Abstract
Pasireotide is a multi-receptor ligand somatostatin analogue approved for medical treatment of Cushing’s disease and acromegaly. The liquid-phase total synthesis of pasireotide-a 18-membered cyclic hexapeptide-was achieved by the 3 + 2 + 1 strategy, and the Pro1-Phe6 peptide bond was selected as the final cyclization position. Two key fragments were simply synthesized using N,O-bis(trimethylsilyl)acetamide/N-hydroxysuccinimide ester (BSA/NHS) as coupling agents, and processes of the two key fragments were simple without any chromatographic purification. The current synthesis method is easily scalable and produces the target peptide with an overall yield of 15%.
1. Introduction
Pasireotide (SOM230, Signifor) is a multi-receptor ligand somatostatin analogue (SSA) developed as the successor of the first-generation SSAs. Clinically, pasireotide is recommended for the treatment of patients with Cushing’s disease in whom surgery was unsuccessful, and patients with acromegaly who either remain uncontrolled after surgical therapy or in whom tumor resection is not possible [1,2,3,4]. Structurally, it belongs to cyclic hexapeptide featuring six amino acid moieties, including one proline (Pro), one phenylglycine (Phg), one tryptophane (Trp), one lysine (Lys), one tyrosine (Tyr), and one phenylalanine (Phe) (Figure 1). Due to its clinical attractiveness and structural unique, the synthesis of pasireotide has attracted increasing research interest of organic chemists from both academic and industrial laboratories.
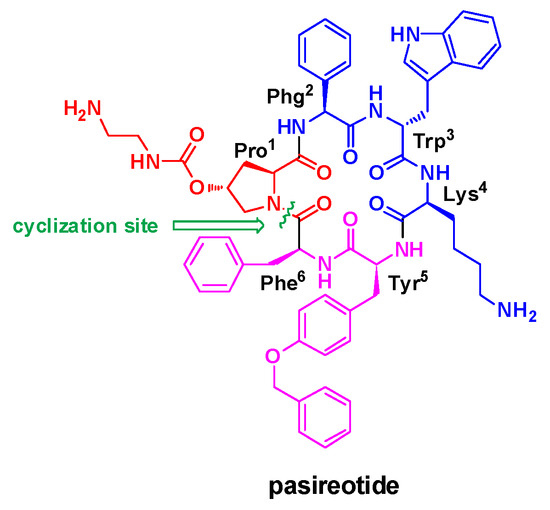
Figure 1.
Structure of pasireotide.
A variety of synthetic process for pasireotide has been developed in recent years [5,6,7,8,9,10,11,12]. In most of these reports, the synthesis routine was performed by solid-phase chemistry, using a Fmoc/tBu protecting scheme and resin as solid support. In solid-phase chemistry, significant shortcomings such as lack of scalability, inadequate in-process controls and low purity of the final products still hamper the development of scale-up synthesis [13]. Alternative approaches consisting of solution-phase reactions have also been developed to ensure well-established isolation, characterization, and purification protocols of the intermediate products [14]. In fact, most peptide-based pharmaceuticals are still synthesized using solution-phase chemistry, preferably employed for small- to medium-sized peptides [15], and pharmaceuticals containing as few as three amino acids (e.g., thyrotropin releasing hormone) and up to 32 residues (e.g., calcitonin) have been synthesized in solution-phase for clinical use [16].
In liquid-phase method, the reported total synthesis of pasireotide was based on a linear strategy in which each amino acid was connected in turn [6]; however, the linear strategy increased the risk of low yield and time consumption. In this regard, the synthesis of pasireotide was conducted by a 3 + 2 + 1 condensation strategy in solution and was expected to increase the yield; and was easily handled in our current study. Based on the structure of pasireotide, the synthesis process comprises a cyclisation step of a corresponding linear peptide in protected form, and there are six cyclisation possibilities in it. According to our experience with peptide synthesis, the cyclisation between amino acids Pro1 and Phe6 is particularly preferred in view of the reduced steric hindrance of these two amino acid moieties relative to other residues in the molecule [17]. In this regard, the corresponding linear peptide was obtained by the 3 + 2 + 1 strategy and disconnected into three fragments, the tripeptide fragment B, dipeptide fragment C, and amino acid fragment D, based on the retrosynthetic analysis as outlined in Scheme 1. Herein, we reported detailed synthesis of pasireotide, the synthesis of building blocks, as well as the cleavage pattern of this product. Additionally, a simplified and easily-handled method using N,O-bis(trimethylsilyl)acetamide/N-hydroxysuccinimide ester (BSA/NHS) as coupling agents for peptide synthesis was carried out in the current study [18]. Additionally, the coupling reaction using BSA/NHS occurred without additional acid/base required, and all byproducts, as well as excessive reactants, were water soluble or hydrolysable and easily eliminated through water-washing at the purification stage.
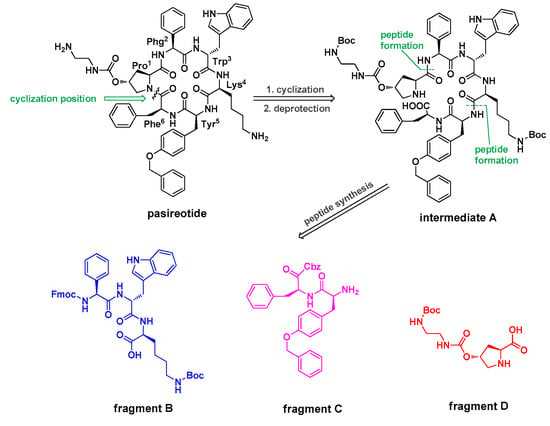
Scheme 1.
Structure of pasireotide A and our retrosynthetic analysis.
2. Results and Discussion
As outlined in the synthetic strategy (Scheme 1), intermediate A could be transformed into pasireotide via cyclisation and deprotection of the amino groups. Additionally, protected key fragments B, C, and D could be prepared effectively from commercially-available protected amino acid moieties. The most important step was the construction of the peptide bond under mild conditions with BSA/NHS as the coupling reagent and no additional acid/base required.
The fragment B was synthesized in five steps using intermediates 1 and 3 (Scheme 2). Firstly, the commercially-available Ne-Boc-l-lysine was coupled with N-Cbz-Trp NHS ester 1 in the presence of BSA to give compound 2 in 82% yield. Subsequently, compound 2 was treated with hydrogen (H2) in the presence of Pd/C, which was deprotected at the amino group to obtain amine intermediate in 94% yield. Then, the amine intermediate was treated with Fmoc-Phg NHS ester 3 in the presence of coupling reagent BSA to give fragment B in 84% yield. The high performance liquid chromatography (HPLC) analysis of fragment B is shown in Figure 2.
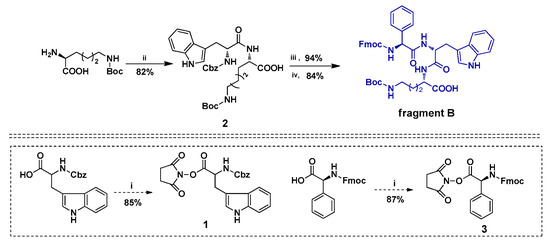
Scheme 2.
Synthesis of fragment B. Reaction condition and reagents: (i) N-hydroxysuccinimide(1.0 equiv.), N,N′-Dicyclohexylcarbodiimide (DCC)(1.1 equiv.), Tetrahydrofuran (THF); (ii) 1(0.83 equiv.), BSA(1.75 equiv.), Dichloromethane (CH2Cl2); (iii) Palladium over activated charcoal (Pd/C10%) (10%, w/w), H2, Methanol (MeOH); (iv) 3(0.83 equiv.), BSA(1.75 equiv.), THF.
Figure 2.
HPLC spectra of fragment B.
In the synthesis of fragment B, the use of BSA in combination with NHS ester was found to be effective for the coupling reaction (Scheme 2, steps i and iii), in which no additional acid/base was required, and all byproducts and excessive reactants were water soluble or hydrolysable and easily eliminated through water washing at the purification stage. Taking the synthesis of compound 2 for example, the coupling conditions and purification process were optimized, as shown in Table 1 (entries 1–4). When no BSA was added, the coupling product 2 was hardly detected (entry 1), and the coupling efficiency and yield were optimal when 1.2 equiv. of Ne-Boc-l-lysine reacted with 2.1 equiv. of BSA first in CH2Cl2 at room temperature, followed by the addition of 1.0 equiv. N-Cbz-Trp NHS ester 2 (entry 2). Significantly, the ratio of BSA was so important that either insufficient or excessive BSA would reduce the coupling yield (entries 3 and 4). Additionally, good yields of fragment B can be obtained in the ratio 2.1/1.2/1.0 of BSA/AA/NHS ester in THF or CH2Cl2 solvent (entries 5 and 8), respectively.

Table 1.
Synthesis of protected peptides via BSA/NHS method.
The fragment C was synthesized in three steps (Scheme 3). Firstly, N-Boc-Tyr NHS ester 4 was treated with commercially-available phenylalanine methyl ester in the presence of triethylamine (TEA) to obtain compound 5 in 94% yield. Then, compound 5 was treated with trifluoroacetic acid (TFA) to deprotect the amino group, giving fragment C in 98% yield. The HPLC analysis of fragment C is shown in Figure 3.
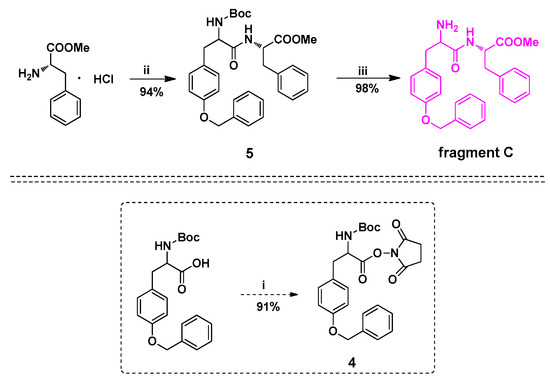
Scheme 3.
Synthesis of fragment C. Reaction condition and reagents: (i) N-hydroxysuccinimide(1.0 equiv.), DCC(1.1 equiv.), THF; (ii) 4(1.0 equiv.), TEA(1.0 equiv.), THF; (iii) TFA(10.0 equiv.), CH2Cl2.
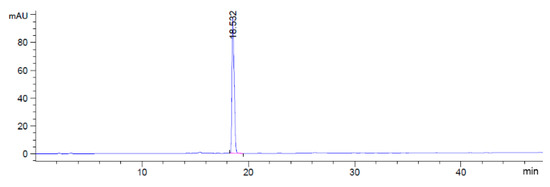
Figure 3.
HPLC spectra of fragment C.
The synthesis of fragment D was accomplished in three steps as shown in Scheme 4. The commercially-available l-hydroxyproline methyl ester was protected by Fmoc N-hydroxysuccinimide ester (Fmoc-OSu) in the presence of Na2CO3, providing compound 6 in 94% yield. Condensing of 6 with triphosgene gas followed by N-boc-ethylenediamine procedure, produced compound 7 in 69% yield. Removal of methyl ester protecting group of 7 by treating with sodium hydroxide (NaOH) gave fragment D in 88% yield. The HPLC analysis of fragment D is shown in Figure 4.
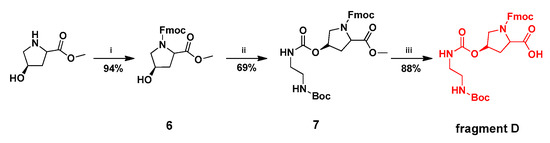
Scheme 4.
Synthesis of fragment D. Reaction condition and reagents: (i) Fmoc-OSu(1.0 equiv.), 1N Na2CO3 (1.0 equiv.), Dioxane/H2O; (ii) triphosgene(0.6 equiv.), N-boc-ethylenediamine(5.0 equiv.), THF; (iii) 1M NaOH(aq) (2.0 equiv.), Fmoc-OSu(1.0 equiv.), Dioxane.
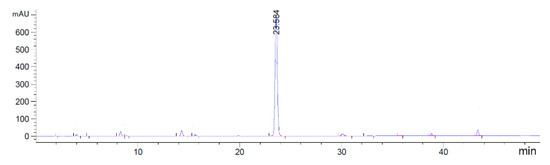
Figure 4.
HPLC spectra of fragment D.
With the two key fragments B and C in hand, firstly we were ready to assemble intermediate 8 by choosing the formation of the Lys4-Tyr5 amide bond in the presence of TBTU/4-methylmorpholine (Scheme 5, step i). Subsequently, removal of the Fmoc protecting group in compound 8, by treating with piperidine, was followed by condensing with fragment D in the presence of TBTU/4-methylmorpholine, yielding compound 9 in 85% yield (Scheme 5, steps ii and iii).
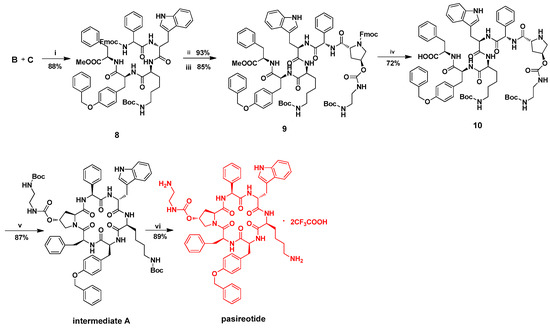
Scheme 5.
Synthesis of pasireotide. Reaction condition and reagents: (i) O-(1H-Benzo-1,2,3-Triazol-1-yl)-N,N,N′,N′-Tetramethyl-Uronium Tetrafluoroborate (TBTU) (1.5 equiv.), 4-methylmorpholine(2.0 equiv.), THF; (ii) piperidine(50.0 equiv.), THF; (iii) fragment D(1.0 equiv.), TBTU(1.5 equiv.), 4-methylmorpholine(2.0 equiv.), THF; (iv) piperidine(50.0 equiv.), THF; Lithium bromide (LiBr) (5.0 equiv.), NaOH(5.0 equiv.); (v) O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-Tetramethyluronium Hexafluorophosphate (HATU) (2.5 equiv.), 1-Hydroxybenzotriazole (HOBt) (2.5 equiv.) anhydrous, N,N-Dimethylformamide (DMF); (vi) TFA(200 equiv.).
Then, hydrolysis of the methyl ester protecting group of compound 9 afforded the carboxylic acid of compound 10 (Scheme 5, step iv). It is well-known that hydrolysis of ester is under alkaline conditions which has a significant impact on the conformation of peptide [19,20,21,22,23,24]. However, the macrocyclization step is critical in the synthesis of cyclic peptide, and may depend on the peptide sequence, structural constraints, and the resulting ring [25,26,27]. In the synthesis of intermediate A, a series of optimization experiments were carried out to achieve an acceptable yield (Table S1, Supporting Information).
After attempting various alkaline conditions, we found that the best result could be achieved by using LiBr/NaOH conditions (Table 2, entry 3), leading to compound 10 in 72% yield with no epimerization product according to the HPLC analysis (Figure S31, Supporting Information). In contrast, the hydrolysis of compound 9 under NaOH solutions (Table 2, entry 1) resulted in epimerization of compound 10 according to the HPLC analysis (Figure S32, Supporting Information).
Table 2.
Hydrolysis of compound 9.
After the liner pentapeptide 10 was obtained, the macrocyclization was accomplished by using HATU/ anhydrous HOBt in DMF to generate the key intermediate A in 87% yield (Scheme 5, step v). Finally, treatment of precursor A with TFA furnished the expected target pasireotide A in 89% yield (Scheme 5, step vi). Finally, the purification was carried out on recycling preparative reversed-phase HPLC (RP-HPLC) using a Jaigel Polyamine column and 1:1 ratio of water:acetonitrile as the solvent system (Figure S33, Supporting Information). The MS spectrum of the major peak in HPLC showed the molecular ion peak and doubly protonated peak of pasireotide A at m/z 1047.50 and 524.25, respectively.
The high resolution mass spectrometry (HRMS) of pasireotide displayed a protonated molecular ion peak [M + H]+ at m/z 1047.5094, corresponding to the molecular formula C58H67N10O9 (calculated 1047.5092), as well as doubly protonated peak [M + 2H]+ at m/z 524.2568 (calculated 524.2585) (Figure 5a). Under collision-induced decomposition (CID) experiment [28] (MS/MS), the protonated molecular ion peak at m/z 1047.5094 produced ions at m/z 1029.4932[M + H − H2O]+ and at m/z 1019.5117 [M + H − CO]+ (Figure 5b). It is reported that an amide bond with a higher proton affinity may be more readily opened due to the basicities of the amide nitrogen [29]. What is more, this is the case with proline-containing cyclic peptides when collision-activated under energy conditions; they afforded selectivity by undergoing selective ring cleavage at the proline residue [30]. Hence, the linear peptide [Pro1-Phg2-Trp3-Lys4-Tyr5-Phe6]H+ was produced by the cleavage of Pro1-Phe6 amide bond (Scheme 6) and subsequently produced a main series of adjacent peaks at m/z 882.4278, 647.3276, 519.2360, and 333.1555 (Figure 2b) corresponding to the successive loss of Phe6, Tyr5, Lys4, and Trp3, respectively (Scheme 6).
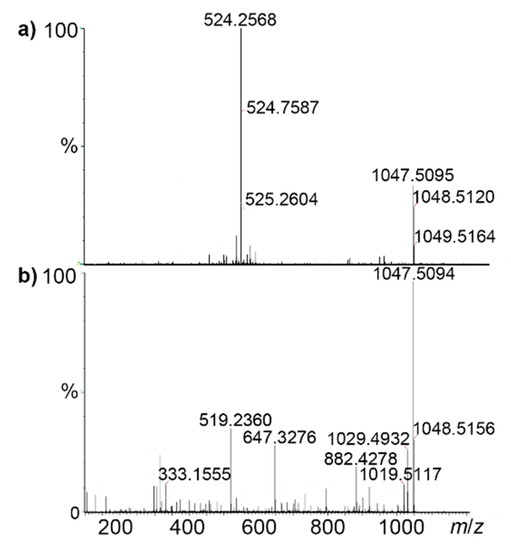
Figure 5.
(a) Mass Spectrometry (MS) and (b) MS/MS spectra of protonated pasireotide.
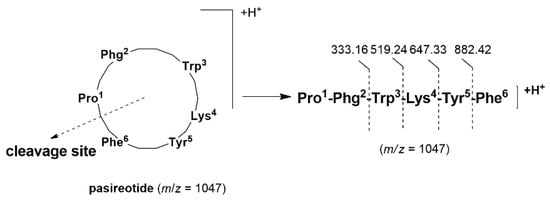
Scheme 6.
Cleavage site of protonated pasireotide by MS/MS spectrum.
The structure of pasireotide was further confirmed through nuclear magnetic resonance spectroscopy (NMR) (see Experimental Section), and it was found to be spectroscopically identical with that of the standard product.
3. Conclusion
In summary, we successfully applied a liquid-phase synthetic route to access pasireotide, a cyclic hexapeptide used to treat Cushing’s disease, through decentralized 3 + 2 + 1 strategy, and the overall yield of pasireotide was about 15% in our study, which is comparable to that reported 20% in solid-phase chemistry [5]. Our synthesis formed the Pro1-Phe6 peptide bond as the final macrocyclization site, and three key fragments can be easily obtained without any chromatographic purification. The spectroscopic data of all of the synthetic products were in good agreement with those reported for the product.
4. Experimental Section
General Experimental Procedures. All reactions were performed under a nitrogen atmosphere using anhydrous techniques unless otherwise noted. 1H and 13C-NMR on a Varian Mercury 500 spectrometer were recorded in DMSO-d6 or CDCl3. Chemical shifts were reported in δ(ppm) units relative to the internal standard tetramethylsilane (TMS). All the reactions were monitored by thin layer chromatography (TLC) analysis on pre-coated silica gel G plates at 254 nm under UV lamp or HPLC analysis.
Chromatographic Methods. Analysis for fragments B, C, and D was achieved on the Agilent model 1260-DAD high-performance liquid chromatographsystem along with the detective wavelength of 220, 225, and 280 nm. The mobile phase consisted of acetonitrile (A) and deionized water (B), both with 0.1% TFA (trifluoroacetic acid) (vol./vol.), using gradient elution. The flow rate was 1.0 mL·min−1, and the column temperature was maintained at 25 °C. The concrete gradient elution conditions are displayed in Table S2. Analysis for compound 10 was achieved on the Waters UPLC, using gradient elution. The preparative column chromatography for crude pasireotide was performed on SHIMADZU Shim-pack PRC-ODS (H) (20 × 250 mm), using gradient elution.
Materials. Protected amino acids, PyBop, and anhydrous HOBt were purchased from Aldrich. All other reagents and chemicals were purchased from Beijing Chemistry Works (Beijing, Chnia), and were used without further purification. Anhydrous organic solvents were obtained either from Aldrich or distilled from the drying agents: Na or CaH2. All reactions were carried out under a N2 atmosphere employing oven- or flame-dried glassware. All solvents were either distilled or obtained from passing through activated alumina. Acetonitrile and trifluoroacetic acid (HPLC grade) were purchased from Sigma-Aldrich (Milwaukee, Germany). Ultrapure water was purchased from Hangzhou Wahaha Co., Ltd. (Hangzhou, China)
Synthesis of Compound 1. To a stirred solution of N-Cbz-d-Trp (10.0 g, 29.6 mmol) in THF (100 mL) was added N-hydroxysuccinimide (3.4 g, 29.6 mmol) at 0 °C. The resulting solution was then added dropwise to a solution of N,N’-dicyclohexylcarbodiimide (DCC) (6.7 g, 32.6 mmol) in THF (20 mL). The mixture was stirred at room temperature for 10 h. After the reaction was completed, DCU was removed by filtration through celite and the crude product was obtained by removing the solvent under vacuum. Pure compound 1, as a slight yellow solid, was obtained after recrystallization in isopropyl alcohol (80% yield). HRMS: m/z Calcd. 436.1509 For C23H22N3O6, Found 436.1504; 1H-NMR(500 MHz, DMSO-d6) δ/ppm m/z 10.94(s, 1H), 8.14(d, J = 8.0 Hz, 1H), 7.56(d, J = 8.0 Hz, 1H), 7.38–7.26(m, 6H), 7.09(t, J = 16.0 Hz, 2H), 7.04(t, J = 12.0 Hz, 1H), 4.99(q, J = 12.6 Hz, 2H), 4.69–4.63(m, 1H), 3.35(dd, J = 4.0, 16.0 Hz, 1H), 3.17(dd, J = 12.0, 8.0 Hz, 1H), 2.84(s, 4H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 170.74, 169.08,156.55,137.28, 136.80, 129.02, 128.50, 128.27, 127.45, 124.97, 121.78, 119.31, 118.51, 112.25, 109.38, 60.44, 66.36, 27.71, 26.18.
Synthesis of Compound 2. To a solution of Ne-Boc-l-lysine (5.0 g, 20.3 mmol) in CH2Cl2 (50 mL) was added, dropwise, BSA (7.2 g, 35.5 mmol) at 0 °C followed by compound 1 (7.3 g, 16.8 mmol) in CH2Cl2 (50 mL). The mixture was stirred at room temperature for 12 h and then concentrated in vacuo. The residue was dissolved in ether (150 mL) and extracted with 5% sodium hydrocarbonate (NaHCO3) solution (50 mL × 3). The combined water layer was acidified with citric acid to pH 6–7 and extracted with EtOAc (100 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and the solvent was removed by a rotary evaporator, under reduced pressure, to yield 2 as a yellow power in 82% yield. HRMS: m/z Calcd. 567.2918 For C30H39N4O7, Found 567.2915; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 12.58(s, 1H), 10.79(s, 1H), 8.24(d, J = 8.0 Hz, 1H), 7.66(d, J = 8.0 Hz, 1H), 7.35–7.28(m, 4H), 7.25–7.24(d, J = 4.0 Hz, 1H), 7.15(s, 1H), 7.05(t, J = 4.0 Hz, 1H), 6.96(t, J = 7.5 Hz, 1H), 6.74(s, 1H), 4.94(s, 2H), 4.38(td, J = 8.0, 12.0 Hz, 1H), 4.15(q, J = 7.6 Hz, 1H), 3.07(dd, J = 8.0, 16.0 Hz, 1H), 2.94–2.88(m, 1H), 2.85(d, J = 4.0 Hz, 2H), 1.65–1.63(m, 1H), 1.55–1.51(m, 1H), 1.35–1.30(m, 11H), 1.17(t, J = 8.0 Hz, 2H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 174.31, 172.36, 156.21, 137.68, 136.68, 128.95, 128.30, 128.08, 127.92, 124.55, 121.44, 119.27, 118.79, 111.89, 110.74, 77.99, 65.83, 60.43, 56.06, 31.68, 29.77, 28.91, 23.19, 21.43, 14.74.
Synthesis of Compound 3. To a stirred solution of Fmoc-l-phenylglycine (15.0 g, 40.0 mmol) in THF (150 mL) was added N-hydroxysuccinimide (4.6 g, 40.0 mmol), dropwise, at 0 °C followed by DCC (9.1 g, 44.0 mmol) in THF dropwise at 5 °C. The mixture was stirred at room temperature for 6 h. After the reaction was completed, DCU was removed by filtration through celite and the crude product was obtained by removing the solvent under vacuum. Pure compound 3, as a white solid, was obtained after recrystallization in EtOAc (87% yield). HRMS: m/z Calcd. 470.1478 For C27H23N2O6, Found 470.1474; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.74–7.75(d, 2H, J = 5.0 Hz), 7.56(s, 2H), 7.37–7.46(m, 7H), 7.28(s, 2H), 5.79–5.80(d, 1H, J = 5.0 Hz), 5.68(s, 1H), 4.42–4.45(t, 2H, J = 7.5 Hz), 4.21(s, 1H), 2.78(s, 4H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 168.33, 166.91, 155.22, 143.76, 143.57, 141.30, 134.41, 129.40, 129.31, 127.75, 127.62, 127.12, 125.07, 120.00, 67.45, 56.44, 47.07, 25.55.
Synthesis of fragment B. To a stirred solution of 2 (5.0 g, 8.8 mmol) in MeOH (50 mL) was added Pd/C10% (0.5 g). A hydrogen atmosphere (1 atm) was applied and the mixture was stirred at room temperature for 5 h. The mixture was filtered through celite and the filtrate was concentrated in vacuo. Next, 5% NaHCO3 solution (30 mL) was added to dissolve the concentrated filtrate and then citric acid was added to adjust the pH to ≈ 6. After that, a white solid precipitated out and was collected by filtration to afford amine intermediate in 94%. HRMS: m/z Calcd. 433.2451 For C25H33N4O5, Found 433.2453. To a solution of the above amine (3.6 g, 8.3 mmol) in dry THF (30 mL) at 0 °C was added BSA (2.9 g, 14.5 mmol), dropwise, and the mixture was stirred for 2 h at room temperature, after which 3 (3.0 g, 6.9 mmol) in THF (20 mL) was added, dropwise, and the resulting mixture was stirred for 10 h at room temperature. After the reaction was completed, the solvents were evaporated. The residue was then dissolved in ether (50 mL) and extracted with 5% NaHCO3 solution (50 mL × 3). The combined water layer was acidified with citric acid to pH 6–7 and extracted with EtOAc (100 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and filtered through celite. The organic solution was evaporated in vacuo and 4.57 g of fragment B was obtained after recrystallization in MeOH (84% yield). HRMS: m/z Calcd. 788.3659 For C45H50N5O8, Found 788.3650; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 12.53(s, 1H), 10.68(d, J = 2.4 Hz, 1H), 8.50(d, J = 8.1 Hz, 1H), 8.25(d, J = 7.8 Hz, 1H), 8.02(d, J = 8.5 Hz, 1H), 7.86(d, J = 7.5 Hz, 2H), 7.75(dd, J = 11.3, 7.6 Hz, 2H), 7.55(d, J = 7.9 Hz, 1H), 7.39(td, J = 7.4, 3.0 Hz, 2H), 7.31–7.17(m, 7H), 7.01(t, J = 7.4 Hz, 1H), 6.91(s, 1H), 6.71(t, J = 5.8 Hz, 1H), 5.36(d, J = 8.4 Hz, 1H), 4.57(q, J = 8.1 Hz, 1H), 4.22(dd, J = 13.5, 7.1 Hz, 2H), 3.04(dd, J = 14.5, 5.2 Hz, 1H), 2.88(d, J = 9.8 Hz, 1H), 2.82(q, J = 6.7, 6.1 Hz, 2H), 1.58(s, 1H), 1.49(t, J = 7.5 Hz, 1H), 1.33(s, 9H), 1.26(q, J = 7.2 Hz, 2H), 1.09(dt, J = 13.9, 7.6 Hz, 2H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 174.25, 171.87, 170.20, 156.42, 156.21, 144.60, 144.32, 141.32, 138.99, 136.62, 128.70, 128.29, 127.94, 127.72, 126.25, 126.12, 124.52, 121.36, 120.73, 119.08, 118.74, 111.81, 110.15, 77.98, 66.67, 58.49, 54.19, 52.61, 47.23, 31.59, 30.58, 29.74, 28.91, 23.19.
Synthesis of Compound 4. To a stirred solution of N-Boc-O-benzyl-d-tyrosine (10.0 g, 27.0 mmol) in dry THF (100 mL) was added N-hydroxysuccinimide (3.1 g, 27.0 mmol) at 0 °C. The resulting solution was then added a solution of DCC (6.1 g, 29.7 mmol) in THF, dropwise, at 5 °C. The mixture was stirred at room temperature for 4 h and monitored by TLC. After the reaction was completed, DCU was removed by filtration through celite. The solvent was evaporated off, after which 11.5 g of 4 was obtained after recrystallization in EtOAc in 91% yield. HRMS: m/z Calcd. 469.1975 For C25H29N2O7, Found 469.1971; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.41–7.42(d, 2H, J = 5.0 Hz), 7.37–7.39(t, 2H, J = 5.0 Hz), 7.20–7.21(d, 2H, J = 5.0 Hz), 6.92–6.94(d, 2H, J = 10.0 Hz), 5.04(s, 2H), 4.89(s, 1H), 3.13–3.25(m, 2H), 2.84(s, 4H), 1.42(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 168.65, 167.77, 158.18, 154.62, 136.97, 130.82, 128.58, 127.96, 127.47, 126.86, 80.52, 70.00, 52.71, 37.28, 28.24, 25.59.
Synthesis of Compound 5. To a stirred solution of phenylalanine methyl ester hydrochloride (5 g, 23.3 mmol) in dry THF (50 mL) was added trimethylamine (2.4 g, 23.3 mmol), dropwise, followed by addition of 4 (10.9 g, 23.3 mmol) in THF (20 mL). The mixture was stirred at room temperature for 5 h and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Filtration followed by concentration in vacuo afforded 11.6 g of 5, as a white solid, in 94% yield. HRMS: m/z Calcd. 533.2652 For C31H37N2O6, Found 533.2647; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.40–7.42(d, 2H, J = 10.0 Hz), 7.35–7.38(t, 2H, J = 7.50 Hz), 7.31–7.32(d, 1H, J = 5.0 Hz), 7.22–7.25(t, 3H, J = 7.50 Hz), 7.09–7.10(d, 2H, J = 5.0 Hz), 6.97–6.99(d, 2H, J = 10.0 Hz), 6.88–6.89(d, 2H, J = 5.0 Hz), 6.27(s, 1H), 5.02(s, 2H), 4.95(s, 1H), 4.77(s, 1H), 4.82(s, 1H), 3.66(s, 3H), 2.96–3.05(m, 4H), 1.40(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 171.36, 170.81, 157.89, 136.98, 135.65, 130.42, 129.22, 128.58, 128.54, 127.96, 127.42, 127.11, 115.03, 80.16, 70.01, 55.80, 53.26, 52.25, 37.99, 37.43, 28.26.
Synthesis of fragment C. To a stirred solution of 5 (6.0 g, 11.6 mmol) in CH2Cl2 (25 mL) was added trifluoroacetic acid (TFA) (8.6 mL, 116 mmol), dropwise, at 5 °C. The mixture was stirred at room temperature for 4 h and monitored by TLC. The solvent was evaporated off, after which 5.9 g of fragment C was obtained after recrystallization in ether (98% yield). HRMS: m/z Calcd. 433.2127 For C26H29N2O4, Found 433.2124; 1H-NMR(500 MHz, CDCl3) δ/ppm = 8.97–8.99(d, 1H, J = 10.0 Hz), 8.10(s, 3H), 7.44–7.45(d, 2H, J = 5.0 Hz), 7.38–7.41(t, 2H, J = 7.5 Hz), 7.29–7.34(m, 3H), 7.22–7.25(t, 3H, J = 7.5 Hz), 7.17–7.18(d, 2H, J = 5.0 Hz), 6.96–6.98(d, 2H, J = 10.0 Hz), 5.08(s, 2H), 4.55–4.60(q, 1H, J = 8.3 Hz), 3.98(s, 1H), 3.61(s, 3H), 3.02–3.09(m, 2H), 2.55–3.00(dd, 1H, J1 = 10.0, J2 = 15.0Hz), 2.85–2.89(dd, 1H, J1 = 5.0, J2 = 15Hz); 13C-NMR(125 MHz, CDCl3) δ/ppm = 171.64, 168.76, 158.08, 137.58, 137.14, 131.12, 129.53, 128.89, 128.86, 128.09, 127.21, 127.15, 115.27, 69.63, 54.27, 53.76, 52.50, 37.10, 36.51.
Synthesis of Compound 6. To a stirred solution of l-hydroxyproline methyl ester (10.0 g, 69.9 mmol) in 1,4-dioxane/H2O (v:v = 2:1) was added Na2CO3 (7.3 g, 69.9 mmol), slowly, followed by Fmoc N-hydroxysuccinimide ester (Fmoc-OSu) (23.6 g, 69.9 mmol). The resulting solution was stirred at room temperature for 10 h and then concentrated in vacuo. The residue was dissolved in EtOAc (100 mL), washed with brine, dried over Na2SO4, and concentrated to yield 6, as a colorless oil, in 94% yield. HRMS: m/z Calcd. 368.1498 For C21H22NO5, Found 368.1494; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.80–7.79(d, J = 4 Hz, 2H), 7.66–7.58(m, 2H), 7.45–7.42(t, J = 6.0 Hz, 2H), 7.36–7.34(t, J = 4.0 Hz, 2H), 4.59–4.44(m, 3H), 4.41–4.35(m, 1H), 4.31–4.15(m, 1H), 3.79(s, 2H), 3.70–3.60(m, 3H), 2.74(s, 1H), 2.41–2.32(m, 1H), 2.15–2.13(m, 1H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 173.16, 155.10, 144.09, 141.33, 127.74, 127.12, 125.15, 119.99, 70.08, 69.22, 67.64, 60.51, 57.98, 57.62, 55.31, 54.68, 52.38, 47.27, 39.33, 38.42.
Synthesis of Compound 7. To a stirred solution of triphosgene (3.2 g, 10.6 mmol) in dry THF (40 mL) was added 6 (6.5 g, 17.7 mmol) in THF (10 mL), slowly, and the mixture was stirred at room temperature for 2 h. Next, N-boc-ethylenediamine (14.2 g, 88.5 mmol) in THF (20 mL) and 4-dimethylaminopyridine (2.2 g, 17.7 mmol) was added. The resulting mixture was stirred at room temperature for 6 h. After the reaction was completed, the solvents were evaporated and then the residue was dissolved in EtOAc (100 mL), washed with 0.1 M HCl solution (100 mL × 3), brine (100 mL × 3), and dried over Na2SO4. The solvent was evaporated off, after which 6.7 g of 7 was obtained after recrystallization in EtOAc in 69% yield. HRMS: m/z Calcd. 576.2322 For C29H35N3O8Na, Found 576.2313; 1H-NMR(500 MHz, CDCl3) δ/ppm = 7.77(d, J = 8.0 Hz, 2H), 7.57(dd, J = 12.0, 4.0 Hz, 2H), 7.40(t, J = 8.0 Hz, 2H), 7.31(t, J = 6.0 Hz, 2H), 5.26(d, J = 20.0 Hz, 1H), 5.14(d, J = 20.0 Hz, 1H), 4.79(s, 1H), 4.51–4.47(m, 1H), 4.43–4.39(m, 1H), 4.34(t, J = 8.0 Hz, 1H), 4.29–4.15(m, 1H), 3.85–3.72(m, 4H), 3.66(s, 1H), 3.27(s, 4H), 2.46(dd, J = 12.0, 20.0 Hz, 2H), 2.22(s, 1H), 1.44(s, 9H); 13C-NMR(125 MHz, CDCl3) δ/ppm = 172.53, 155.78, 154.75, 143.91, 143.59, 141.29, 127.73, 127.08, 125.12, 120.00, 79.75, 73.13, 72.23, 67.73, 60.40, 52.43, 47.27, 28.35, 21.04, 14.20.
Synthesis of fragment D. To a stirred solution of 7 (5.0 g, 9.0 mmol) in dry MeOH (30 mL) was added NaOH solution (18.0 mmol, 10 mL), dropwise, at 0 °C and the mixture was stirred at room temperature for 2 h. Solvents were removed by vacuo and the residue was resolved in 1, 4-dioxane followed by addition of Fmoc-OSu (3.0 g, 9.0 mmol). The resulting mixture was stirred at room temperature for 6 h and solvents were evaporated, after which 4.2 g of fragment D was obtained in 88% yield. HRMS: m/z Calcd. 562.5667 For C28H33N3O8Na, Found 562.2167. 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 7.93–7.89(t, J = 10.0 Hz, 1H)7.77–7.74(dd, J1 = 10.0, J2 = 45 Hz, 1H), 7.66(t, J = 7.5 Hz, 1H), 7.44(q, J = 6.6 Hz, 2H), 7.36(t, J = 5.0 Hz, 2H), 7.27(s, 1H), 6.85(s, 1H), 5.11(s, 1H), 4.28–4.19(m, 3H), 4.05(q, J = 10.0 Hz, 1H), 3.65–3.73(ddd, J = 27.8, 11.9, 5.3 Hz, 1H), 3.46–3.38(m, 1H), 3.43(s, 1H), 3.01(s, 4H), 2.25(s, 1H), 2.15(s, 1H), 1.39(s, 9H). 13C-NMR (125 MHz, DMSO-d6) δ/ppm = 171.00, 156.22, 155.12, 141.34, 140.04, 138.05, 129.57, 127.94, 122.05, 120.69, 78.25, 72.56, 67.37, 60.42, 52.99, 47.22, 28.87, 21.43, 14.74.
Synthesis of Compound 8. To a stirred solution of fragments B (3.5 g, 4.4 mmol) and C (1.4 g, 4.4 mmol) in dry THF (50 mL) was added TBTU (2.1 g, 6.6 mmol) and 4-methylmorpholine (0.9 g, 8.8 mmol), slowly. The mixture was stirred at room temperature for 6 h and solvents were removed by vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Solvents were evaporated off, after which 4.7 g of 8 was obtained after recrystallization in MeOH in 88% yield. HRMS: m/z Calcd. 1202.5603 For C71H76N7O11, Found 1202.5590; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 10.72(s, 1H), 8.64(d, J = 8.0 Hz, 1H), 8.27(d, J = 4.0 Hz, 1H), 8.17(d, J = 8.0 Hz, 1H), 7.97–7.91(dd, J = 16.0, 4.0 Hz, 1H), 7.85(d, J = 8.0 Hz, 2H), 7.74–7.68(dd, J = 16.0, 8.0 Hz, 2H), 7.53(d, J = 4.0 Hz, 1H), 7.37–7.33(m, 5H), 7.32–7.25(m, 7H), 7.23–7.17(m, 8H), 7.13(d, J = 8.0 Hz, 2H), 7.00(t, J = 6.0Hz, 1H), 6.93–6.89(m, 2H), 6.84(d, J = 8.0 Hz, 2H), 6.67(t, J = 6.0 Hz, 1H), 5.38(d, J = 8.0 Hz, 1H), 4.97(s, 2H), 4.48–4.45(m, 3H), 4.22–4.16(m, 3H), 4.09(s, 1H), 3.58–3.51(m, 1H), 3.47(s, 2H), 3.07–2.98(m, 3H), 2.94–2.86(m, 2H), 2.79–2.74(m, 3H), 1.48–1.43(m, 2H), 1.33(s, 9H), 1.20(s, 2H), 0.93(d, J = 8.0 Hz, 2H); 13C-NMR(125 MHz, DMSO-d6) δ/ppm = 171.88, 170.73, 157.52, 156.22, 144.66, 144.22, 141.30, 140.05, 138.97, 138.05, 137.77, 136.62, 130.88, 129.72, 129.61, 129.02, 128.90, 128.30, 127.98, 127.73, 127.62, 127.23, 126.20, 122.05, 120.70, 119.10, 118.83, 114.92, 111.83, 110.48, 110.00, 78.05, 69.71, 66.72, 58.40, 54.52, 53.49, 52.30, 47.17, 37.29, 35.75, 31.93, 29.87, 28.89, 27.19, 22.93.
Synthesis of Compound 9. To a stirred solution of 8 (4.0 g, 3.3 mmol) in dry THF (40 mL) was added piperidine (16.5 mL, 165 mmol), dropwise, at 0 °C and the mixture was stirred at room temperature for 40 min. Solvents were evaporated off, after which 3.0 g of amine intermediate was obtained after recrystallization in MeOH in 93% yield. HRMS: m/z Calcd. 980.4922 For C56H66N7O9, Found 980.4932. To a stirred solution of the above amine (1.7 g, 3.1 mmol) and fragment D (3.0 g, 3.1 mmol) in dry THF (50 mL) was added TBTU (1.5 g, 4.7 mmol) and 4-methylmorpholine (0.9 g, 9.3 mmol), slowly. The mixture was stirred at room temperature for 6 h and solvents were removed by vacuo. The residue was dissolved in EtOAc (100 mL) and washed with 5% NaHCO3 solution (100 mL × 3), 5% citric acid solution (100 mL × 3), brine, and dried over Na2SO4. Solvents were evaporated off, after which 3.96 g of 9 was obtained after recrystallization in MeOH in 85% yield. HRMS: m/z Calcd. 1501.7084 For C84H97N10O16, Found 1501.7087; 1H-NMR(500 MHz, DMSO-d6) δ/ppm = 10.69(dd, J = 27.1, 6.2 Hz, 1H), 8.54(dd, J = 42.4, 12.0 Hz, 2H), 8.36(dd, J = 21.2, 8.6 Hz, 1H), 8.08(dd, J = 19.8, 9.3 Hz, 1H), 7.96–7.90(m, 1H), 7.87(d, J = 5.0 Hz, 2H), 7.59(d, J = 7.8 Hz, 2H), 7.56–7.51(m, 1H), 7.42–7.11(m, 26H), 6.88(m, 2H), 5.58(dd, J = 39.8, 8.0 Hz, 1H), 4.96(m, 2H), 4.89–4.70(m, 1H), 4.49(s, 2H), 4.23(d, J = 9.8 Hz, 2H), 4.15(s, 1H), 4.08–4.00(m, 1H), 3.90(s, 1H), 3.80(t, J = 14.3 Hz, 1H), 3.52(d, J = 4.9 Hz, 3H), 3.29(s, 2H), 3.07–2.93(m, 9H), 2.92–2.82(m, 4H), 2.28–2.14(m, 1H),2.02(dd, J = 15.3, 9.7 Hz, 1H), 1.55–1.40(m, 2H), 1.35(s, 18H), 1.30–1.23(m, 2H), 0.97(m, J = 16.8, 10.8 Hz, 2H); 13C-NMR (125 MHz, DMSO-d6) δ/ppm = 172.25, 171.84, 171.77, 171.37, 170.22, 157.60, 156.14, 154.48, 144.47, 141.36, 138.60, 137.58, 130.90, 129.70, 129.00, 128.90, 128.39, 128.27, 128.09, 127.77, 127.32, 127.21, 125.70, 124.82, 122.92, 120.65, 120.12, 116.62, 114.99, 77.93, 73.23, 67.42, 62.14, 58.56, 58.06, 54.54, 54.25, 53.15, 52.42, 47.13, 37.79, 37.35, 36.43, 32.33, 31.43, 29.82, 28.88, 28.36, 23.02,
Synthesis of Compound 10. To a stirred solution of 9 (2.0 g, 1.3 mmol) in dry THF (20 mL) was added piperidine (6.5 mL, 65 mmol), dropwise, at 0 °C and the mixture was stirred at room temperature for 30 min. Solvents were evaporated off and residue was re-dissolved in dry THF (20 mL). Next, LiBr (0.5 g, 6.5 mmol) in H2O (1 mL) was added, dropwise, to the above solution at 0 °C followed by addition of 1M NaOH solution (6.5 mL, 6.5 mmol) over a period of 3 h. The mixture was stirred at room temperature for 12 h and concentrated in vacuo to yield 1.2 g of 10, as a white solid, in 72% yield. HRMS: m/z Calcd. 1265.6247 For C68H85N10O14, Found 1265.6262; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.72(s, 1H), 8.95(s, 1H), 8.74–8.63(m, 1H), 8.16(t, J = 9.4 Hz, 1H), 8.06(d, J = 7.3 Hz, 1H), 7.87(dd, J = 11.8, 8.0 Hz, 1H), 7.54(d, J = 7.9 Hz, 1H), 7.39(d, J = 7.2 Hz, 2H), 7.37–7.32(m, 2H), 7.32–7.28(m, 1H), 7.29–7.14(m, 10H), 7.14–6.98(m, 4H), 6.92–6.88(m, 1H), 6.85(d, J = 8.3 Hz, 2H), 6.79(dd, J = 18.0, 7.3 Hz, 1H), 6.68(t, J = 5.7 Hz, 1H), 5.56(d, J = 40.3, 8.0 Hz, 1H), 5.00(s, 2H), 4.54(dq, J = 35.6, 7.4 Hz, 1H), 4.46–4.34(m, 2H), 4.07(s, 2H), 3.09–2.88(m, 9H), 2.74(dt, J = 31.7, 10.3 Hz, 3H), 2.64(d, J = 15.3 Hz, 1H), 2.55(d, J = 15.3 Hz, 1H), 2.26(d, J = 44.3 Hz, 1H), 1.94–1.83(m, 1H), 1.46–1.37(m, 1H), 1.36–1.31(m, 18H), 1.27(t, J = 7.6 Hz, 1H), 1.19(dt, J = 17.5, 8.5 Hz, 2H), 0.98–0.79(m, 2H); 13C-NMR(150 MHz, DMSO-d6) δ/ppm = 173.02, 171.60, 171.16, 169.77, 157.28, 156.09, 139.56, 139.27, 138.16, 137.58, 136.65, 136.48, 130.59, 130.15, 130.05, 128.81, 128.72, 128.12, 128.03, 127.81, 127.06, 125.98, 124.16, 121.24, 118.99, 118.65, 114.75, 111.70, 110.77, 110.38, 77.77, 76.08, 69.51, 59.74, 55.92, 55.35, 55.10, 53.90, 53.07, 38.35, 37.75, 37.13, 32.22, 31.65, 29.59, 28.68, 27.97, 23.02.
Synthesis of intermediate A. To a stirred solution of 10 (1.0 g, 0.8 mmol) in dry DMF (250 mL) was added HATU (0.8 g, 2.0 mmol) and anhydrous HOBt (0.3 g, 2.0 mmol), slowly, and the mixture was stirred at −5 °C for 2 h. After the reaction was completed, H2O (20 mL) was added to the above solution and white solid precipitated out. The mixture was filtered through celite and the solid residue was obtained to give 0.86 g of intermediate A in 87% yield. HRMS: m/z Calcd. 1247.6141 For C68H83N10O13, Found 1247.6098; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.85(s, 1H), 9.34(d, J = 7.4 Hz, 1H), 8.33(d, J = 6.8 Hz, 1H), 7.90(d, J = 12.6 Hz, 1H), 7.71(d, J = 9.8 Hz 1H), 7.55(d, J = 16.2 Hz, 1H), 7.43(d, J = 18.4 Hz, 2H), 7.33(ddt, J = 36.4, 21.3, 7.4 Hz, 10H), 7.22(dd, J = 15.1, 7.6 Hz, 3H), 7.19–7.14(m,1H), 7.06–6.91(m, 3H), 6.88(d, J = 6.5 Hz, 1H), 6.83–6.74(m, 3H), 6.72–6.61(m, 1H), 5.39(d, J = 8.3 Hz, 1H), 5.18(s, 1H), 5.05–4.92(m, 3H), 4.60–4.46(m, 1H), 4.49–4.32(m, 2H), 3.69(dd, J = 34.6, 7.6 Hz, 1H), 3.62–3.51(m, 1H), 3.04–2.85(m, 7H), 2.83–2.55(m, 5H), 2.15(d, J = 7.0 Hz, 1H), 2.08–1.89(m, 1H), 1.44(s, 1H), 1.33(s, 18H), 1.26–1.08(m, 3H), 1.03–0.86(m, 2H); 13C-NMR(150 MHz, DMSO-d6) δ/ppm = 172.60, 171.70, 171.17, 169.87, 157.41, 156.06, 139.95, 137.64, 137.56, 136.76, 136.45, 136.41, 131.19, 130.67, 130.28, 129.73, 129.31, 129.16, 128.85, 128.50, 128.24, 128.12, 127.43, 126.28, 124.21, 121.27, 118.62, 114.50, 111.65, 109.67, 78.22, 77.89, 69.58, 59.91, 57.41, 54.89, 53.76, 51.74, 51.11, 36.62, 36.29, 31.70, 30.95, 29.54, 28.69, 27.95, 22.98.
Synthesis of pasireotide. TFA (6.0 mL) was added to intermediate A (0.5 g, 0.4 mmol) and the mixture was stirred at 0 °C for 2 h. After the reaction was completed, excess TFA was evaporated off. Diethyl ether was added to the residue and a white solid was precipitated off to give 0.45 g of pasireotide after filtration (89% yield). ESI-MS(m/z): 524.04[M/2 + H−CF3COOH]+; 1047.86[M + H−2CF3COOH]+; 1069.81[M + Na−2CF3COOH]+; HRMS: m/z Calcd. 1047.5092 For C58H67N10O9, Found 1047.5107; 1H-NMR(600 MHz, DMSO-d6) δ/ppm = 10.87(s, 1H), 7.82(d, J = 42 Hz, 10H), 7.42–7.38(m, 5H), 7.36–7.25(m, 10H), 7.23–7.21(t, J = 6.0 Hz, 3H), 7.07–7.04(t, J = 9.0 Hz, 1H), 7.0(s, 1H), 6.96–6.93(t, J = 9.0 Hz, 3H), 6.82(d, J = 12 Hz, 2H), 5.48(d, J = 6.0 Hz, 1H), 5.22(s, 1H), 5.01(s, 2H), 4.59(d, J = 6.0 Hz, 1H), 4.46–4.43(dd, J = 10.2, 7.0 Hz, 1H), 4.41–4.39(m, 1H), 3.98(s, 1H), 3.78(d, J = 12.0 Hz, 1H), 3.70(q, J = 6.0 Hz, 1H), 3.60(d, J = 6.0 Hz, 1H), 3.28–3.24(m, 2H), 3.22–3.16(m, 2H), 3.03–3.01(m, 2H), 2.90–2.84(m, 4H), 2.69(d, J = 12.0 Hz, 2H), 2.20(d, J = 6.0 Hz, 1H), 2.05–2.01(t, J = 12.0 Hz, 1H), 1.43–1.27(m, 2H), 1.22–1.17(m, 2H), 0.92(s, 1H), 0.72(s, 1H); 13C-NMR (150 MHz, DMSO-d6) δ/ppm = 172.57, 170.58, 170.30, 170.10, 168.90, 157.33, 156.03, 138.32, 137.61, 137.01, 136.43, 130.50, 130.32, 130.27, 128.92, 128.88, 128.50, 128.26, 128.20, 128.15, 128.08, 127.04, 124.29, 121.30, 120.68, 118.88, 118.70, 116.71, 114.80, 111.70, 109.77, 74.13, 69.60, 59.97, 57.72, 57.55, 56.71, 56.14, 54.79, 53.11, 40.52, 39.12, 38.96, 38.45, 37.54, 36.25, 34.49, 31.09, 26.93, 22.22.
Supplementary Materials
The following are available online.
Author Contributions
C.M. and W.F. conceived and designed the experiments. C.M., M.C., W.C., J.T., D.K., and M.Z. performed the experiments. C.M. wrote the manuscript. J.T. and M.Z. revised the manuscript. All authors read and approved the final manuscript.
Funding
Financial support was provided by CAMS Innovation Fund for Medical Sciences (CAMS-2017-I2M-1-011).
Conflicts of Interest
There are no conflicts to declare.
References
- Nadia, S.G.; Maciej, O.; Marek, R. Pasireotide-Mechanism of Action and Clinical Applications. Curr. Drug Metab. 2018, 19, 876–882. [Google Scholar] [CrossRef]
- Wauters, L.; Vanuytsel, T. Applications of peptide hormone ligands for the treatment of dumping and short bowel syndrome. Curr. Opin. Pharmacol. 2018, 43, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Lesche, S.; Lehmann, D.; Nagel, F.; Schmid, H.A.; Schulz, S. Differential Effects of Octreotide and Pasireotide on Somatostatin Receptor Internalization and Trafficking in Vitro. J. Clin. Endocrinol. Metab. 2009, 94, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, A.; Melmed, S. Drug profile: Pasireotide—A somatostatin analog for the potential treatment of acromegaly, neuroendocrine tumors and Cushing’s disease. IDrugs 2007, 10, 885–895. [Google Scholar] [PubMed]
- Lewis, I.; Bauer, W.; Albert, R.; Chandramouli, N.; Pless, J.; Weckbecker, G.; Bruns, C. A Novel somatostatin mimic with broad somatotropin release inhibitory factor receptor binding and superior therapeutic potential. J. Med. Chem. 2003, 46, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Hellstern, H.; Pachinger, W.; Prikoszovich, W.; Wietfeld, B. Preparation of somatostatin peptides. WO2005014624A2, 17 February 2005. [Google Scholar]
- Zhang, W.; Liu, J.; Ma, Y.; Yuan, J. Process for Preparation of Pasireotide. CN103467575A, 25 December 2013. [Google Scholar]
- Mi, P.; Pan, J.; Ma, Y.; Yuan, J. Process for Preparation of Polypeptide Pasireotide Diaspartate. CN103641894A, 19 March 2014. [Google Scholar]
- Guo, D.; Zeng, D.; Dong, H.; Wen, Y. Process for Preparation of Pasireotide. CN104447962A, 25 March 2015. [Google Scholar]
- Goud, A.N.; Shafee, M.A.; Kumar, V.S.; Patil, N.D.; Rao, K.M. A process for the Preparation of Pasireotide. WO2016097962A1, 23 June 2016. [Google Scholar]
- Pullagurla, M.R.; Nagarapu, R.; Rangisetty, J.B. Novel Improved Process for the Preparation of Pasireotide. WO2016207912A1, 29 December 2016. [Google Scholar]
- Sun, L.; Chen, W.; Zhang, X.; Zheng, F.; Guo, J. Process for Preparation of Pasireotide. CN106432429A, 22 February 2017. [Google Scholar]
- Bruckdorfer, T.; Marder, O.; Albericio, F. From production of peptides in milligram amounts for research to multi-tons quantities for drugs of the future. Curr. Pharm. Biotechnol. 2004, 5, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Blomberg, L.; Flegel, M.; Lepsa, L.; Nilsson, B.; Verlander, M. Large-scale synthesis of peptides. Biopolymers 2000, 55, 227–250. [Google Scholar] [CrossRef]
- Uhlig, T.; Kyprianou, T.; Martinelli, F.G.; Oppici, C.A.; Heiligers, D.; Hills, D.; Calvo, X.R.; Verhaert, P. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. Eupa Open Proteom. 2014, 4, 58–69. [Google Scholar] [CrossRef]
- Goodwin, D.; Simerska, P.; Toth, I. Peptides As Therapeutics with Enhanced Bioactivity. Curr. Med. Chem. 2012, 19, 4451–4461. [Google Scholar] [CrossRef]
- Li, Y.; Chang, Q.; Wu, M.; Zhao, X. Total synthesis of five proline-enriched cyclic heptapeptides from the marine sponge Stylissa carteri. Tetrahedron Lett. 2018, 59, 1828–1831. [Google Scholar] [CrossRef]
- Huang, Y.; Feng, W.H. N,O-Bis(trimethylsilyl)acetamide/N-hydroxysuccinimide ester (BSA/NHS) as coupling agents for dipeptide synthesis. Chin. Chem. Lett. 2016, 27, 357–360. [Google Scholar] [CrossRef]
- Kuwata, S.; Tachibana, S.; Moriyama, Y.; Takatsuki, E.; Yanagihara, R.; Miyazawa, T.; Yamada, T. Side reaction at the hydrolysis of the methyl ester of Aib-containing peptide. Pept. Chem. 1996, 33, 77–80. [Google Scholar]
- Basse, N.; Piguel, S.; Papapostolou, D.; Ferrier-Berthelot, A.; Richy, N.; Pagano, M.; Sarthou, P.; Sobczak-Thepot, J.; Reboud-Ravaux, M.; Vidal, J. Linear TMC-95-Based Proteasome Inhibitors. J. Med. Chem. 2007, 50, 2842–2850. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, A.; Dutot, L.; Barbeau, O.; Hamchaoui, W.; Wakselman, M.; Mazaleyrat, J.-P. Synthesis of terminally protected (S)-β3-H-DOPA by Arndt-Eistert homologation: An approach to crowned β-peptides. Tetrahedron Asymmetry 2005, 16, 857–864. [Google Scholar] [CrossRef]
- Zhou, T.; Hider, R.C.; Jenner, P.; Campbell, B.; Hobbs, C.J.; Rose, S.; Jairaj, M.; Tayarani-Binazir, K.A.; Syme, A. Design, synthesis and biological evaluation of l-dopa amide derivatives as potential prodrugs for the treatment of Parkinson’s disease. Eur. J. Med. Chem. 2010, 45, 4035–4042. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Singh, E.K.; Wahyudi, H.; Alexander, L.D.; Kunicki, J.B.; Nazarova, L.A.; Fairweather, K.A.; Giltrap, A.M.; Jolliffe, K.A.; McAlpine, S.R. Synthesis of sansalvamide A peptidomimetics: Triazole, oxazole, thiazole, and pseudoproline containing compounds. Tetrahedron 2012, 68, 1029–1051. [Google Scholar] [CrossRef] [PubMed]
- Fink, B.E.; Kym, P.R.; Katzenellenbogen, J.A. Design, Synthesis, and Conformational Analysis of a Proposed Type I β-Turn Mimic. J. Am. Chem. Soc. 1998, 120, 4334–4344. [Google Scholar] [CrossRef]
- Yep, Y.-h.; Gao, X.-m.; Liu, M.; Tang, Y.-C.; Tian, G.-l. Studies on the synthetic methodology of head to tail cyclization of linear peptides. Lett. Pept. Sci. 2004, 10, 571–579. [Google Scholar] [CrossRef]
- Tang, Y.C.; Xie, H.B.; Tian, G.L.; Ye, Y.H. Synthesis of cyclopentapeptides and cycloheptapeptides by DEPBT and the influence of some factors on cyclization. J. Pept. Res. 2002, 60, 95–103. [Google Scholar] [CrossRef]
- Peng, L.; Peter, P.R.; Jiecheng, X. Current Synthetic Approaches to Peptide and Peptidomimetic Cyclization. Curr. Org. Chem. 2002, 6, 411–440. [Google Scholar] [CrossRef]
- Williams, J.D.; Flanagan, M.; Lopez, L.; Fischer, S.; Miller, L.A.D. Using accurate mass electrospray ionization-time-of-flight mass spectrometry with in-source collision-induced dissociation to sequence peptide mixtures. J. Chromatogr. A 2003, 1020, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Tomer, K.B.; Crow, F.W.; Gross, M.L.; Kopple, K.D. Fast-atom bombardment combined with tandem mass spectrometry for the determination of cyclic peptides. Anal. Chem. 1984, 56, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Qi, W.; He, Z.; Qiao, B. Sequencing peptides by electrospray ion-trap mass spectrometry: A useful tool in synthesis of axinastatin 3. Eur. J. Chem. 2006, 4, 620–631. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–10, fragment B, fragment C, fragment D, intermediate A, and pasireotide are available from the authors. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).