Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives

 ,
,
Abstract
1. Introduction

2. Results and Discussion
2.1. Chemistry
2.2. Biological Study
2.2.1. Antibacterial Activity
2.2.2. Anti-Proliferative Activity Screening
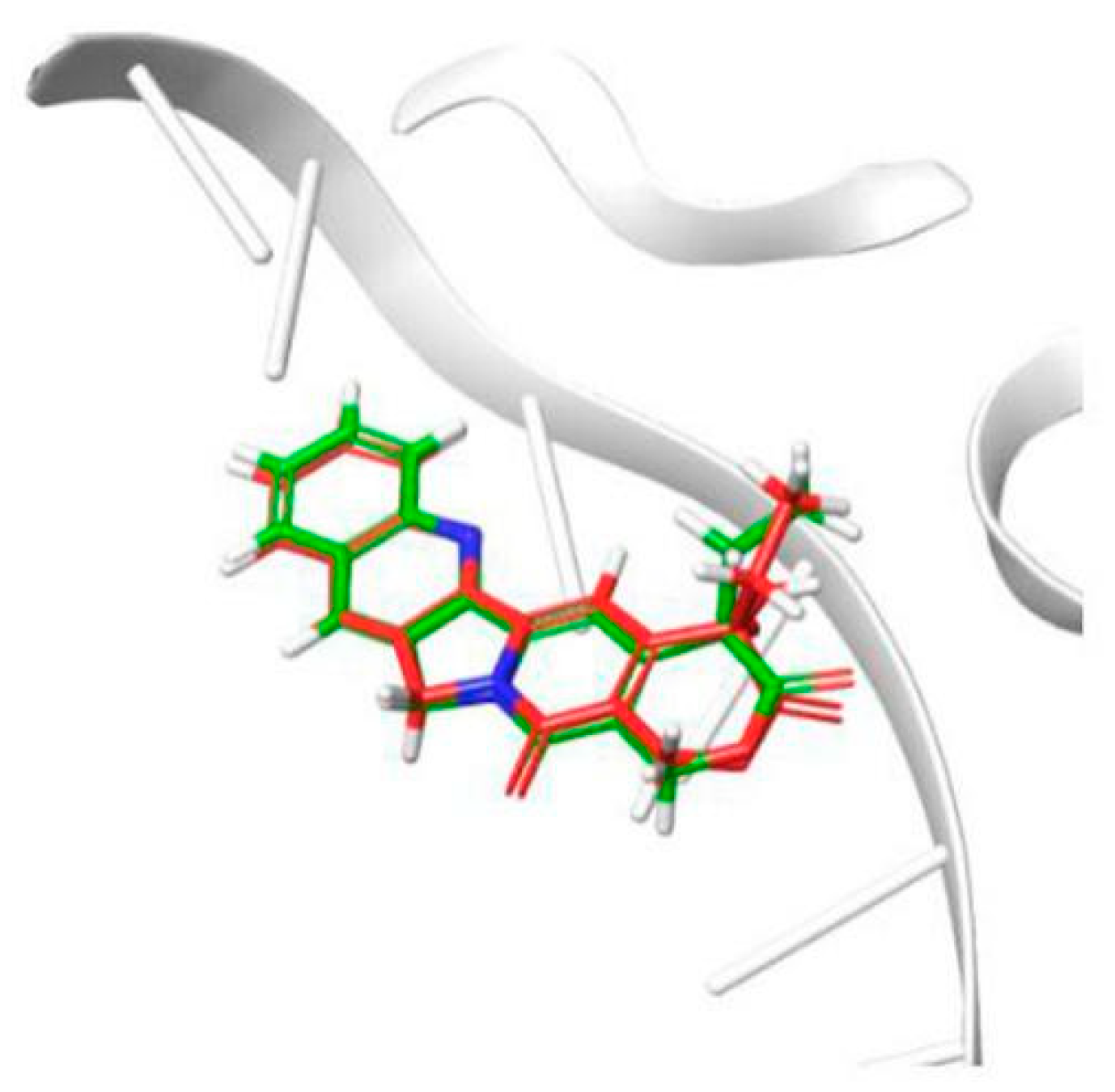
2.2.3. Evaluation of Topo I Inhibitory Activity and Molecular Docking Study
2.2.4. Apoptosis and Cell-Cycle Analysis
3. Materials and Methods
3.1. Synthesis Methods
3.1.1. Synthesis of N-naphthyl-o-aminobenzoic Acid (1)
3.1.2. Synthesis of 7-chlorine benz[c]acridine (2)
3.1.3. Synthesis of 7-benz[c]acridine Isothiocyanate (3)
3.1.4. General Procedure for the Synthesis of 1-Aryl-4-(7-benz[c]acridinyl) Thiosemicarbazides Derivatives 4a–e
- 1-Pyridyl-4-(7-benz[c]acridinyl) Thiosemicarbazides (4a): Orange-yellow powder, yield 46.7%, m.p. 177.4–180.9 °C.
- 1-(4-chlorin-phenyl)-4-(7-benz[c]acridinyl) Thiosemicarbazides (4b): Orange-yellow powder, yield 34.0%, m.p. 203.3–203.9 °C.
- 1-(4-nitro-phenyl)-4-(7-benz[c]acridinyl) Thiosemicarbazides (4c): Orange-red powder, yield 31.0%, m.p. 238.7–240.1 °C.
- 1-(4-methoxy-phenyl)-4-(7-benz[c]acridinyl) Thiosemicarbazides (4d): Orange-yellow powder, yield 68.2%, m.p. 184.2–187.4 °C.
- 1-phenyl-4-(7-benz[c]acridinyl) Thiosemicarbazides (4e): Orange-yellow powder, yield 58.6%, m.p. 181.4–182.9 °C
3.2. Biological Activity
3.2.1. Antimicrobial Activity
3.2.2. Antiproliferative Activity
MTS Assay
3.3. Topo I Inhibitory Activity
3.4. Molecular Docking
3.5. General Procedure for AO/EB Staining
3.6. General Procedure for Apoptosis Ratio Determination
3.7. General Procedure Cell Cycle
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lang, X.; Luan, X.; Gao, C.; Jiang, Y. Recent Progress of Acridine Derivatives with Antitumor Activity. Prog. Chem. 2012, 24, 1497–1505. [Google Scholar]
- Wang, S.S.; Lee, Y.J.; Hsu, S.C.; Chang, H.O.; Yin, W.K.; Chang, L.S.; Chou, S.Y. Linker-modified triamine-linked acridine dimers: Synthesis and cytotoxicity properties in vitro and in vivo. Bioorganic Med. Chem. 2007, 15, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.; Reches, R.; Caignard, D.H.; Renard, P.; Pujol, M.D. Synthesis andbiological evaluation ofmodified acridines: The effect of N- and O- substituent inthenitrogenated ring onantitumor activity. Cheminform 2006, 41, 340–352. [Google Scholar]
- Bacherikov, V.A.; Chang, J.Y.; Lin, Y.W.; Chen, C.H.; Pan, W.Y.; Dong, H.; Lee, R.Z.; Chou, T.C.; Su, T.L. Synthesis and antitumor activity of 5-(9-acridinylamino) anisidine derivatives. Bioorganic Med. Chem. 2005, 23, 6513–6520. [Google Scholar] [CrossRef]
- Vispé, S.; Vandenberghe, I.; Robin, M.; Annereau, J.P.; Créancier, L.; Pique, V.; Galy, J.P.; Kruczynski, A.; Barret, J.M.; Bailly, C. Novel tetra-acridine derivatives as dual inhibitors of topoisomerase II and the human proteasome. Biochem. Pharmacol. 2007, 73, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.; Bosson, J.; Godet, T.; Tiano, M. Acridine and Acridone Derivatives, Anticancer Properties and Synthetic Methods: Where Are We Now? Anti-Cancer Agents Med. Chem. 2007, 2, 139–169. [Google Scholar] [CrossRef]
- Oppegard, L.M.; Ougolkov, A.V.; Luchini, D.N.; Schoon, R.A.; Goodell, J.R.; Kaur, H.; Billadeau, D.D.; Ferguson, D.M.; Hiasaa, H. Novel acridine-based compounds that exhibit an anti-pancreatic cancer activity are catalytic inhibitors of human topoisomerase II. Eur. J. Pharmacol. 2009, 602, 223–229. [Google Scholar] [CrossRef]
- Cain, B.F.; Atwell, G.J.; Denny, W.A. Potential antitumor agents 17. 9-Anilino-10-Methylacridinium Salts. J. Med. Chem. 1976, 19, 772–777. [Google Scholar] [CrossRef]
- Kimura, M.; Okabayashi, I.; Kato, A. Acridine derivatives. III. Preparation and antitumor activity of the novel acridinyl-substituted uracils. Chem. Pharm. Bull. 1989, 37, 697. [Google Scholar] [CrossRef]
- Gamage, S.A.; Spicer, J.A.; Rewcastle, G.W.; Denny, W.A. A new synthesis of substituted acridine-4-carboxylic acids and the anticancer drug N-[2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA). Tetrahedron Lett. 1997, 38, 699–702. [Google Scholar] [CrossRef]
- Abbas, S.Y.; Elsharief, M.A.; Basyouni, W.M.; Fakhr, I.M.; Elgammal, E.W. Thiourea derivatives incorporating a hippuric acid moiety: Synthesis and evaluation of antibacterial and antifungal activities. Eur. J. Med. Chem. 2013, 64, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Rashid, N.; Jones, P.G.; Ali, M.; Hussain, R. Synthesis, characterization and biological evaluation of some thiourea derivatives bearing benzothiazole moiety as potential antimicrobial and anticancer agents. Eur. J. Med. Chem. 2010, 45, 1323–1331. [Google Scholar] [CrossRef]
- Zhong, Z.; Xing, R.; Liu, S.; Wang, L.; Cai, S.; Li, P. Synthesis of acyl thiourea derivatives of chitosan and their antimicrobial activities in vitro. Carbohydr. Res. 2008, 343, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.P.; Wu, Y.; Song, Z.Q.; Shang, S.B.; Wang, Z.D. Synthesis and antitumor activities of unsymmetrically disubstituted acylthioureas fused with hydrophenanthrene structure. Med. Chem. Res. 2011, 20, 333–338. [Google Scholar] [CrossRef]
- Burgeson, J.R.; Moore, A.L.; Boutilier, J.K.; Cerruti, N.R.; Gharaibeh, D.N.; Lovejoy, C.E.; Amberg, S.M.; Hruby, D.E.; Tyavanagimatt, S.R.; AllenIII, R.D. SAR analysis of a series of acylthiourea derivatives possessing broad-spectrum antiviral activity. Bioorganic Med. Chem. Lett. 2012, 22, 4263–4272. [Google Scholar] [CrossRef] [PubMed]
- Limban, C.; Vasile, A.; Chirita, I.C.; Caproiu, M. Preparation of new thiourea derivatives with potential anti-parasitic and antimicrobial activity. Rev. De Chim. 2010, 61, 946–950. [Google Scholar]
- Burgess, S.J.; Audrey, S.; Jane Xu, K.; Smilkstein, M.J.; Riscoe, M.K.; Peyton, D.H. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 2006, 49, 5623. [Google Scholar] [CrossRef]
- Solinas, A.; Faure, H.; Roudaut, H.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Manetti, F.; Taddei, M.; Ruat, M. Acylthiourea, acylurea, and acylguanidine derivatives with potent hedgehog inhibiting activity. J. Med. Chem. 2012, 55, 1559–1571. [Google Scholar] [CrossRef]
- Pommier, Y. Diversity of DNA topoisomerases I and inhibitors. Biochimie 1998, 80, 255–270. [Google Scholar] [CrossRef]
- Zhao, L.X.; Moon, Y.S.; Basnet, A.; Kim, E.K.; Jahng, Y.; Park, J.G.; Jeong, T.C.; Cho, W.J.; Choi, S.U.; Chong, O.L. Synthesis, topoisomerase I inhibition and structure–activity relationship study of 2,4,6-trisubstituted pyridine derivatives. Bioorganic Med. Chem. Lett. 2004, 14, 1333–1337. [Google Scholar] [CrossRef]
- Lill, M. Virtual screening in drug design. Silico Models Drug Discov. 2013, 993, 1–12. [Google Scholar]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Modeling 2009, 2, 444–460. [Google Scholar] [CrossRef]
- Maddika, S.; RaoAnde, S.; Panigrahi, S.; Paranjothy, T.; Weglarczyk, K.; Zuse, A.; Eshraghi, M.; Manda, K.D.; Wiechec, E.; Los, M. Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resist. Updates 2007, 10, 13–29. [Google Scholar] [CrossRef]
- Bunz, F. Cell death and cancer therapy. Curr. Opin. Pharmacol. 2001, 1, 337–341. [Google Scholar] [CrossRef]
- Liu, K.; Liu, P.; Liu, R.; Wu, X. Dual AO/EB staining to detect apoptosis in osteosarcoma cells compared with flow cytometry. Med. Sci. Monit. Basic Res. 2015, 21, 15–20. [Google Scholar] [PubMed]
- Huang, X.C.; Wang, M.; Wang, H.S.; Chen, Z.F.; Zhang, Y.; Pan, Y.M. Synthesis and antitumor activities of novel dipeptide derivatives derived from dehydroabietic acid. Bioorganic Med. Chem. Lett. 2014, 24, 1511–1518. [Google Scholar] [CrossRef]
- Cappuccino, J.; Sherman, N. Microbiology: A Laboratory Manual, 4th ed.; Addison Wesley Longman Inc.: Reading, MA, USA, 1999; p. 263. [Google Scholar]
- Ministry of Health & Family welfare, Government of india. Indian Pharmacopoeia 2007, 5th ed.; The Indian Pharmacopoeia Commission: Ghaziabad, India, 2007; Volume 1, p. 37.
- Jin, Y.; Chen, Q.; Shi, X.; Lu, Z.; Cheng, C.; Lai, Y.; Zheng, Q.; Pan, J. Activity of triptolide against human mast cells harboring the kinase domain mutant KIT. Cancer Sci. 2009, 100, 1335–1343. [Google Scholar] [CrossRef]
- Xu, Y.; Jing, D.; Chen, R.; Haroon, U.R.; Jiang, J.; Liu, X.; Wang, L.; Wang, P. Design, synthesis and evaluation of novel sophoridinic imine derivatives containing conjugated planar structure as potent anticancer agents. Bioorgnic Med. Chem. 2018, 26, 4136–4144. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 4a–4e are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Ar | Yield (%) | 1H NMR (DMSO-d6) δ ppm (J in Hz) | 13C NMR (DMSO-d6) | ESI-MS: m/z: [(M − H)]− | Melting Point (°C) |
|---|---|---|---|---|---|---|
| 4a |  | 46.7% | 11.36 (s, 1H, -NH), 10.55 (s, 1H, -NH), 10.31 (s, 1H, -NH), 9.38~9.41 (m, 1H, ArH), 8.81 (d, 2H, J = 4.4Hz, ArH), 8.33 (d, 1H, J = 8.8Hz, ArH), 8.21 (d, 1H, J = 8.4Hz, ArH), 8.04 (t, 1H, ArH), 7.90~7.97 (m, 5H, ArH), 7.82~7.84 (m, 2H, ArH), 7.73 (t, 1H, ArH) | 183.36, 165.46, 150.71, 147.98, 147.88, 142.04, 139.99, 133.88, 131.24, 130.68, 129.99, 129.83, 128.65, 128.09, 128.01, 126.80, 125.34, 125.11, 124.94, 123.22, 122.51, 122.32. | 422 | 177.4–180.9 °C |
| 4b |  | 34.0% | 11.09 (s, 1H, -NH), 10.52 (s, 1H, -NH), 10.23 (s, 1H, -NH), 9.40 (t, 1H, ArH), 8.33 (d, 1H, J = 8.4Hz, ArH), 8.22 (d, 1H, J = 8.8Hz, ArH), 8.03~8.09 (m, 3H, ArH), 7.90~7.97 (m,3H, ArH), 7.82~7.84 (m, 2H, ArH), 7.73 (t, 1H, ArH), 7.64 (d, 2H, J = 8.4, ArH) | 183.49, 165.96, 147.98, 147.87, 142.22, 137.27, 133.89, 131.78, 131.26, 130.62, 130.47,129.95, 129.79, 128.85, 128.62, 127.96, 126.71, 125.39, 125.11, 125.02, 123.25, 122.61. | 455 | 203.3–203.9 °C |
| 4c |  | 31.0% | 11.32 (s, 1H, -NH), 10.57 (s, 1H, -NH), 10.32 (s, 1H, -NH), 9.40 (t, 1H, ArH), 8.40 (d, 2H, J = 8.0Hz, ArH), 8.34 (d, 1H, J = 8.8Hz, ArH), 8.28 (d, 2H, J = 8.4Hz, ArH), 8.21 (d, 1H, J = 8.4Hz, ArH), 8.05 (t, 1H, ArH), 7.91~7.97 (m, 3H, ArH), 7.82~7.84 (m, 2H, ArH), 7.74 (t, 1H, ArH) | 183.29, 165.42, 149.88, 147.99, 147.88, 142.09, 138.76, 133.89, 131.25, 130.68, 130.05, 129.98, 129.84, 128.65, 128.09, 128.00, 126.80, 125.37, 125.12, 124.95, 123.93, 123.24, 122.52, | 466 | 238.7–240.1 °C |
| 4d |  | 68.2% | 10.82 (s, 1H, -NH), 10.49 (s, 1H, -NH), 10.15 (s, 1H, -NH), 9.40 (t, 1H, ArH), 8.33 (d, 1H, J = 8.8Hz, ArH), 8.23 (d, 1H, J = 8.4Hz, ArH), 7.90~8.05 (m, 6H, ArH), 7.90~7.93 (m, 2H, ArH), 7.82~7.84 (m, 1H, ArH), 7.73 (t, 2H, ArH), 3.83 (s, 3H, OCH3). | 183.60, 166.39, 147.99, 142.22, 147.86, 142.41, 133.90, 131.27, 130.58, 130.47, 129.92, 128.77, 128.61, 127.93, 126.64, 126.63, 125.45, 125.11, 123.28, 122.72, 113.97, 55.92. | 451 | 184.2–187.4 °C |
| 4e |  | 58.6% | 11.09 (s, 1H, -NH), 10.52 (s, 1H, -NH), 10.19 (s, 1H, -NH), 9.40 (dd, J = 6.1, 3.5 Hz, 1H, ArH), 8.32 (d, J = 8.6 Hz, 1H, ArH), 8.25 (d, J = 8.5 Hz, 1H, ArH), 8.10 (d, J = 7.7 Hz, 2H, ArH), 8.04 (dd, J = 6.0, 3.2 Hz, 1H, ArH), 7.99 (d, J = 9.3 Hz, 1H, ArH), 7.95~7.88 (m, 2H, ArH), 7.83 (dt, J = 6.1, 3.6 Hz, 2H, ArH), 7.72 (t, J = 7.6 Hz, 1H, ArH), 7.60 (d, J = 7.2 Hz, 1H, ArH), 7.54 (t, J = 7.5 Hz, 2H, ArH). | 183.51, 166.82, 147.98, 147.86, 142.39, 133.90, 132.93, 131.26, 130.59, 129.92, 129.75, 128.71, 128.61, 127.93, 126.64, 125.43, 125.09, 123.26, 123.26, 122.74 | 421 | 181.4–182.9 °C |
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | Antifungal Activity | ||
|---|---|---|---|---|---|
| Staphylococcus aureus | Shigella Castellani | Escherichia coli | Pseudomonas aeruginosa | Candida albicans | |
| 4a | 10 | >100 | >100 | 10 | >100 |
| 4b | 40 | 20 | >100 | 20 | 80 |
| 4c | 10 | 10 | 20 | 10 | 10 |
| 4d | >100 | >100 | >100 | >100 | 40 |
| 4e | 40 | 80 | >100 | 20 | 20 |
| Streptomycin | 2 | 2 | 2 | 2 | 2 |
| Compounds | HL-60 | MT-4 | Hela | HepG2 | MCF-7 |
|---|---|---|---|---|---|
| 4a | >100 | 18.42 ± 1.18 | 30.93 ± 1.78 | 32.96 ± 0.81 | >100 |
| 4b | >100 | 15.73 ± 0.90 | 45.68 ± 2.14 | 54.80 ± 2.95 | >100 |
| 4c | >100 | 57.59 ± 2.37 | 59.39 ± 2.56 | 51.60 ± 2.46 | >100 |
| 4d | 59.56 ± 2.54 | 10.96 ± 0.62 | 42.04 ± 2.07 | 29.05 ± 1.87 | >100 |
| 4e | 70.19 ± 3.72 | 11.63 ± 0.11 | 68.18 ± 3.92 | 58.99 ± 3.11 | >100 |
| cisplatin | 3.66 ± 0.14 | 5.99 ± 0.12 | 6.65 ± 0.12 | 14.45 ± 0.49 | 25.92 ± 1.68 |
| Compound | Amino Acid | Type | Hydrophobic Residue | Binding Affinity (Total Score) |
|---|---|---|---|---|
| 4a | TGP11 | π-π | DA113, ARG364, TGP11, DT10, DT9, DC112 | 8.82 |
| DA113 | π-π | |||
| DC112 | π-π | |||
| ASN722 | Hydrogen Bond | |||
| 4b | TGP11 | π-π | ARG364, DC112, TGP11, DT10 | 7.82 |
| DA113 | π-π | |||
| DT10 | π-π | |||
| DC112 | π-π | |||
| DA113 | Hydrogen Bond | |||
| 4c | TGP11 | π-π | TGP11, THR718, DT10,DT9, DA113, DC112 | 8.32 |
| DA113 | π-π | |||
| DC112 | π-π | |||
| ARG364 | Hydrogen Bond | |||
| 4d | TGP11 | π-π | ARG364, DT10, DA113, ASN352, TRP416, LYS374, GLU418, DC112, TGP11 | 10.31 |
| DA113 | π-π | |||
| DC112 | π-π | |||
| LYS452 | Hydrogen Bond | |||
| GLU356 | Hydrogen Bond | |||
| 4e | TGP11 | π-π | DT10, TGP11, ARG364, DC112, TRP416, GLU418, ASN352,DA113 | 9.62 |
| DA113 | π-π | |||
| DC112 | π-π | |||
| LYS425 | Hydrogen Bond | |||
| GLU356 | Hydrogen Bond | |||
| CPT | TGP11 | π-π | DC112, DA113, TGP11, DT10, DG12 | 10.27 |
| DA113 | π-π | |||
| DC112 | π-π | |||
| ASP533 | Hydrogen Bond | |||
| ARP364 | Hydrogen Bond |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Huo, L.; Jaiswal, Y.; Huang, J.; Zhong, Z.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y.; Yan, Z. Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules 2019, 24, 2065. https://doi.org/10.3390/molecules24112065
Chen R, Huo L, Jaiswal Y, Huang J, Zhong Z, Zhong J, Williams L, Xia X, Liang Y, Yan Z. Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules. 2019; 24(11):2065. https://doi.org/10.3390/molecules24112065
Chicago/Turabian StyleChen, Rui, Lini Huo, Yogini Jaiswal, Jiayong Huang, Zhenguo Zhong, Jing Zhong, Leonard Williams, Xing Xia, Yan Liang, and Zhenshuo Yan. 2019. "Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives" Molecules 24, no. 11: 2065. https://doi.org/10.3390/molecules24112065
APA StyleChen, R., Huo, L., Jaiswal, Y., Huang, J., Zhong, Z., Zhong, J., Williams, L., Xia, X., Liang, Y., & Yan, Z. (2019). Design, Synthesis, Antimicrobial, and Anticancer Activities of Acridine Thiosemicarbazides Derivatives. Molecules, 24(11), 2065. https://doi.org/10.3390/molecules24112065