Novel Nucleic Acid Binding Small Molecules Discovered Using DNA-Encoded Chemistry

Abstract
1. Introduction
2. Results
2.1. DNA-Encoded Chemical Libraries
2.2. Oligonucleotide Synthesis
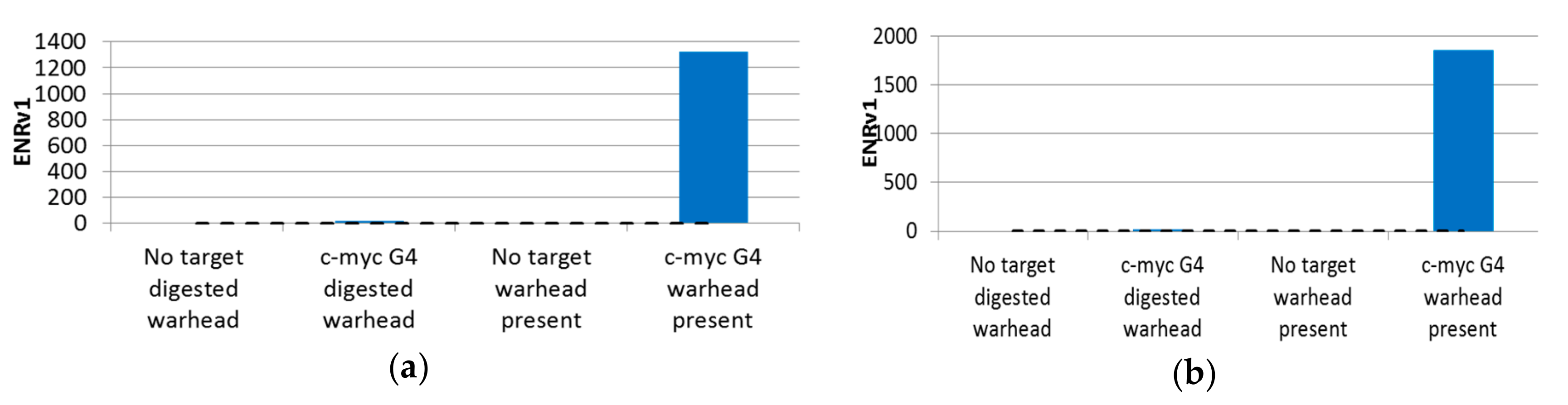
2.3. Affinity-Mediated Selection
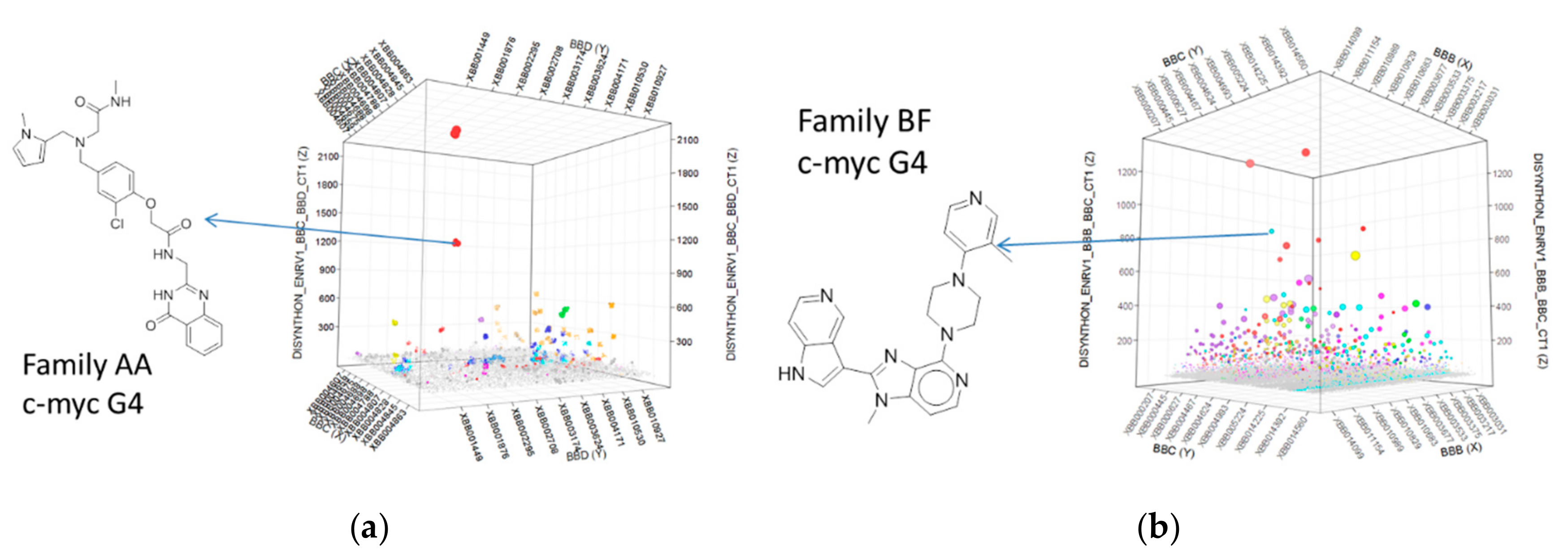
2.4. Selection Output Analysis
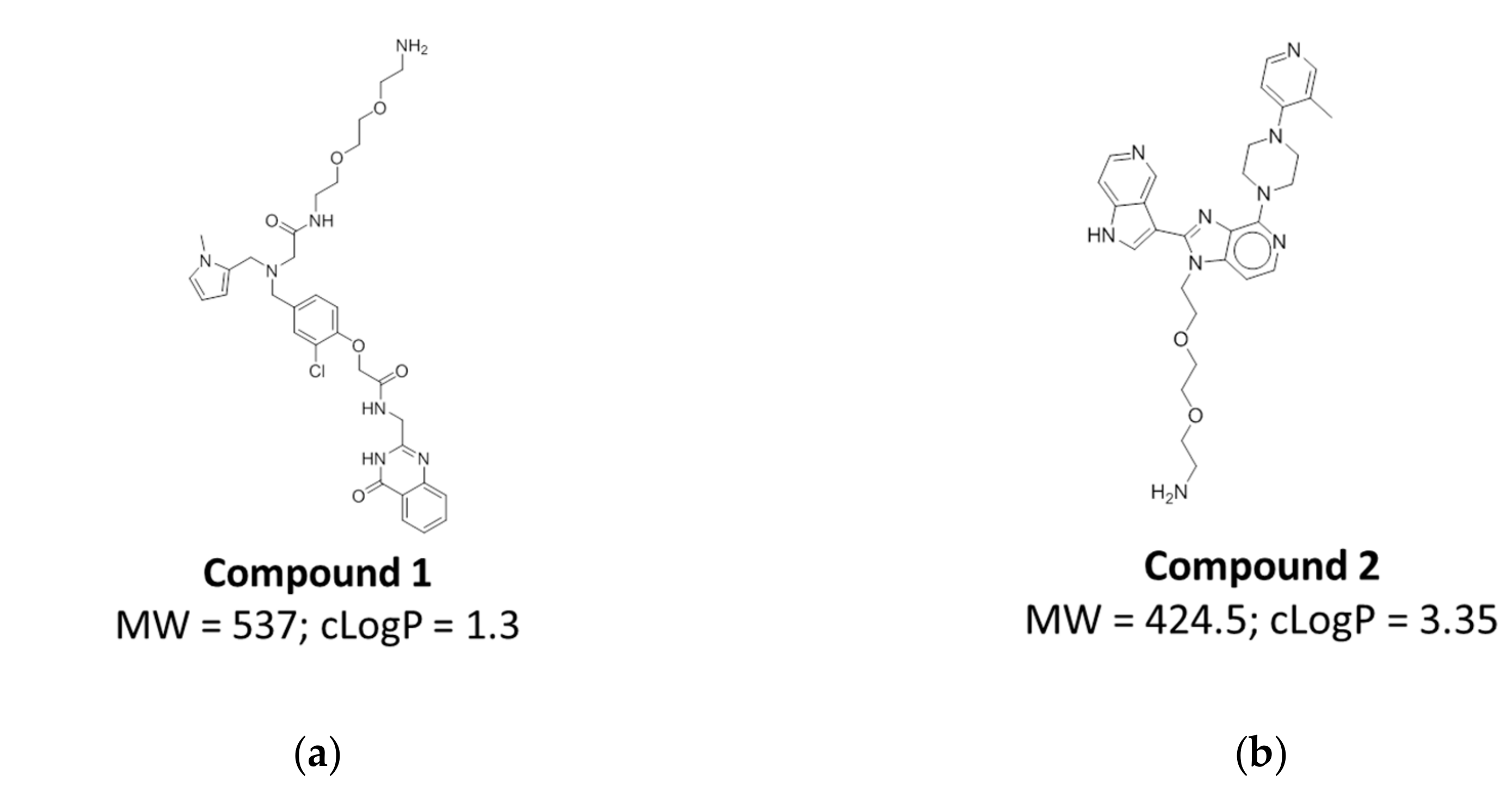
2.5. Compound Design and Synthesis
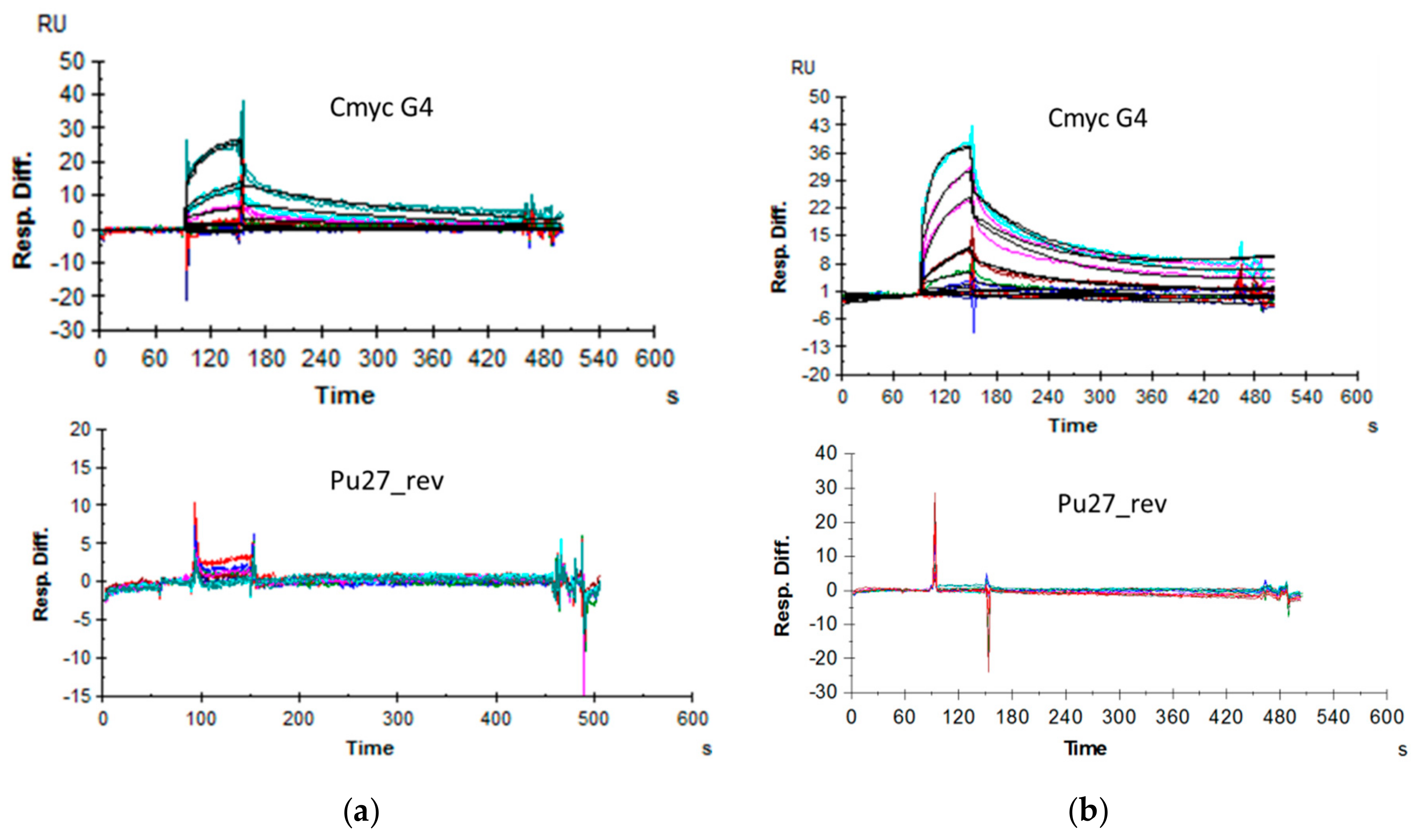
2.6. Affinity Assessment Using Surface Plasmon Resonance
2.7. PCR-Stop Assay
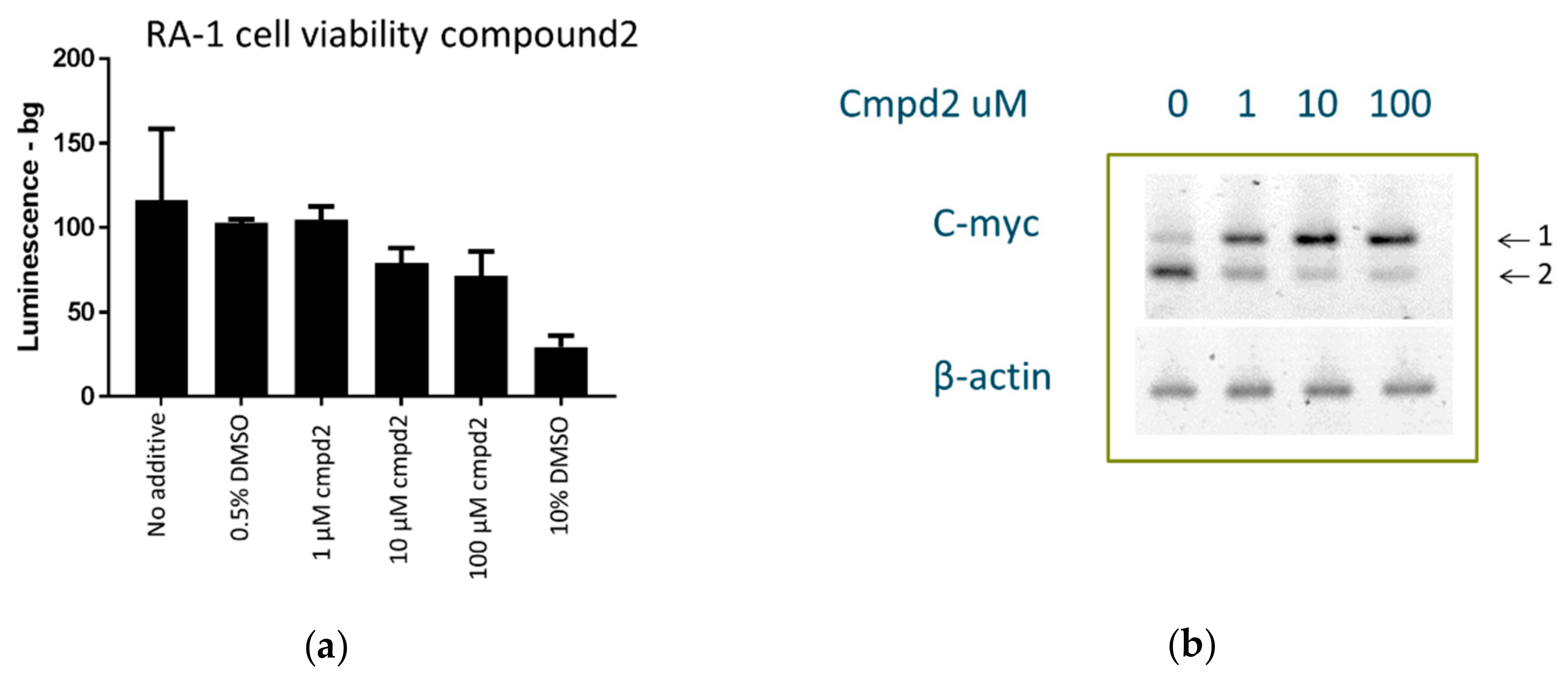
2.8. Cell Culture Experiments
3. Discussion
4. Materials and Methods
4.1. Library Synthesis
4.2. Oligonucleotide Synthesis
4.3. Affinity-Mediated Selection
4.4. Surface Plasmon Resonance Experiments
4.4.1. Surface Plasmon Resonance
Compound Biotinylation
Affinity Assessment through SPR
4.5. PCR-Stop Assay
4.6. Cell Viability Assay
4.7. RT-PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Goodnow, R.A., Jr.; Dumelin, C.E.; Keefe, A.D. DNA-encoded chemistry: Enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2016, 16, 131. [Google Scholar] [CrossRef]
- Zimmermann, G.; Neri, D. DNA-encoded chemical libraries: Foundations and applications in lead discovery. Drug Discov. Today 2016, 21, 1828–1834. [Google Scholar] [PubMed]
- Cuozzo, J.W.; Centrella, P.A.; Gikunju, D.; Habeshian, S.; Hupp, C.D.; Keefe, A.D.; Sigel, E.A.; Soutter, H.H.; Thomson, H.A.; Zhang, Y.; et al. Discovery of a Potent BTK Inhibitor with a Novel Binding Mode by Using Parallel Selections with a DNA Encoded Chemical Library. Chem. Biochem. 2017, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Brown, G.A.; Centrella, P.; Certel, K.; Cooke, R.M.; Cuozzo, J.W.; Dekker, N.; Dumelin, C.E.; Ferguson, A.; Fiez-Vandal, C.; et al. Agonists and Antagonists of Protease-Activated Receptor 2 Discovered within a DNA-Encoded Chemical Library Using Mutational Stabilization of the Target. SLAS Discov. 2018, 23, 429–436. [Google Scholar] [CrossRef]
- Litovchick, A.; Dumelin, C.E.; Habeshian, S.; Gikunju, D.; Guie, M.A.; Centrella, P.; Zhang, Y.; Sigel, E.A.; Cuozzo, J.W.; Keefe, A.D.; et al. Encoded Library Synthesis Using Chemical Ligation and the Discovery of sEH Inhibitors from a 334-Million Member Library. Sci. Rep. 2015, 5, 10916. [Google Scholar]
- Soutter, H.H.; Centrella, P.; Clark, M.A.; Cuozzo, J.W.; Dumelin, C.E.; Guie, M.A.; Habeshian, S.; Keefe, A.D.; Kennedy, K.M.; Sigel, E.A.; et al. Discovery of cofactor-specific, bactericidal Mycobacterium tuberculosis InhA inhibitors using DNA-encoded library technology. Proc. Natl. Acad. Sci. USA 2016, 113, E7880–E7889. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Montalván, A.E.; Berger, M.; Kuropka, B.; Koo, S.J.; Badock, V.; Weiske, J.; Puetter, V.; Holton, S.J.; Stöckigt, D.; Ter Laak, A.; et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem. Biol. 2017, 12, 2730–2736. [Google Scholar] [CrossRef]
- Johannes, J.W.; Bates, S.; Beigie, C.; Belmonte, M.A.; Breen, J.; Cao, S.; Centrella, P.A.; Clark, M.A.; Cuozzo, J.W.; Dumelin, C.E.; et al. Structure Based Design of Non-Natural Peptidic Macrocyclic Mcl-1 Inhibitors. ACS Med. Chem. Lett. 2016, 8, 239–244. [Google Scholar] [CrossRef]
- Petter, J. RNA-Targeted Small Molecules: Opportunities in Drug Discovery. In Proceedings of the RNA-Targeted Drug Discovery Summit, Boston, MA, USA, 3–5 December 2018. [Google Scholar]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 50-end of NHE III1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef]
- Szostak, J.W.; Blackburn, E.H. Cloning yeast telomeres on linear plasmid vectors. Cell 1982, 29, 245–255. [Google Scholar] [CrossRef]
- Yang, D.; Okamoto, K. Structural insights into G-quadruplexes: Towards new anticancer drugs. Future Med. Chem. 2010, 2, 619–646. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef]
- Neidle, S. Human telomeric G-quadruplex: The current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2010, 277, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC Gquadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Bemark, M.; Neuberger, M.S. The c-MYC allele that is translocated into the IgH locus undergoes constitutive hypermutation in a Burkitt’s lymphoma line. Oncogene 2000, 19, 3404–3410. [Google Scholar] [CrossRef]
- Watt, R.; Nishikura, K.; Sorrentino, J.; ar-Rushdi, A.; Croce, C.M.; Rovera, G. The structure and nucleotide sequence of the 5’end of the human c-myc oncogene. Proc. Natl. Acad. Sci. USA 1983, 80, 6307–6311. [Google Scholar] [CrossRef]
- Simonsson, T.; Pecinka, P.; Kubista, M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998, 26, 1167–1172. [Google Scholar] [CrossRef]
- Gonzalez, V.; Guo, K.; Hurley, L.; Sun, D. Identification and Characterization of Nucleolin as a c-myc G-quadruplex-binding Protein. J. Biol. Chem. 2009, 284, 23622–23635. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Tian, T.; Chen, Y.; Yan, S.; Xing, X.; Zhang, Z.; Zhai, Q.; Xu, L.; Wang, S.; Weng, X.; et al. Existence of G-quadruplex structures in promoter region of oncogenes confirmed by G-quadruplex DNA cross-linking strategy. Sci. Rep. 2013, 3, 1811. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, T.; Fiaschetti, G.; Nagasawa, K.; Shin-ya, K.; Baumgartner, M.; Grotzer, M. G-Quadruplexes as Potential Therapeutic Targets for Embryonal Tumors. Molecules 2013, 18, 12500–12537. [Google Scholar] [CrossRef]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.M.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S., Jr. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Litovchick, A.; Clark, M.A.; Keefe, A.D. Universal strategies for the DNA-encoding of libraries of small molecules using the chemical ligation of oligonucleotide tags. Artif. DNA PNA XNA 2016, 5, e27896. [Google Scholar] [CrossRef]
- Keefe, A.D.; Clark, M.A.; Hupp, C.D.; Litovchick, A.; Zhang, Y. Chemical ligation methods for the tagging of DNA-encoded chemical libraries. Curr. Opin. Chem. Biol. 2015, 2015, 80–88. [Google Scholar] [CrossRef]
- Ou, T.-M.; Lu, Y.-J.; Zhang, C.; Huang, Z.-S.; Wang, X.-D.; Tan, J.-H.; Chen, Y.; Ma, D.-L.; Wong, K.-Y.; Tang, J.C.-O.; et al. Stabilization of G-Quadruplex DNA and Down-Regulation of Oncogene c-myc by Quindoline Derivatives. J. Med. Chem. 2007, 50, 1465–1474. [Google Scholar] [CrossRef]
- Lemarteleur, T.; Gomez, D.; Paterski, R.; Mandine, E.; Mailliet, P.; Riou, J.-F. Stabilization of the c-myc gene promoter quadruplex by specific ligands’ inhibitors of telomerase. Biochem. Biophys. Res. Commun. 2004, 323, 802–808. [Google Scholar] [CrossRef]
- Brown, R.V.; Danford, F.L.; Gokhale, V.; Hurley, L.H.; Brooks, T.A. Demonstration that Drug-targeted Down-regulation of MYC in Non-Hodgkins Lymphoma Is Directly Mediated through the Promoter G-quadruplex. J. Biol. Chem. 2011, 286, 41018–41027. [Google Scholar] [CrossRef]
- Sedoris, K.C.; Thomas, S.D.; Miller, D.M. c-myc Promoter Binding Protein Regulates the Cellular Response to an Altered Glucose Concentration. Biochemistry 2007, 46, 8659–8668. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Description |
|---|---|---|
| Tel1xG4 | 5′-AAAGGGTTAGGGTTAGGGTTAGGGAA-3′ | Single G-quartet forming four telomere repeats |
| Tel1xG4-biotin | 5′-Biotin-C18-C18-AAAGGGTTAGGGTTAGGGTTAGGGAA-3′ | Biotinylated single G-quartet forming four telomere repeats |
| Tel3xG4 | 5′-AAAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAA-3′ | Triple G-quartet forming 12 telomere repeats |
| Tel3xG4-biotin | 5′-Biotin-C18-C18-AAAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAA-3′ | Biotinylated triple G-quartet forming 12 telomere repeats |
| C-mycG4 (Pu-27) | 5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′ | C-myc G4 |
| Biotin-C-mycG4 (biotin-Pu-27) | 5′-Biotin-C18-C18-TGGGGAGGGTGGGGAGGGTGGGGAAGG-3′ | Biotinylated C-myc G4 |
| Pu27rev | ATCGATCGCTTCTTCGTCCTTCCCCA | Non-G-quartet control sequence, Complement to Pu-27 for PCR-stop assay |
| c-myc_G4_1234 | 5′-TGGGGAGGGTGGGGAGGGTGG-3′ | 3′-end truncated C-myc G4 |
| c-myc_G4_2345 | 5′-GAGGGTGGGGAGGGTGGGGAA-3′ | 5′-end truncated C-myc G4 |
| C-kit1 | 5′-AGGGAGGGCGCTGGGAGGAGGG-3′ | First G-quartet of C-kit promoter |
| C-kit2 | 5′-GGGCGGGCGCGAGGGAGGGG-3′ | Second G-quartet of C-kit promoter |
| Target | Compound 1 1 | Compound 2 1 |
|---|---|---|
| C-myc G4 | 1.05 µM | 328 nM |
| C-myc G4 1234 | 338 nM | 59 nM |
| C-myc G4 2345 | 308 nM | 197 nM |
| C-kit1 G4 | 1.1 uM | 807 nM |
| C-kit2 G4 | 1.7 uM | 1.4 uM |
| Telomere G4 | 8.4 uM | 5.25 uM |
| HP006 | NB | NB |
| Pu27rev | NB | NB |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litovchick, A.; Tian, X.; Monteiro, M.I.; Kennedy, K.M.; Guié, M.-A.; Centrella, P.; Zhang, Y.; Clark, M.A.; Keefe, A.D. Novel Nucleic Acid Binding Small Molecules Discovered Using DNA-Encoded Chemistry. Molecules 2019, 24, 2026. https://doi.org/10.3390/molecules24102026
Litovchick A, Tian X, Monteiro MI, Kennedy KM, Guié M-A, Centrella P, Zhang Y, Clark MA, Keefe AD. Novel Nucleic Acid Binding Small Molecules Discovered Using DNA-Encoded Chemistry. Molecules. 2019; 24(10):2026. https://doi.org/10.3390/molecules24102026
Chicago/Turabian StyleLitovchick, Alexander, Xia Tian, Michael I. Monteiro, Kaitlyn M. Kennedy, Marie-Aude Guié, Paolo Centrella, Ying Zhang, Matthew A. Clark, and Anthony D. Keefe. 2019. "Novel Nucleic Acid Binding Small Molecules Discovered Using DNA-Encoded Chemistry" Molecules 24, no. 10: 2026. https://doi.org/10.3390/molecules24102026
APA StyleLitovchick, A., Tian, X., Monteiro, M. I., Kennedy, K. M., Guié, M.-A., Centrella, P., Zhang, Y., Clark, M. A., & Keefe, A. D. (2019). Novel Nucleic Acid Binding Small Molecules Discovered Using DNA-Encoded Chemistry. Molecules, 24(10), 2026. https://doi.org/10.3390/molecules24102026