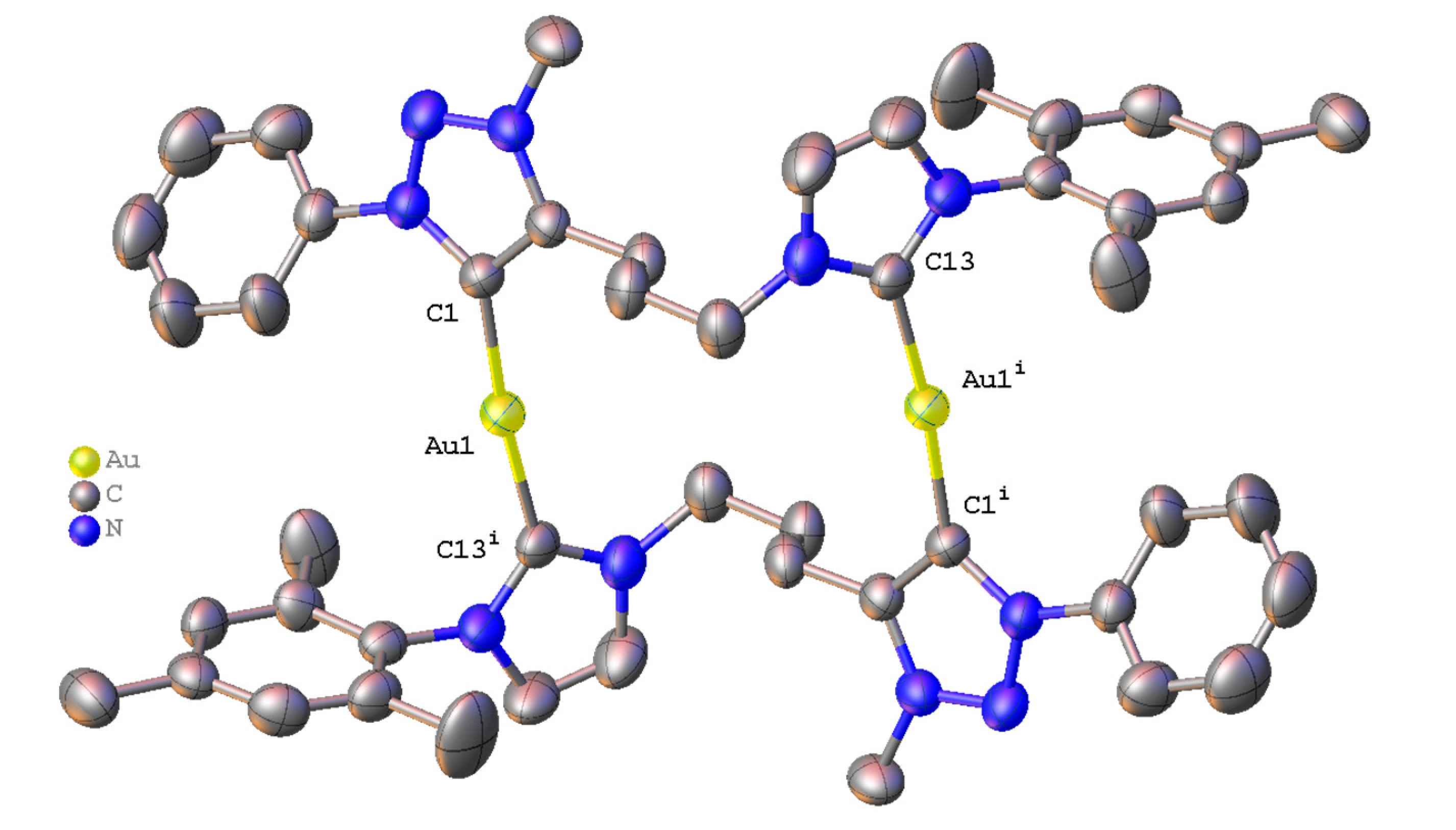
2.3. Synthesis and Properties of Dinuclear Complexes
2.4. Luminescence Properties
3. Materials and Methods
3.1. Synthesis of the Imidazolium Proligands
3.1.1. Synthesis of the Alkynyl Imidazolium Salts 1a and 1b
3.1.2. Synthesis of Proligands 2a and 2b
3.1.3. Synthesis of Proligand 3b
3.2. Synthesis of the Gold(I) Mononuclear Complexes
3.2.1. Synthesis of Complex 4
3.2.2. Synthesis of Complex 5 and Characterization of Complex 7
3.2.3. Synthesis of Complex 6
3.2.4. Synthesis of Complex 8 by Methylation of Complex 6
3.3. Synthesis of the Dinuclear Complexes with Bridging Dicarbene Ligands
3.3.1. Synthesis of Complex 9
3.3.2. Synthesis of Complex 10
3.3.3. Synthesis of Complexes 11/11′
3.4. Crystal Structure Determination
3.5. Luminescence Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Huynh, H.V. The Organometallic Chemistry of N-Heterocyclic Carbenes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Peris, E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P. N-Heterocyclic Carbenes, Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Diez-Gonzalez, S. N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, 2nd ed.; RSC Catalysis Series; RSC: Cambridge, UK, 2017. [Google Scholar]
- Biffis, A.; Tubaro, C.; Baron, M. Advances in Transition-Metal-Catalysed Alkyne Hydroarylations. Chem. Rec. 2016, 16, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Vivancos, A.; Segarra, C.; Albrecht, M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118, 9493–9586. [Google Scholar] [CrossRef]
- Visbal, R.; Gimeno, M.C. N-heterocyclic carbene metal complexes: Photoluminescence and applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef] [PubMed]
- Mercs, L.; Albrecht, M. Beyond catalysis: N-heterocyclic carbene complexes as components for medicinal, luminescent, and functional materials applications. Chem. Soc. Rev. 2010, 39, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- Oehninger, L.; Rubbiani, R.; Ott, I. N-Heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3284. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gust, R. Update on metal N-heterocyclic carbene complexes as potential anti-tumor metallodrugs. Coord. Chem. Rev. 2016, 329, 191–213. [Google Scholar] [CrossRef]
- Mora, M.; Gimeno, M.C.; Visbal, R. Recent advances in gold–NHC complexes with biological properties. Chem. Soc. Rev. 2019, 48, 447–462. [Google Scholar] [CrossRef]
- Biffis, A.; Baron, M.; Tubaro, C. Chapter five-Poly-NHC complexes of transition metals: recent applications and new trends. In Advances in Organometallic Chemistry; Pérez, P.J., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 203–288. [Google Scholar]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef]
- Baron, M.; Tubaro, C.; Biffis, A.; Basato, M.; Graiff, C.; Poater, A.; Cavallo, L.; Armaroli, N.; Accorsi, G. Blue-Emitting Dinuclear N-heterocyclic Dicarbene Gold(I) Complex Featuring a Nearly Unit Quantum Yield. Inorg. Chem. 2012, 51, 1778–1784. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, M.M.; Nesterov, V.N.; Omary, M.A. Remarkable Aurophilicity and Photoluminescence Thermochromism in a Homoleptic Cyclic Trinuclear Gold(I) Imidazolate Complex. Inorg. Chem. 2017, 56, 12086–12089. [Google Scholar] [CrossRef] [PubMed]
- Penney, A.A.; Sizov, V.V.; Grachova, E.V.; Krupenya, D.V.; Gurzhiy, V.V.; Starova, G.L.; Tunik, S.P. Aurophilicity in Action: Fine-Tuning the Gold(I)–Gold(I) Distance in the Excited State to Modulate the Emission in a Series of Dinuclear Homoleptic Gold(I)–NHC Complexes. Inorg. Chem. 2016, 55, 4720–4732. [Google Scholar] [CrossRef] [PubMed]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Böhmer, M.; Kampert, F.; Tan, T.T.Y.; Guisado-Barrios, G.; Peris, E.; Hahn, F.E. IrIII/AuI and RhIII/AuI Heterobimetallic Complexes as Catalysts for the Coupling of Nitrobenzene and Benzylic Alcohol. Organometallics 2018, 37, 4092–4099. [Google Scholar] [CrossRef]
- Böhmer, M.; Guisado-Barrios, G.; Kampert, F.; Roelfes, F.; Tan, T.T.Y.; Peris, E.; Hahn, F.E. Synthesis and Catalytic Applications of Heterobimetallic Carbene Complexes Obtained via Sequential Metalation of Two Bisazolium Salts. Organometallics 2019, 38, 2120–2131. [Google Scholar] [CrossRef]
- Schulte to Brinke, C.; Hahn, F.E. Trinuclear Heterobimetallic Complexes by Stepwise Metalation of a Macrocyclic Tetraimidazolium Salt. Eur. J. Inorg. Chem. 2015, 3227–3231. [Google Scholar] [CrossRef]
- Gonell, S.; Poyatos, M.; Peris, E. Pincer-CNC mononuclear, dinuclear and heterodinuclear Au(III) and Pt(II) complexes supported by mono- and poly-N-heterocyclic carbenes: Synthesis and photophysical properties. Dalton Trans. 2016, 45, 5549–5556. [Google Scholar] [CrossRef]
- Majumder, A.; Naskar, R.; Roy, P.; Maity, R. Homo- and Heterobimetallic Complexes Bearing NHC Ligands: Applications in α-Arylation of Amide, Suzuki–Miyaura Coupling Reactions, and Tandem Catalysis. Eur. J. Inorg. Chem. 2019, 1810–1815. [Google Scholar] [CrossRef]
- Teng, Q.; Huynh, H.V. (Hetero)bimetallic and Tetranuclear Complexes of Pincer-Bridged N-Heterocyclic Carbene Ligands. Organometallics 2018, 37, 4119–4127. [Google Scholar] [CrossRef]
- Zanardi, A.; Corberán, R.; Mata, J.A.; Peris, E. Homo- and Heterodinuclear Complexes with Triazolyl-diylidene. An Easy Approach to Tandem Catalysts. Organometallics 2008, 27, 3570–3576. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, L.; Lv, H.; Zhang, G.; Xia, C.; Hahn, F.E.; Li, F. Modular “Click” Preparation of Bifunctional Polymeric Heterometallic Catalysts. Angew. Chem. Int. Ed. 2016, 55, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Bente, S.; Kampert, F.; Tan, T.T.Y.; Hahn, F.E. Site-selective metallation of dicarbene precursors. Chem. Commun. 2018, 54, 12887–12890. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Baron, M.; Tubaro, C.; Bellemin-Laponnaz, S.; Graiff, C.; Bottaro, G.; Armelao, L.; Orian, L. Structural and Luminescent Properties of Homoleptic Silver(I), Gold(I), and Palladium(II) Complexes with nNHC-tzNHC Heteroditopic Carbene Ligands. ACS Omega 2019, 4, 4192–4205. [Google Scholar] [CrossRef]
- Zamora, M.T.; Ferguson, M.J.; McDonald, R.; Cowie, M. Unsymmetrical Dicarbenes Based on N -Heterocyclic/Mesoionic Carbene Frameworks: A Stepwise Metalation Strategy for the Generation of a Dicarbene-Bridged Mixed-Metal Pd/Rh Complex. Organometallics 2012, 31, 5463–5477. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef] [PubMed]
- Chardon, E.; Puleo, G.L.; Dahm, G.; Fournel, S.; Guichard, G.; Bellemin-Laponnaz, S. Easy Derivatisation of Group 10 N-Heterocyclic Carbene Complexes and In Vitro Evaluation of an Anticancer Oestradiol Conjugate. ChemPlusChem 2012, 77, 1028–1038. [Google Scholar] [CrossRef]
- Mendoza-Espinosa, D.; Alvarez-Hernández, A.; Angeles-Beltrán, D.; Negrón-Silva, G.E.; Suárez-Castillo, O.R.; Vásquez-Pérez, J.M. Bridged N -Heterocyclic/Mesoionic (NHC/MIC) Heterodicarbenes as Ligands for Transition Metal Complexes. Inorg. Chem. 2017, 56, 2092–2099. [Google Scholar] [CrossRef]
- Gaillard, S.; Nun, P.; Slawin, A.M.Z.; Nolan, S.P. Expeditious Synthesis of [Au(NHC)(L)]+ (NHC = N-Heterocyclic Carbene; L = Phosphine or NHC) Complexes. Organometallics 2010, 29, 5402–5408. [Google Scholar] [CrossRef]
- Visbal, R.; Laguna, A.; Gimeno, M.C. Simple and efficient synthesis of [MCI(NHC)] (M = Au, Ag) complexes. Chem. Commun. 2013, 49, 5642–5644. [Google Scholar] [CrossRef]
- Baron, M.; Tubaro, C.; Basato, M.; Isse, A.A.; Gennaro, A.; Cavallo, L.; Graiff, C.; Dolmella, A.; Falivene, L.; Caporaso, L. Insights into the Halogen Oxidative Addition Reaction to Dinuclear Gold(I) Di(NHC) Complexes. Chem. Eur. J. 2016, 22, 10211–10224. [Google Scholar] [CrossRef] [PubMed]
- Pell, T.P.; Wilson, D.J.D.; Skelton, B.W.; Dutton, J.L.; Barnard, P.J. Heterobimetallic N -Heterocyclic Carbene Complexes: A Synthetic, Spectroscopic, and Theoretical Study. Inorg. Chem. 2016, 55, 6882–6891. [Google Scholar] [CrossRef] [PubMed]
- Tubaro, C.; Baron, M.; Costante, M.; Basato, M.; Biffis, A.; Gennaro, A.; Isse, A.A.; Graiff, C.; Accorsi, G. Dinuclear gold(I) complexes with propylene bridged N-heterocyclic dicarbene ligands: Synthesis, structures, and trends in reactivities and properties. Dalton Trans. 2013, 42, 10952–10963. [Google Scholar] [CrossRef] [PubMed]
- Keske, E.C.; Zenkina, O.V.; Wang, R.; Crudden, C.M. Synthesis and Structure of Silver and Rhodium 1,2,3-Triazol-5-ylidene Mesoionic Carbene Complexes. Organometallics 2012, 31, 456–461. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, Y.-C.; Jin, R.; Chen, B.-L.; Chen, Z. Synthesis of Axially Chiral 1,2,3-Triazol-5-ylidene–Au(I) Complex and Its Application in Enantioselective [2 + 2] Cycloaddition of Alleneamides with Alkenes. Organometallics 2018, 37, 3196–3209. [Google Scholar] [CrossRef]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Crystalline 1H-1,2,3-Triazol-5-ylidenes: New Stable Mesoionic Carbenes (MICs). Angew. Chem. Int. Ed. 2010, 49, 4759–4762. [Google Scholar] [CrossRef]
- Huynh, H.V.; Han, Y.; Jothibasu, R.; Yang, J.A. 13C-NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium(II). Organometallics 2009, 28, 5395–5404. [Google Scholar] [CrossRef]
- Huynh, H.V. Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination. Chem. Rev. 2018, 118, 9457–9492. [Google Scholar] [CrossRef]
- Hemmert, C.; Poteau, R.; dit Dominique, F.J.-B.; Ceroni, P.; Bergamini, G.; Gornitzka, H. Amide-Functionalized Bis(NHC) Systems: Anion Effect on Gold–Gold Interactions. Eur. J. Inorg. Chem. 2012, 2012, 3892–3898. [Google Scholar] [CrossRef]
- Cure, J.; Poteau, R.; Gerber, I.C.; Gornitzka, H.; Hemmert, C. Dimeric Gold Bis(carbene) Complexes by Transmetalation in Water. Organometallics 2012, 31, 619–626. [Google Scholar] [CrossRef]
- Strassner, T.; Ahrens, S. Salts comprising aryl-alkyl-substituted imidazolium and triazolium cations and the use thereof. Patent WO2009095012A1, 6 August 2009. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.a.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Box, H.K.; Howell, T.O.; Kennon, W.E.; Burk, G.A.; Valle, H.U.; Hollis, T.K. An efficient route to unsymmetrical bis(azolium) salts: CCC-NHC pincer ligand complex precursors. Tetrahedron 2017, 73, 2191–2195. [Google Scholar] [CrossRef]
- Aznarez, F.; Sanz Miguel, P.J.; Tan, T.T.Y.; Hahn, F.E. Preparation of Rhodium(III) Di-NHC Chelate Complexes Featuring Two Different NHC Donors via a Mild NaOAc-Assisted C–H Activation. Organometallics 2016, 35, 410–419. [Google Scholar] [CrossRef]
- Monticelli, M.; Bellemin-Laponnaz, S.; Tubaro, C.; Rancan, M. Synthesis, Structure and Antitumoural Activity of Triazole-Functionalised NHC–Metal Complexes. Eur. J. Inorg. Chem. 2017, 2017, 2488–2495. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longhi, A.; Baron, M.; Rancan, M.; Bottaro, G.; Armelao, L.; Sgarbossa, P.; Tubaro, C. Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands. Molecules 2019, 24, 2305. https://doi.org/10.3390/molecules24122305
Longhi A, Baron M, Rancan M, Bottaro G, Armelao L, Sgarbossa P, Tubaro C. Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands. Molecules. 2019; 24(12):2305. https://doi.org/10.3390/molecules24122305
Chicago/Turabian StyleLonghi, Andrea, Marco Baron, Marzio Rancan, Gregorio Bottaro, Lidia Armelao, Paolo Sgarbossa, and Cristina Tubaro. 2019. "Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands" Molecules 24, no. 12: 2305. https://doi.org/10.3390/molecules24122305
APA StyleLonghi, A., Baron, M., Rancan, M., Bottaro, G., Armelao, L., Sgarbossa, P., & Tubaro, C. (2019). Possible Synthetic Approaches for Heterobimetallic Complexes by Using nNHC/tzNHC Heteroditopic Carbene Ligands. Molecules, 24(12), 2305. https://doi.org/10.3390/molecules24122305