
Synthesis and Biological Evaluation of NH2-Sulfonyl Oseltamivir Analogues as Influenza Neuraminidase Inhibitors


Abstract
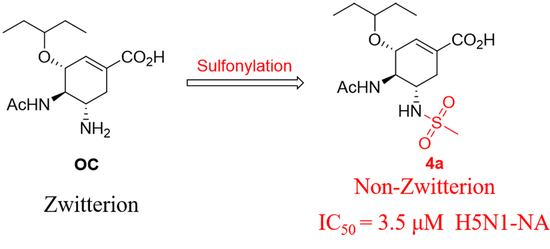
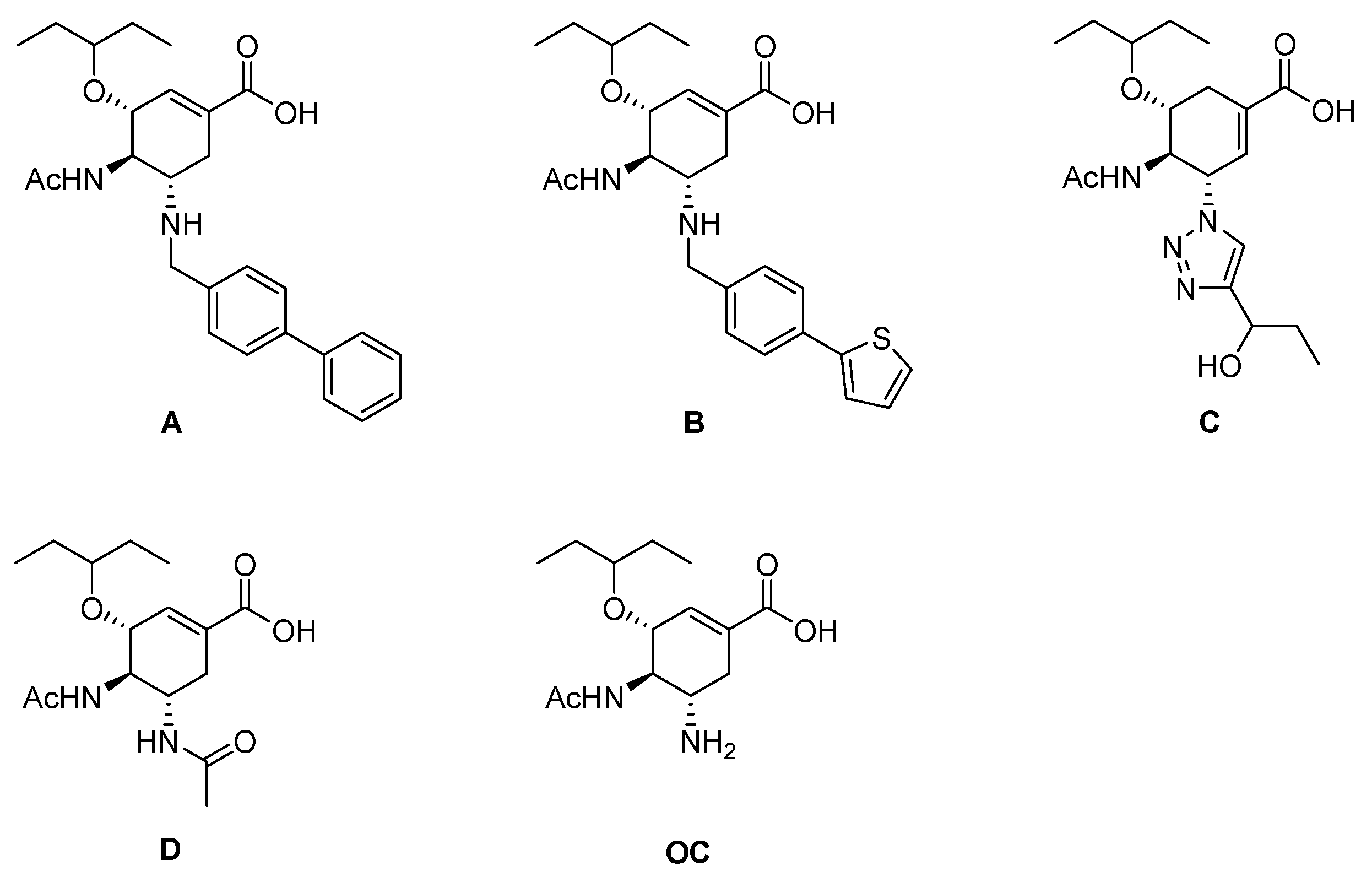
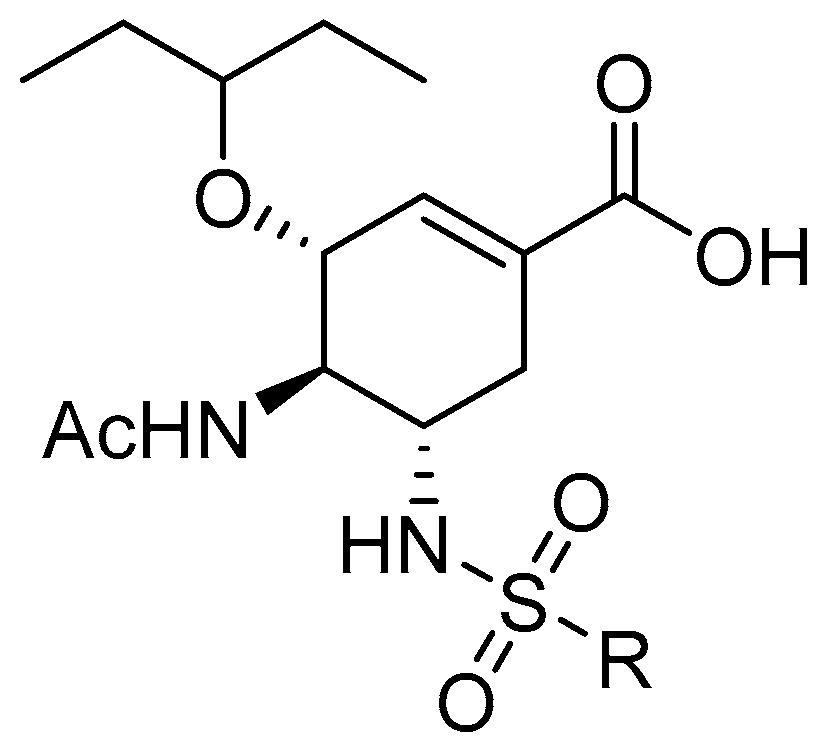
1. Introduction
2. Results and Discussion
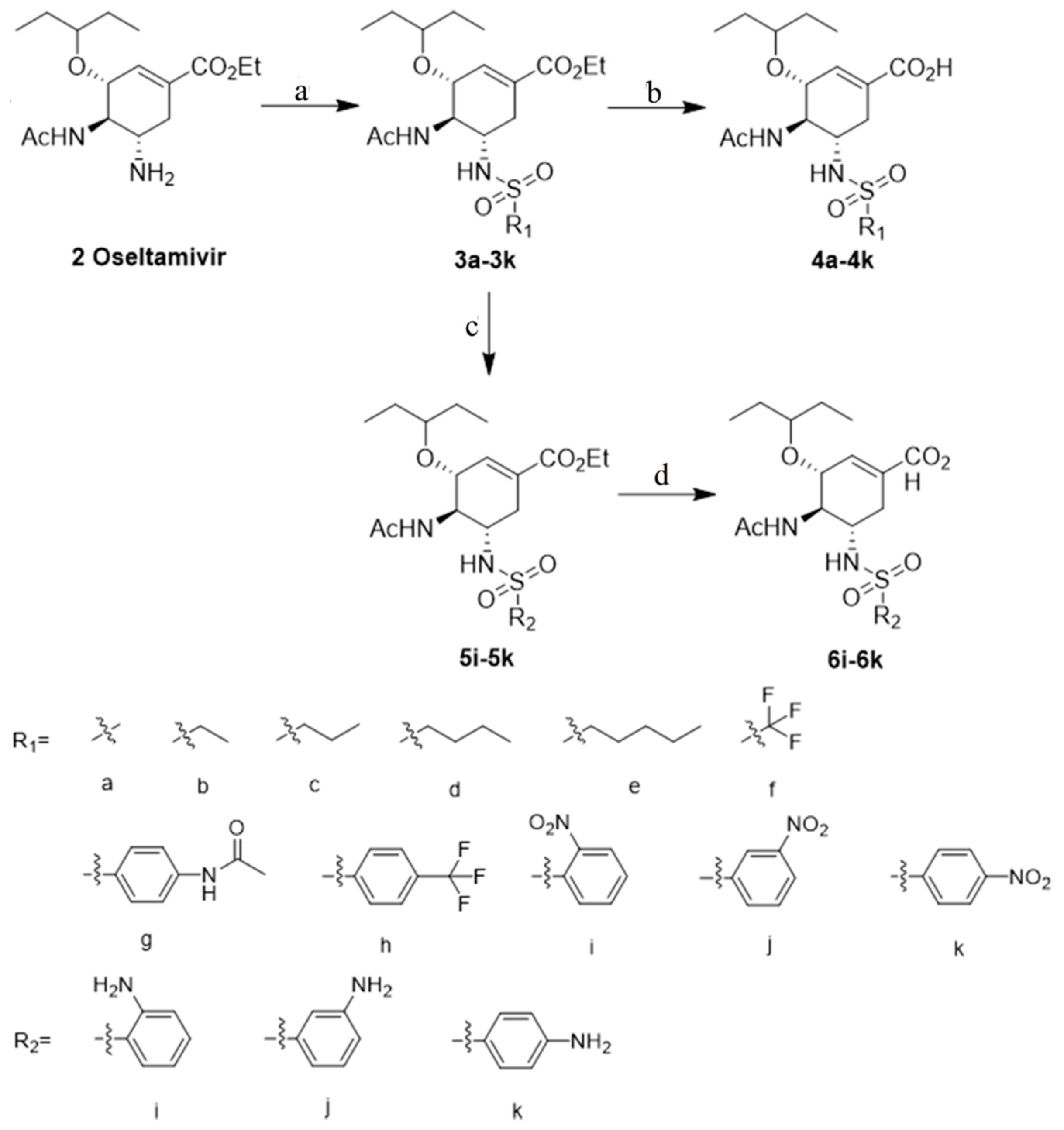
2.1. Synthesis
2.2. Neuraminidase Enzyme Inhibitory Assay
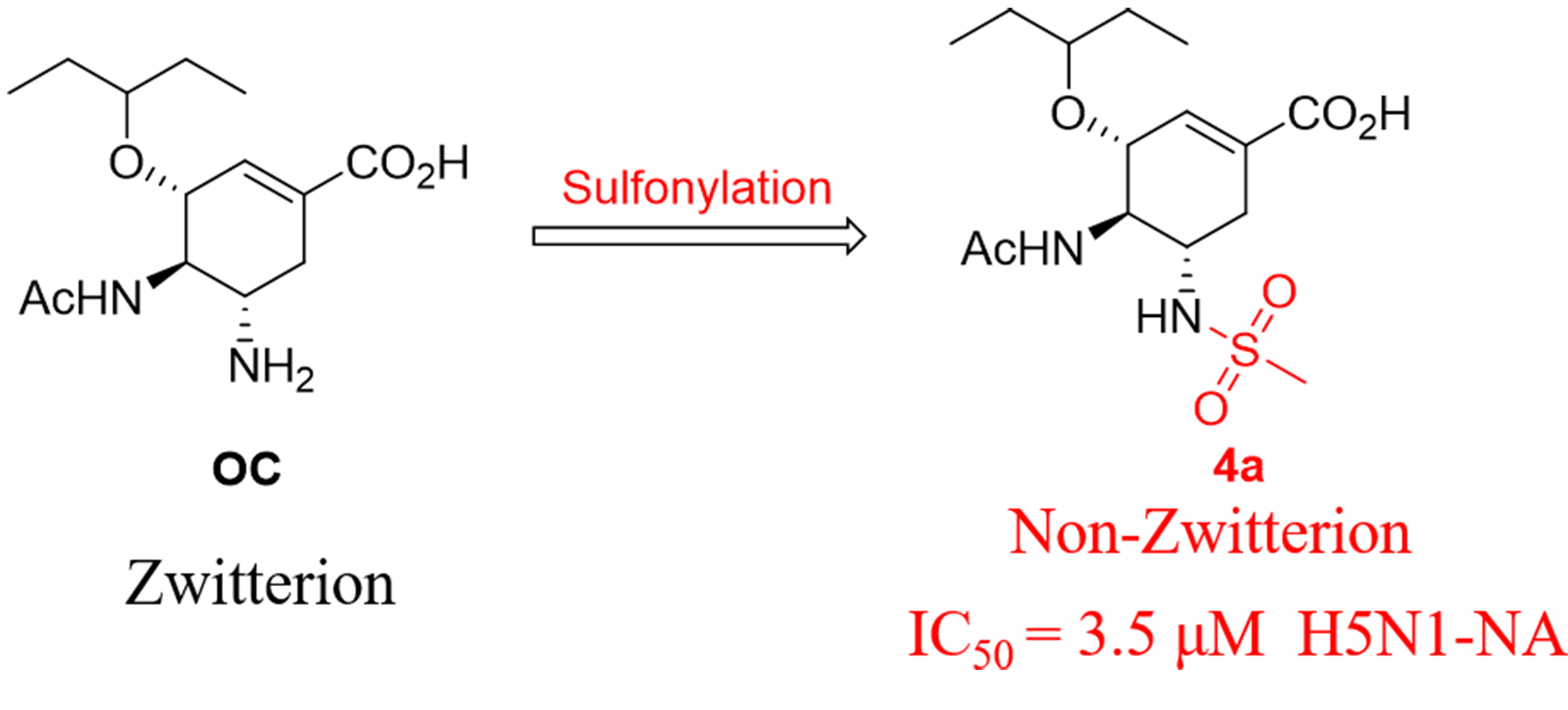
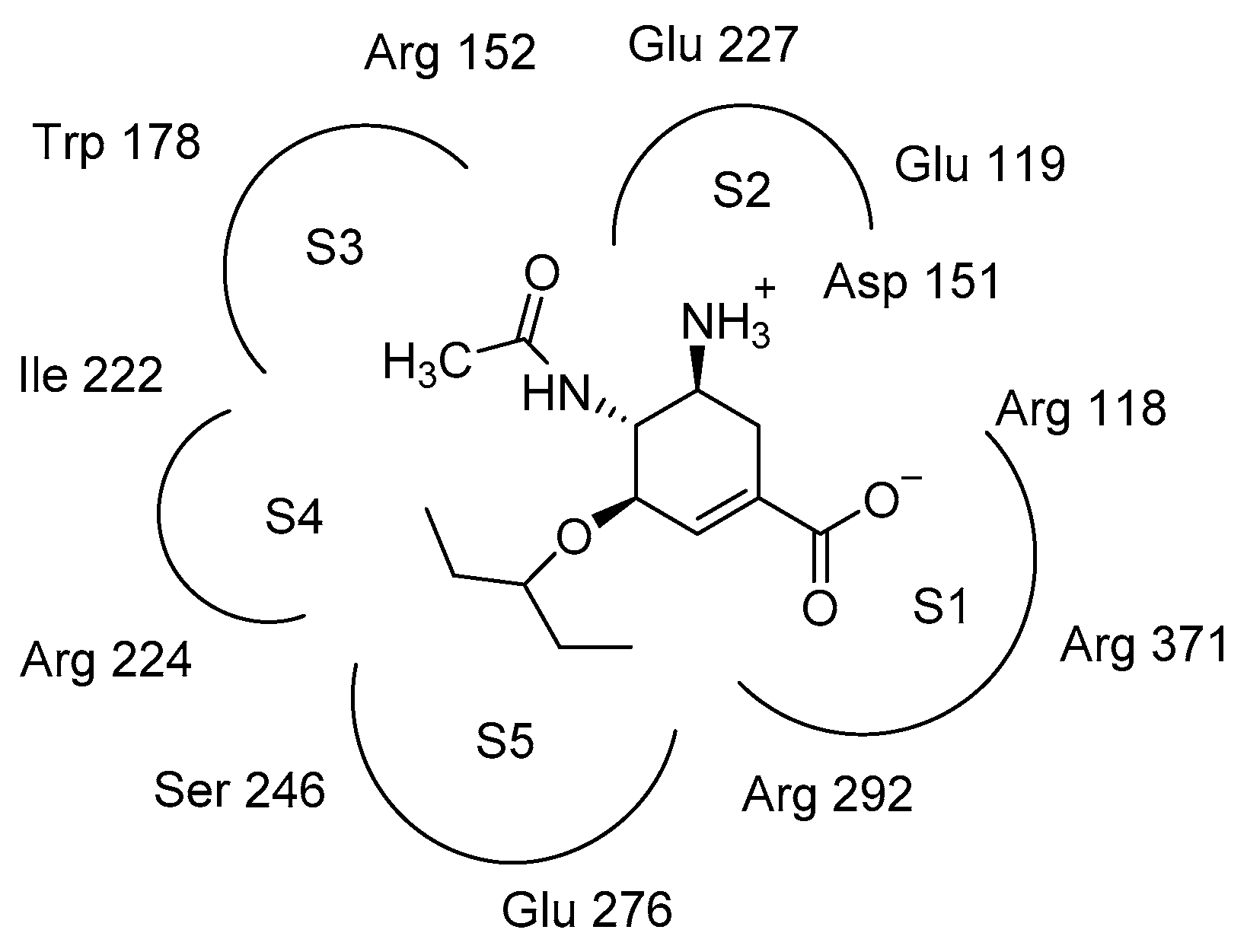
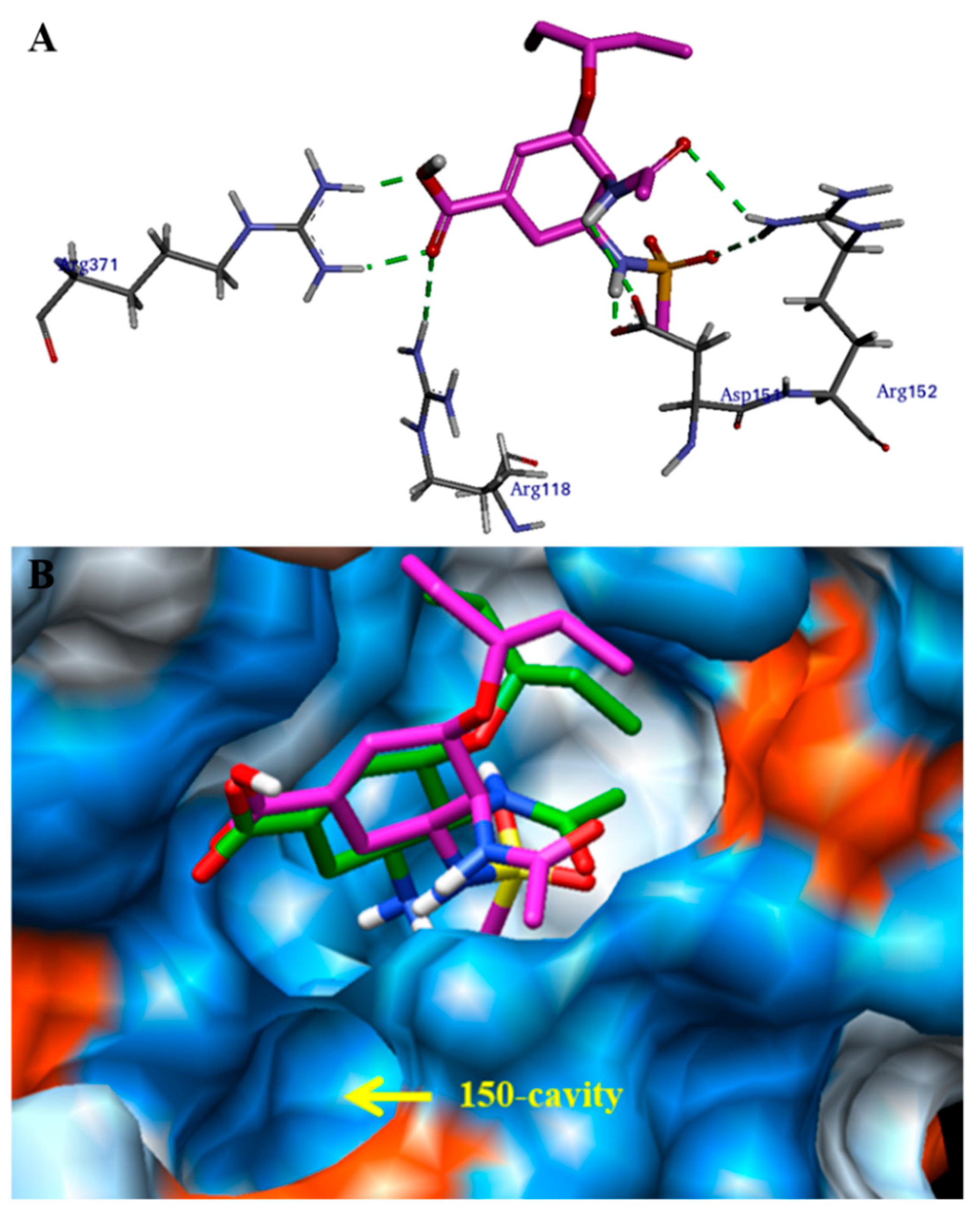
2.3. Molecular Docking Model Analysis
2.4. Metabolic Stability in Human Liver Microsomes In Vitro
3. Materials and Methods
3.1. Chemistry
General Procedure for the Preparation of Compounds 4a–4k and 6i–6k
3.2. Biological Evaluation
3.2.1. Neuraminidase Enzyme Inhibitory Assay
3.2.2. Metabolic Stability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NA | neuraminidase |
| NAs | neuraminidases |
| NAIs | neuraminidase inhibitors |
| OC | oseltamivir carboxylate |
| TEA | triethylamine |
References
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The Mother of all Pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Webster, R.G. The Influenza Virus Enigma. Cell 2009, 136, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Adabala, P.J.P.; LeGresley, E.B.; Bance, N.; Niikura, M.; Pinto, B.M. Exploitation of the Catalytic Site and 150 Cavity for Design of Influenza A Neuraminidase Inhibitors. J. Org. Chem. 2013, 78, 10867–10877. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D. Antiviral agents active against influenza a viruse. Nat. Rev. Drug Discov. 2006, 5, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Kotthaus, J.; Riebling, L.; Kotthaus, J.; Müller-Fielitz, H.; Seidel, N.; Clement, B.; Schade, D.; Raasch, W.; Koch, O.; Schmidtke, M. Development of Novel Potent Orally Bioavailable Oseltamivir Derivatives Active against Resistant Influenza A. J. Med. Chem. 2014, 57, 759–769. [Google Scholar]
- Lew, W.; Chen, X.; Kim, C. Discovery and Development of GS 4104 (oseltamivir) An Orally Active Influenza Neuraminidase Inhibitor. Curr. Med. Chem. 2000, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Das, K. Antivirals Targeting Influenza A Virus. J. Med. Chem. 2012, 55, 6263–6277. [Google Scholar] [CrossRef]
- Ye, D.; Shin, W.-J.; Li, N.; Tang, W.; Feng, E.; Li, J.; He, P.-L.; Zuo, J.-P.; Kim, H.; Nam, K.-Y.; et al. Synthesis of C-4-modified zanamivir analogs as neuraminidase inhibitors and their anti-AIV activities. Eur. J. Med. Chem. 2012, 54, 764–770. [Google Scholar] [CrossRef]
- McLaughlin, M.M.; Skoglund, E.W.; Ison, M.G. Peramivir: An intravenous neuraminidase inhibitor. Expert Opin. Pharmacother. 2015, 16, 1889–1900. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.Y.; Xin, L.; Wei, G.; Xie, X.; Yang, Y.; Zhong, W.; Zheng, A. In Vitro Evaluation of Absorption Characteristics of Peramivir for Oral Delivery. Eur. J. Drug. Metab. Pharmacokinet. 2017, 42, 757–765. [Google Scholar] [CrossRef]
- Toyama, K.; Furuie, H.; Ishizuka, H. Safety and Pharmacokinetics of Nebulized Laninamivir Octanoate, A Long Acting Neuraminidase Inhibitor, In Healthy Subjects. Clin. Ther. 2017, 39, e25–e26. [Google Scholar] [CrossRef][Green Version]
- Mohan, S.; McAtamney, S.; Haselhorst, T.; Von Itzstein, M.; Pinto, B.M. Carbocycles Related to Oseltamivir as Influenza Virus Group-1-Specific Neuraminidase Inhibitors. Binding to N1 Enzymes in the Context of Virus-like Particles. J. Med. Chem. 2010, 53, 7377–7391. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-J.R.; Weinheimer, S.; Tarbet, E.B.; Jan, J.-T.; Cheng, Y.-S.E.; Shie, J.-J.; Chen, C.-L.; Chen, C.-A.; Hsieh, W.-C.; Huang, P.-W.; et al. Development of Oseltamivir Phosphonate Congeners as Anti-Influenza Agents. J. Med. Chem. 2012, 55, 8657–8670. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.J.; Haire, L.F.; Stevens, D.J.; Collins, P.J.; Lin, Y.P.; Blackburn, G.M.; Hay, A.J.; Gamblin, S.J.; Skehel, J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443, 45–49. [Google Scholar] [CrossRef]
- Maring, C.J.; Stoll, V.S.; Zhao, C.; Sun, M.; Krueger, A.C.; Stewart, K.D.; Madigan, D.L.; Kati, W.M.; Xu, Y.; Carrick, R.J.; et al. Structure-Based Characterization and Optimization of Novel Hydrophobic Binding Interactions in a Series of Pyrrolidine Influenza Neuraminidase Inhibitors. J. Med. Chem. 2005, 48, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.R. The Evolution of Synthetic Oral Drug Properties. Bioorg. Med. Chem. Lett. 2005, 15, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xu, D.; Huang, B.; Ma, X.; Qi, W.; Shi, F.; Liu, X.; Zhang, Y.; Xu, W. Discovery of N-Substituted Oseltamivir Derivatives as Potent and Selective Inhibitors of H5N1 Influenza Neuraminidase. J. Med. Chem. 2014, 57, 8445–8458. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, F.; Wang, L.; Liu, K.; Sun, L.; Lin, B.; Hu, Y.; Wang, B.; Cheng, M.; Tian, Y. Synthesis and biological evaluation of NH 2 -acyl oseltamivir analogues as potent neuraminidase inhibitors. Eur. J. Med. Chem. 2017, 141, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Gulçin, I.; Taslimi, P.; Gülçın, I. Sulfonamide inhibitors: A patent review 2013-present. Expert Opin. Ther. Pat. 2018, 28, 541–549. [Google Scholar] [CrossRef]
- Rakesh, K.P.; Wang, S.-M.; Leng, J.; Ravindar, L.; Asiri, A.M.; Marwani, H.M.; Qin, H.-L. Recent development of sulfonyl or sulfonamide hydrids as potential anticancer agents: A key review. Anti-Cancer Agents Med. Chem. 2018, 18, 488–505. [Google Scholar] [CrossRef]
- Magano, J. Synthetic Approaches to the Neuraminidase Inhibitors Zanamivir (Relenza) and Oseltamivir Phosphate (Tamiflu) for the Treatment of Influenza. Chem. Rev. 2009, 109, 4398–4438. [Google Scholar] [CrossRef] [PubMed]
- Shie, J.-J.; Fang, J.-M.; Wang, S.-Y.; Tsai, K.-C.; Cheng, Y.-S.E.; Yang, A.-S.; Hsiao, S.-C.; Su, C.-Y.; Wong, C.-H. Synthesis of Tamiflu and its Phosphonate Congeners Possessing Potent Anti-Influenza Activity. J. Am. Chem. Soc. 2007, 129, 11892–11893. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Maycock, C.D. Formal enantioselective syntheses of oseltamivir and tamiphosphor. Org. Chem. Front. 2017, 4, 236–240. [Google Scholar] [CrossRef]
- Hajzer, V.; Fišera, R.; Latika, A.; Durmis, J.; Kollár, J.; Frecer, V.; Tučeková, Z.; Miertuš, S.; Kostolanský, F.; Varečková, E.; et al. Stereoisomers of oseltamivir—Synthesis, in silico prediction and biological evaluation. Org. Biomol. Chem. 2017, 15, 1828–1841. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhou, D.-G.; He, F.-C.; Chen, J.-X.; Chen, Y.-Z.; Gan, X.-H.; Hu, D.-Y.; Song, B.-A. Synthesis and antiviral bioactivity of novel chalcone derivatives containing purine moiety. Chin. Chem. Lett. 2018, 29, 127–130. [Google Scholar] [CrossRef]
- Wan, Y.; Auberger, N.; Thétiot-Laurent, S.; Bouillère, F.; Zulauf, A.; He, J.; Courtiol-Legourd, S.; Guillot, R.; Kouklovsky, C.; Combes, S.C.D.; et al. Constrained Cyclic β,γ-Diamino Acids from Glutamic Acid: Synthesis of Both Diastereomers and Unexpected Kinetic Resolution. Eur. J. Org. Chem. 2018, 2018, 329–340. [Google Scholar] [CrossRef]
- Shi, D.; Yang, J.; Yang, D.; Lecluyse, E.L.; Black, C.; You, L.; Akhlaghi, F.; Yan, B. Anti-Influenza Prodrug Oseltamivir Is Activated by Carboxylesterase Human Carboxylesterase 1, and the Activation Is Inhibited by Antiplatelet Agent Clopidogrel. J. Pharmacol. Exp. Ther. 2006, 319, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Sweeny, D.J.; Lynch, G.; Bidgood, A.M.; Lew, W.; Wang, K.Y.; Cundy, K.C. Metabolism of the influenza neuraminidase inhibitor prodrug oseltamivir in the rat. Drug Metab. Dispos. 2000, 28, 737–741. [Google Scholar] [PubMed]
- Sławiński, J.; Szafrański, K.; Pogorzelska, A.; Żołnowska, B.; Kawiak, A.; Macur, K.; Belka, M.; Bączek, T. Novel 2-benzylthio-5-(1,3,4-oxadiazol-2-yl)benzenesulfonamides with anticancer activity: Synthesis, QSAR study, and metabolic stability. Eur. J. Med. Chem. 2017, 132, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bai, L.; Liu, L.; McEachern, D.; Stuckey, J.A.; Meagher, J.L.; Yang, C.-Y.; Ran, X.; Zhou, B.; Hu, Y.; et al. Structure-Based Discovery of 4-(6-Methoxy-2-methyl-4-(quinolin-4-yl)-9 H -pyrimido[4,5- b ]indol-7-yl)-3,5-dimethylisoxazole (CD161) as a Potent and Orally Bioavailable BET Bromodomain Inhibitor. J. Med. Chem. 2017, 60, 3887–3901. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 4a–4k, 6i–6k are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | 10 μM | 100 μM | Compounds | 10 μM | 100 μM |
|---|---|---|---|---|---|
| 4a | 73.9% | 91.8% | 4h | 40.4% | 79.2% |
| 4b | 50.3% | 85.1% | 4i | 32.0% | 70.1% |
| 4c | 43.2% | 56.5% | 4j | 20.3% | 33.8% |
| 4d | 30.3% | 53.5% | 4k | 0.5% | 38.9% |
| 4e | 28.5% | 50.5% | 6i | 63.8% | 86.6% |
| 4f | 24.8% | 49.7% | 6j | 37.3% | 52.7% |
| 4g | NDb | ND | 6k | ND | 12.5% |
| OC | 91.0% | 95.0% |
| Compounds | OC | 4a | 4h | 4i | 6i |
|---|---|---|---|---|---|
| IC50/μM | 0.21 ± 0.021 | 3.50 ± 0.17 | 12.00 ± 2.49 | 20.74 ± 1.14 | 8.50 ± 0.63 |
| Compounds | Microsomal Stability T1/2 (min) | Remaining (T = 60 min) |
|---|---|---|
| 4a | >145 | 101.6% |
| Testosterone | 12.5 | 3.8% |
| Diclofenac | 9.2 | 1.1% |
| Propafenone | 5.7 | 0.1% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Chen, B.; Lei, Z.; Zhao, H.; Zhu, H.; Quan, P.; Tian, Y. Synthesis and Biological Evaluation of NH2-Sulfonyl Oseltamivir Analogues as Influenza Neuraminidase Inhibitors. Molecules 2019, 24, 2176. https://doi.org/10.3390/molecules24112176
Hu Y, Chen B, Lei Z, Zhao H, Zhu H, Quan P, Tian Y. Synthesis and Biological Evaluation of NH2-Sulfonyl Oseltamivir Analogues as Influenza Neuraminidase Inhibitors. Molecules. 2019; 24(11):2176. https://doi.org/10.3390/molecules24112176
Chicago/Turabian StyleHu, Yaping, Binfeng Chen, Zaiqiang Lei, Hongqian Zhao, Hongxi Zhu, Peng Quan, and Yongshou Tian. 2019. "Synthesis and Biological Evaluation of NH2-Sulfonyl Oseltamivir Analogues as Influenza Neuraminidase Inhibitors" Molecules 24, no. 11: 2176. https://doi.org/10.3390/molecules24112176
APA StyleHu, Y., Chen, B., Lei, Z., Zhao, H., Zhu, H., Quan, P., & Tian, Y. (2019). Synthesis and Biological Evaluation of NH2-Sulfonyl Oseltamivir Analogues as Influenza Neuraminidase Inhibitors. Molecules, 24(11), 2176. https://doi.org/10.3390/molecules24112176