Recent Advances and New Perspectives in Capillary Electrophoresis-Mass Spectrometry for Single Cell “Omics”


Abstract
1. Introduction
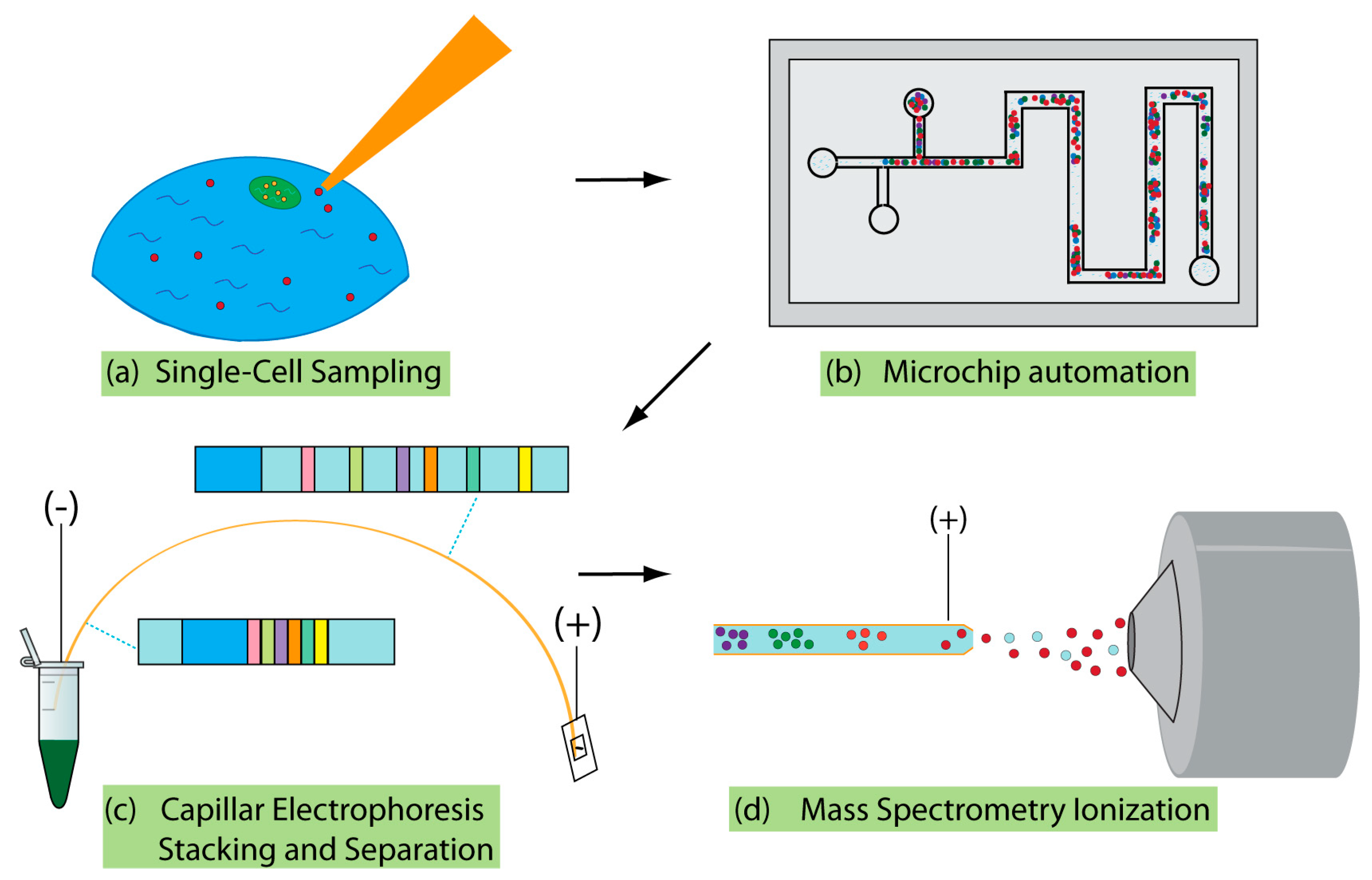
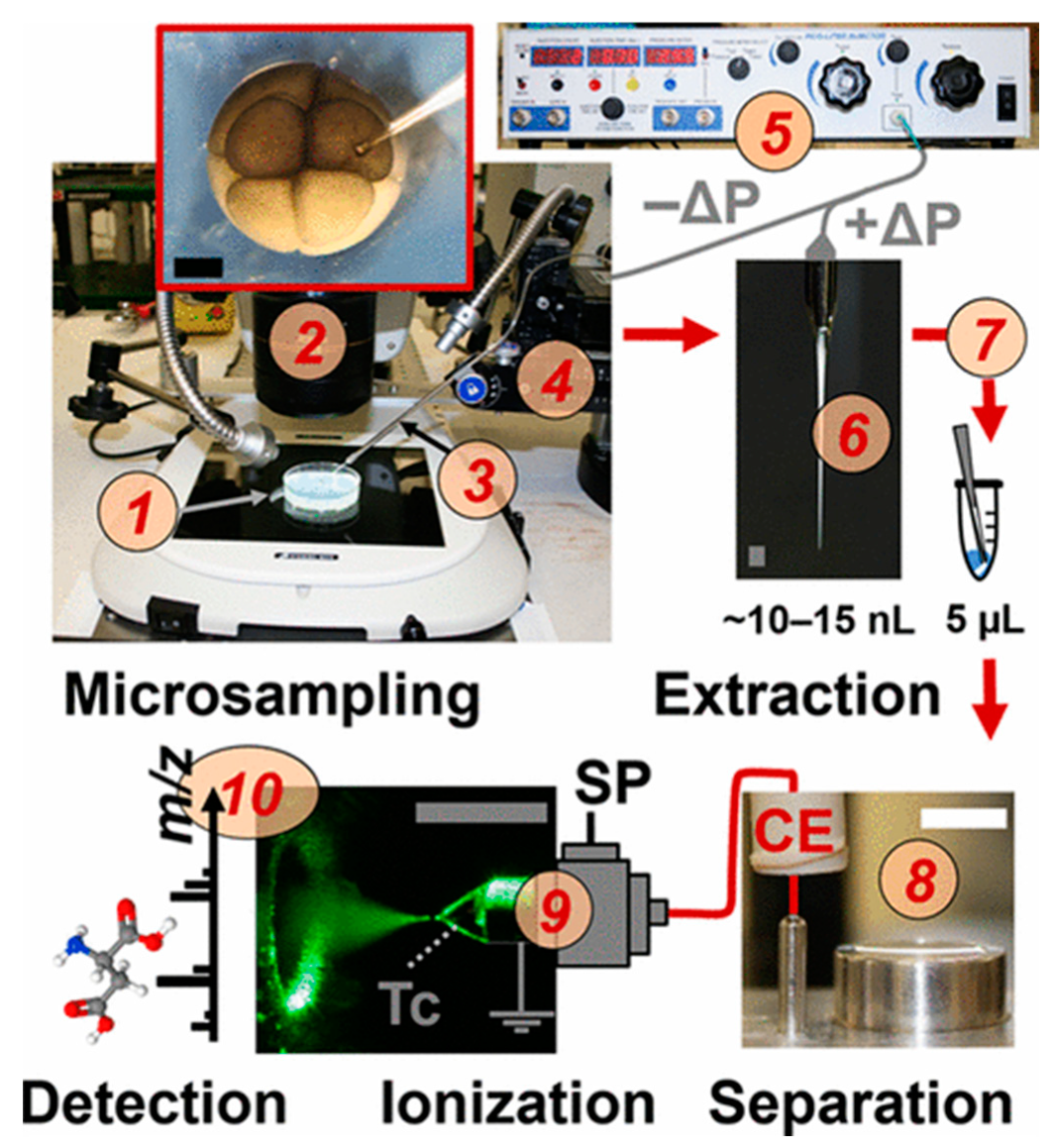
2. Addressing the Challenges of Single-Cell Sampling
3. Microchip Single-Cell Analysis—Toward the Automation of Sample Handling
4. Separations Prior to MS
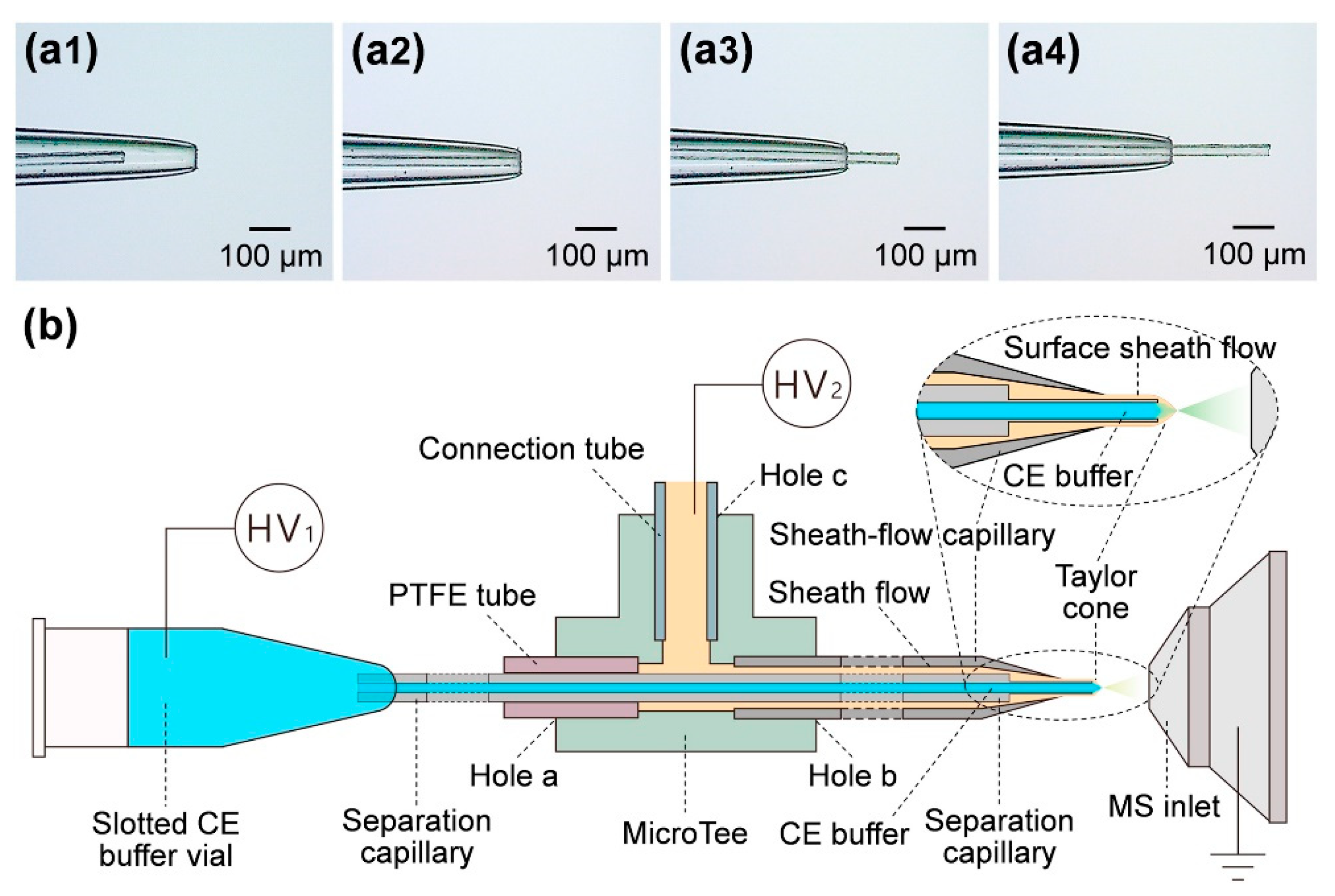
5. Interfacing CE with MS
6. Novel Applications of Single-Cell CE-MS
6.1. Metabolites
6.2. Proteins
6.3. Other Types of Analytes
7. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Altschuler, S.J.; Wu, L.F. Cellular heterogeneity: When do differences make a difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Grima, R. Single-cell variability in multicellular organisms. Nat. Commun. 2018, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by shrinking: Empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet. 2017, 18, 345. [Google Scholar] [CrossRef]
- Zhang, L.; Vertes, A. Single-Cell Mass Spectrometry Approaches to Explore Cellular Heterogeneity. Angew. Chem. Int. Ed. Engl. 2018, 57, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Prabhakaran, V.; Laskin, J. Quantitative Extraction and Mass Spectrometry Analysis at a Single-Cell Level. Anal. Chem. 2018, 90, 7937–7945. [Google Scholar] [CrossRef]
- Onjiko, R.M.; Portero, E.P.; Moody, S.A.; Nemes, P. In Situ Microprobe Single-Cell Capillary Electrophoresis Mass Spectrometry: Metabolic Reorganization in Single Differentiating Cells in the Live Vertebrate (Xenopus laevis) Embryo. Anal. Chem. 2017, 89, 7069–7076. [Google Scholar] [CrossRef]
- Comi, T.J.; Makurath, M.A.; Philip, M.C.; Rubakhin, S.S.; Sweedler, J.V. MALDI MS Guided Liquid Microjunction Extraction for Capillary Electrophoresis–Electrospray Ionization MS Analysis of Single Pancreatic Islet Cells. Anal. Chem. 2017, 89, 7765–7772. [Google Scholar] [CrossRef]
- Do, T.D.; Ellis, J.F.; Neumann, E.K.; Comi, T.J.; Tillmaand, E.G.; Lenhart, A.E.; Rubakhin, S.S.; Sweedler, J.V. Optically Guided Single Cell Mass Spectrometry of Rat Dorsal Root Ganglia to Profile Lipids, Peptides and Proteins. Chemphyschem 2018, 19, 1180–1191. [Google Scholar] [CrossRef]
- Zhang, L.; Khattar, N.; Kemenes, I.; Kemenes, G.; Zrinyi, Z.; Pirger, Z.; Vertes, A. Subcellular Peptide Localization in Single Identified Neurons by Capillary Microsampling Mass Spectrometry. Sci. Rep. 2018, 8, 12227. [Google Scholar] [CrossRef]
- Deng, J.; Li, W.; Yang, Q.; Liu, Y.; Fang, L.; Guo, Y.; Guo, P.; Lin, L.; Yang, Y.; Luan, T. Biocompatible Surface-Coated Probe for in Vivo, in Situ, and Microscale Lipidomics of Small Biological Organisms and Cells Using Mass Spectrometry. Anal. Chem. 2018, 90, 6936–6944. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Yang, Z.; Wawrik, B. Metabolomic Fingerprints of Individual Algal Cells Using the Single-Probe Mass Spectrometry Technique. Front. Plant Sci. 2018, 9, 571. [Google Scholar] [CrossRef] [PubMed]
- Pan, N.; Rao, W.; Kothapalli, N.R.; Liu, R.; Burgett, A.W.; Yang, Z. The single-probe: A miniaturized multifunctional device for single cell mass spectrometry analysis. Anal. Chem. 2014, 86, 9376–9380. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.T.; Wang, C.; Rausch, S.J.; Lee, C.S.; Tang, K. Pneumatic Microvalve-Based Hydrodynamic Sample Injection for High-Throughput, Quantitative Zone Electrophoresis in Capillaries. Anal. Chem. 2014, 86, 6723–6729. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Q.; Chen, P.; Li, Z.; Chen, Z.; Tang, B. Consecutive Gated Injection-Based Microchip Electrophoresis for Simultaneous Quantitation of Superoxide Anion and Nitric Oxide in Single PC-12 Cells. Anal. Chem. 2016, 88, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, S.; Hu, H.; Liu, Y.M. A microchip electrophoresis-mass spectrometric platform with double cell lysis nano-electrodes for automated single cell analysis. J. Chromatogr. A 2016, 1451, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Misevic, G.N.; BenAssayag, G.; Rasser, B.; Sales, P.; Simic-Krstic, J.; Misevic, N.J.; Popescu, O. Design and construction of wall-less nano-electrophoretic and nano in micro array high throughput devices for single cell ‘omics’ single molecule detection analyses. J. Mol. Struct. 2014, 1073, 142–149. [Google Scholar] [CrossRef]
- Huang, Q.; Mao, S.; Khan, M.; Zhou, L.; Lin, J.-M. Dean flow assisted cell ordering system for lipid profiling in single-cells using mass spectrometry. Chem. Commun. 2018, 54, 2595–2598. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhao, L.; Guo, J.; Yang, S.; Ding, H.; Wang, X.; Pu, Q. Double-helix micro-channels on microfluidic chips for enhanced continuous on-chip derivatization followed by electrophoretic separation. Biosens. Bioelectron. 2015, 72, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.H.; Li, M.; Huang, Y.; Hahn, J.H. A hydrophobic ionic liquid compartmentalized sampling/labeling and its separation techniques in polydimethylsiloxane microchip capillary electrophoresis. Electrophoresis 2016, 38, 372–379. [Google Scholar] [CrossRef]
- Jang, L.-W.; Razu, M.E.; Jensen, E.C.; Jiao, H.; Kim, J. A fully automated microfluidic micellar electrokinetic chromatography analyzer for organic compound detection. Lab Chip 2016, 16, 3558–3564. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Huang, M.; Wang, X.K.; Zhu, Y.; Li, J.S.; Wong, C.C.L.; Fang, Q. Nanoliter-Scale Oil-Air-Droplet Chip-Based Single Cell Proteomic Analysis. Anal. Chem. 2018, 90, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Sun, L.; Yan, X.; Dovichi, N.J. Single-shot proteomics using capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry with production of more than 1250 Escherichia coli peptide identifications in a 50 min separation. Anal. Chem. 2013, 85, 2569–2573. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hebert, A.S.; Yan, X.; Zhao, Y.; Westphall, M.S.; Rush, M.; Zhu, G.; Champion, M.; Coon, J.; Dovichi, N. Over 10000 Peptide Identifications from the HeLa Proteome by Using Single-Shot Capillary Zone Electrophoresis Combined with Tandem Mass Spectrometry. Angew. Chem. Int. Ed. 2014, 53, 13931–13933. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhu, G.; Mou, S.; Zhao, Y.; Champion, M.M.; Dovichi, N.J. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry for quantitative parallel reaction monitoring of peptide abundance and single-shot proteomic analysis of a human cell line. J. Chromatogr. A 2014, 1359, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.R.; Sun, L.; Zhu, G.; Dovichi, N.J.; Hummon, A.B. Over 2300 phosphorylated peptide identifications with single-shot capillary zone electrophoresis-tandem mass spectrometry in a 100 min separation. Anal. Chem. 2015, 87, 9532–9537. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.B.; Lombard-Banek, C.; Munoz, L.P.; Manzini, M.C.; Nemes, P. Enhanced Peptide Detection Toward Single-Neuron Proteomics by Reversed-Phase Fractionation Capillary Electrophoresis Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 913–922. [Google Scholar] [CrossRef]
- Liu, J.X.; Aerts, J.T.; Rubakhin, S.S.; Zhang, X.X.; Sweedler, J.V. Analysis of endogenous nucleotides by single cell capillary electrophoresis-mass spectrometry. Analyst 2014, 139, 5835–5842. [Google Scholar] [CrossRef]
- Lombard-Banek, C.; Reddy, S.; Moody, S.A.; Nemes, P. Label-free Quantification of Proteins in Single Embryonic Cells with Neural Fate in the Cleavage-Stage Frog (Xenopus laevis) Embryo using Capillary Electrophoresis Electrospray Ionization High-Resolution Mass Spectrometry (CE-ESI-HRMS). Mol. Cell. Proteom. 2016, 15, 2756–2768. [Google Scholar] [CrossRef]
- Lubeckyj, R.A.; McCool, E.N.; Shen, X.; Kou, Q.; Liu, X.; Sun, L. Single-Shot Top-Down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia coli Proteoforms. Anal. Chem. 2017, 89, 12059–12067. [Google Scholar] [CrossRef]
- Yang, Z.; Shen, X.; Chen, D.; Sun, L. Microscale Reversed-Phase Liquid Chromatography/Capillary Zone Electrophoresis-Tandem Mass Spectrometry for Deep and Highly Sensitive Bottom-Up Proteomics: Identification of 7500 Proteins with Five Micrograms of an MCF7 Proteome Digest. Anal. Chem. 2018, 90, 10479–10486. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.B.; Zamarbide, M.; Manzini, C.; Nemes, P. Tapered-Tip Capillary Electrophoresis Nano-Electrospray Ionization Mass Spectrometry for Ultrasensitive Proteomics: The Mouse Cortex | SpringerLink. J. Am. Soc. Mass Spectrom. 2017, 28, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Pan, J.Z.; Fang, Q. A robust and extendable sheath flow interface with minimal dead volume for coupling CE with ESI-MS. Talanta 2018, 180, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Comi, T.J.; Do, T.D.; Rubakhin, S.S.; Sweedler, J.V. Categorizing Cells on the Basis of their Chemical Profiles: Progress in Single-Cell Mass Spectrometry. J. Am. Chem. Soc. 2017, 139, 3920–3929. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-H.; Ai, F.; Wang, Z.-L.; Cheng, J.-K. Recent advances in single-cell analysis using capillary electrophoresis and microfluidic devices. J. Chromatogr. B 2008, 866, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Voeten, R.L.C.; Ventouri, I.K.; Haselberg, R.; Somsen, G.W. Capillary Electrophoresis: Trends and Recent Advances. Anal. Chem. 2018, 90, 1464–1481. [Google Scholar] [CrossRef] [PubMed]
- Sans, M.; Feider, C.L.; Eberlin, L.S. Advances in mass spectrometry imaging coupled to ion mobility spectrometry for enhanced imaging of biological tissues. Curr. Opin. Chem. Biol. 2018, 42, 138–146. [Google Scholar] [CrossRef]
- Buchberger, A.R.; DeLaney, K.; Johnson, J.; Li, L. Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem. 2017, 90, 240–265. [Google Scholar] [CrossRef]
- Zhang, L.; Sevinsky, C.J.; Davis, B.M.; Vertes, A. Single-Cell Mass Spectrometry of Subpopulations Selected by Fluorescence Microscopy. Anal. Chem. 2018, 90, 4626–4634. [Google Scholar] [CrossRef]
- Jha, R.R.; Singh, C.; Pant, A.B.; Patel, D.K. Ionic liquid based ultrasound assisted dispersive liquid-liquid micro-extraction for simultaneous determination of 15 neurotransmitters in rat brain, plasma and cell samples. Anal. Chim. Acta 2018, 1005, 43–53. [Google Scholar] [CrossRef]
- Pan, Q.; Herr, A.E. Geometry-induced injection dispersion in single-cell protein electrophoresis. Anal. Chim. Acta 2018, 1000, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, B.; He, M.; Wang, H.; Hu, B. Chip-based magnetic solid phase microextraction coupled with ICP-MS for the determination of Cd and Se in HepG2 cells incubated with CdSe quantum dots. Talanta 2018, 179, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, B.; Wang, H.; Huang, X.; He, M.; Hu, B. Chip-based monolithic microextraction combined with ICP-MS for the determination of bismuth in HepG2 cells. J. Anal. Atom. Spectrosc. 2016, 31, 1391–1399. [Google Scholar] [CrossRef]
- Sanz-Nebot, V.; Balaguer, E.; Benavente, F.; Barbosa, J. Comparison of sheathless and sheath-flow electrospray interfaces for the capillary electrophoresis-electrospray ionization-mass spectrometry analysis of peptides. Electrophoresis 2005, 26, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Method. 2014, 11, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Onjiko, R.M.; Morris, S.E.; Moody, S.A.; Nemes, P. Single-cell mass spectrometry with multi-solvent extraction identifies metabolic differences between left and right blastomeres in the 8-cell frog (Xenopus) embryo. Analyst 2016, 141, 3648–3656. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, G.; Yang, Z. Towards rapid prediction of drug-resistant cancer cell phenotypes: Single cell mass spectrometry combined with machine learning. Chem. Commun. 2018. [Google Scholar] [CrossRef]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteom. Res. 2003, 2, 488–494. [Google Scholar] [CrossRef]
- Chen, Y.; Arriaga, E.A. Individual electrophoretic mobilities of liposomes and acidic organelles displaying pH gradients across their membranes. Langmuir 2007, 23, 5584–5590. [Google Scholar] [CrossRef]
- Lapainis, T.; Rubakhin, S.S.; Sweedler, J.V. Capillary Electrophoresis with Electrospray Ionization Mass Spectrometric Detection for Single Cell Metabolomics. Anal. Chem. 2009, 81, 5858–5864. [Google Scholar] [CrossRef]
- Nemes, P.; Knolhoff, A.M.; Rubakhin, S.S.; Sweedler, J.V. Single-Cell Metabolomics: Changes in the Metabolome of Freshly Isolated and Cultured Neurons. ACS Chemical. Neurosci. 2012, 3, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Vertes, A. Energy Charge, Redox State, and Metabolite Turnover in Single Human Hepatocytes Revealed by Capillary Microsampling Mass Spectrometry—PubMed—NCBI. Anal. Chem. 2015, 87, 10397–10405. [Google Scholar] [CrossRef]
- Onjiko, R.M.; Portero, E.P.; Moody, S.A.; Nemes, P. Microprobe Capillary Electrophoresis Mass Spectrometry for Single-cell Metabolomics in Live Frog (Xenopus laevis) Embryos. J. Vis. Exp. 2017, 130, e56956. [Google Scholar] [CrossRef] [PubMed]
- Onjiko, R.M.; Plotnick, D.O.; Moody, S.A.; Nemes, P. Metabolic Comparison of Dorsal versus Ventral Cells Directly in the Live 8-cell Frog Embryo by Microprobe Single-cell CE-ESI-MS. Anal. Methods 2017, 9, 4964–4970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, Z.; Gong, X.; Si, X.; Zhao, Y.; Yang, C.; Zhang, S.; Zhang, X. Integrated Droplet-Based Microextraction with ESI-MS for Removal of Matrix Interference in Single-Cell Analysis. Sci. Rep. 2016, 6, 24730. [Google Scholar] [CrossRef]
- Patel, A.V.; Kawai, T.; Wang, L.; Rubakhin, S.S.; Sweedler, J.V. Chiral Measurement of Aspartate and Glutamate in Single Neurons by Large-Volume Sample Stacking Capillary Electrophoresis. Anal. Chem. 2017, 89, 12375–12382. [Google Scholar] [CrossRef]
- Lombard-Banek, C.; Moody, S.A.; Nemes, P. Single-Cell Mass Spectrometry for Discovery Proteomics: Quantifying Translational Cell Heterogeneity in the 16-Cell Frog (Xenopus) Embryo. Angew. Chem. Int. Ed. Engl. 2016, 55, 2454–2458. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Dubiak, K.M.; Peuchen, E.H.; Zhang, Z.; Zhu, G.; Huber, P.W.; Dovichi, N.J. Single cell proteomics using frog (Xenopus laevis) blastomeres isolated from early stage embryos, which form a geometric progression in protein content. Anal. Chem. 2016, 88, 6653–6657. [Google Scholar] [CrossRef]
- Mainz, E.R.; Wang, Q.; Lawrence, D.S.; Allbritton, N.L. An Integrated Chemical Cytometry Method: Shining a Light on Akt Activity in Single Cells. Angew. Chem. Int. Ed. Engl. 2016, 55, 13095–13098. [Google Scholar] [CrossRef]
- Geng, X.; Shi, M.; Ning, H.; Feng, C.; Guan, Y. A compact and low-cost laser induced fluorescence detector with silicon based photodetector assembly for capillary flow systems. Talanta 2018, 182, 279–284. [Google Scholar] [CrossRef]
- Chen, D.; Fan, F.; Zhao, X.; Xu, F.; Chen, P.; Wang, J.; Ban, L.; Liu, Z.; Feng, X.; Zhang, Y.; et al. Single Cell Chemical Proteomics with Membrane-Permeable Activity-Based Probe for Identification of Functional Proteins in Lysosome of Tumors. Anal. Chem. 2016, 88, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Proctor, A.; Sims, C.E.; Allbritton, N.L. Chemical fixation to arrest phospholipid signaling for chemical cytometry. J. Chromatogr. A 2017, 1523, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Mironov, G.; Berezovski, M.V. Direct deposition of endogenous MicroRNAs and their post-transcriptional modifications in cancer serum by capillary electrophoresis-mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 2891–2899. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hebert, A.S.; Westphall, M.S.; Qu, Y.; Coon, J.J.; Dovichi, N.J. Production of Over 27000 Peptide and Nearly 4400 Protein Identifications by Single-Shot Capillary-Zone Electrophoresis-Mass Spectrometry via Combination of a Very-Low-Electroosmosis Coated Capillary, a Third-Generation Electrokinetically-Pumped Sheath-Flow Nanospray Interface, an Orbitrap Fusion Lumos Tribrid Mass Spectrometer, and an Advanced-Peak-Determination Algorithm. Anal. Chem. 2018, 90, 12090–12093. [Google Scholar] [PubMed]
- Faserl, K.; Sarg, B.; Sola, L.; Lindner, H.H. Enhancing Proteomic Throughput in Capillary Electrophoresis-Mass Spectrometry by Sequencial Sample Injection. Proteomics 2017, 17, 1700310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, N.; Zeng, H.; Nakajima, H.; Lin, J.M.; Uchiyama, K. Inkjet Printing Based Separation of Mammalian Cells by Capillary Electrophoresis. Anal. Chem. 2017, 89, 8674–8677. [Google Scholar] [CrossRef] [PubMed]
- Rodogiannis, K.; Duong, J.T.; Kovarik, M.L. Microfluidic single-cell analysis of oxidative stress in Dictyostelium discoideum. Analyst 2018, 143, 3643–3650. [Google Scholar] [CrossRef]
- Artner, C.; Holtkamp, H.U.; Hartinger, C.G.; Meier-Menches, S.M. Characterizing activation mechanisms and binding preferences of ruthenium metallo-prodrugs by a competitive binding assay. J. Inorg. Biochem. 2017, 177, 322–327. [Google Scholar] [CrossRef]
- Zhong, X.; Zhang, Z.; Jiang, S.; Li, L. Recent advances in coupling capillary electrophoresis based separation techniques to ESI and MALDI MS. Electrophoresis 2014, 35, 1214–1225. [Google Scholar] [CrossRef]
- Wang, J.H.; Ye, H.; Zhang, Z.C.; Xiang, F.; Girdaukas, G.; Li, L.J. Advancing Matrix-Assisted Laser Desorption/Ionization-Mass Spectrometric Imaging for Capillary Electrophoresis Analysis of Peptides. Anal. Chem. 2011, 83, 3462–3469. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, H.; Wang, J.; Hui, L.; Li, L. Pressure-assisted capillary electrophoresis coupling with matrix-assisted laser desorption/ionization-mass spectrometric imaging for quantitative analysis of complex peptide mixtures. Anal. Chem. 2012, 84, 7684–7691. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Xiang, F.; Zhang, Z.; Li, L. Combining Capillary Electrophoresis Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry and Stable Isotopic Labeling Techniques for Comparative Crustacean Peptidomics. J. Chromatogr. A 2010, 1217, 4463–4470. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Key Challenges | Strategies | References |
|---|---|---|---|
| Sampling | Small-volume handling | Localized electroosmotic extraction | [6] |
| Microprobe aspiration | [7] | ||
| Cell discrimination/selection | MALDI-MS for guided sampling prior to CE-ESI-MS | [8,9] | |
| Novel probes for capillary microsampling | [10,11,12,13] | ||
| Microchip automation | Small-volume precision | Microvalve or gated sample injection | [14,15] |
| On-chip high-voltage cell lysis | [16] | ||
| Increasing throughput, reducing sample loss | Novel device geometries | [17,18] | |
| Including sample preparation steps on device | [16,19,20,21,22] | ||
| Separation | Increasing resolution and sensitivity | Reduce EOF and lengthen capillary to provide longer separation | [23,24,25,26] |
| Fractionation prior to separation | [27] | ||
| Limited sample amounts | Sample stacking for analyte pre-concentration | [28,29,30,31] | |
| Analyte enrichment via NiMA device | [17] | ||
| MS Interface | Sample dilution | Tapered-tip emitter enabling lower sheath flow rate | [32] |
| Etched capillary allows for closer placement to the emitter orifice | [25,33] | ||
| Increasing throughput | Optical microscopy and MALDI-MS cell screening | [8] | |
| Rapid separation with NEA microchip | [9] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeLaney, K.; Sauer, C.S.; Vu, N.Q.; Li, L. Recent Advances and New Perspectives in Capillary Electrophoresis-Mass Spectrometry for Single Cell “Omics”. Molecules 2019, 24, 42. https://doi.org/10.3390/molecules24010042
DeLaney K, Sauer CS, Vu NQ, Li L. Recent Advances and New Perspectives in Capillary Electrophoresis-Mass Spectrometry for Single Cell “Omics”. Molecules. 2019; 24(1):42. https://doi.org/10.3390/molecules24010042
Chicago/Turabian StyleDeLaney, Kellen, Christopher S. Sauer, Nhu Q. Vu, and Lingjun Li. 2019. "Recent Advances and New Perspectives in Capillary Electrophoresis-Mass Spectrometry for Single Cell “Omics”" Molecules 24, no. 1: 42. https://doi.org/10.3390/molecules24010042
APA StyleDeLaney, K., Sauer, C. S., Vu, N. Q., & Li, L. (2019). Recent Advances and New Perspectives in Capillary Electrophoresis-Mass Spectrometry for Single Cell “Omics”. Molecules, 24(1), 42. https://doi.org/10.3390/molecules24010042