Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors



Abstract
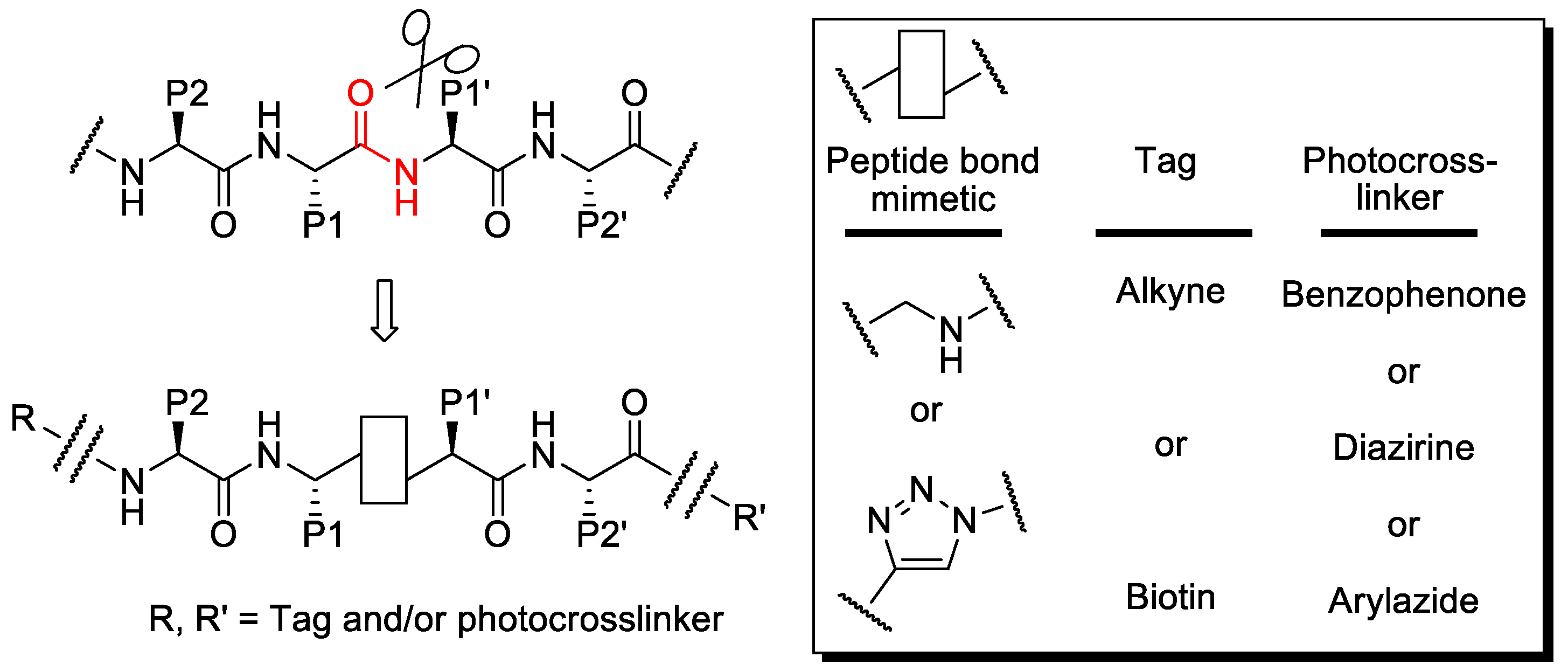
1. Introduction
2. Results
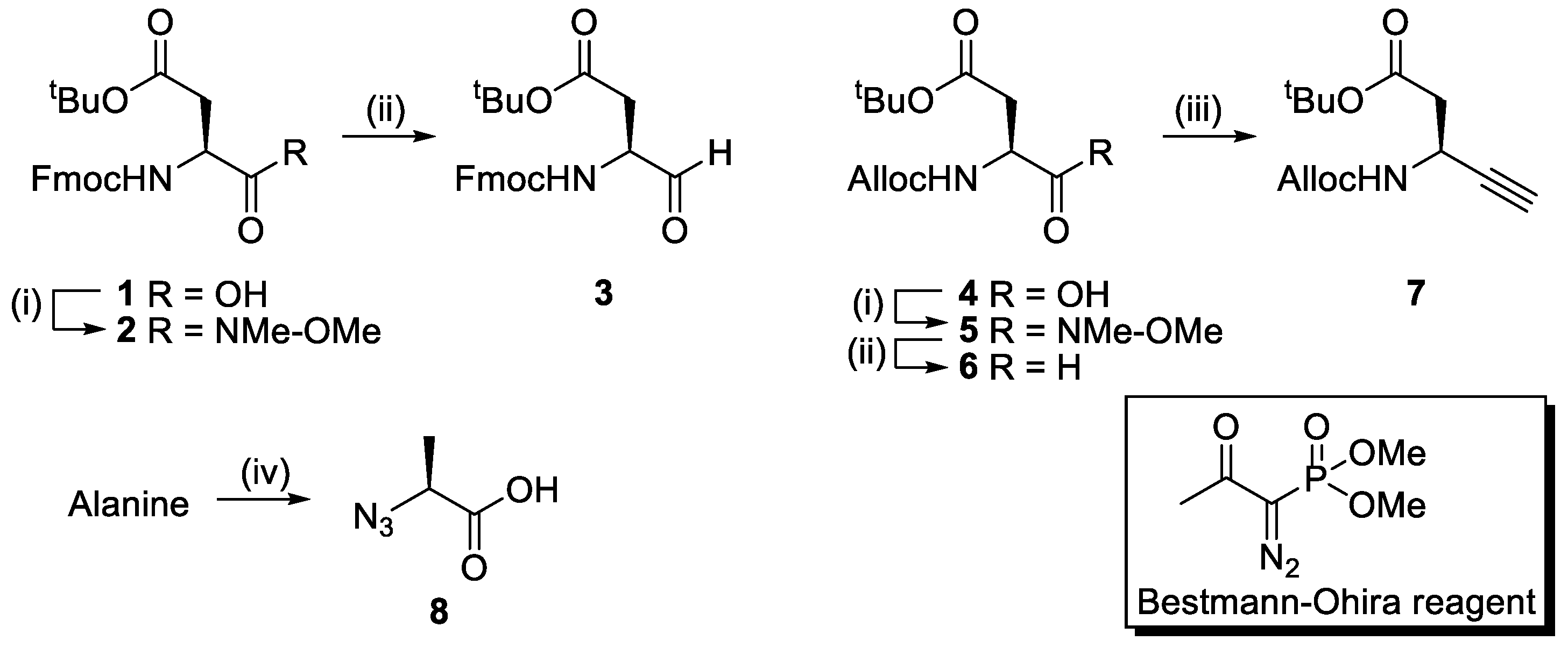
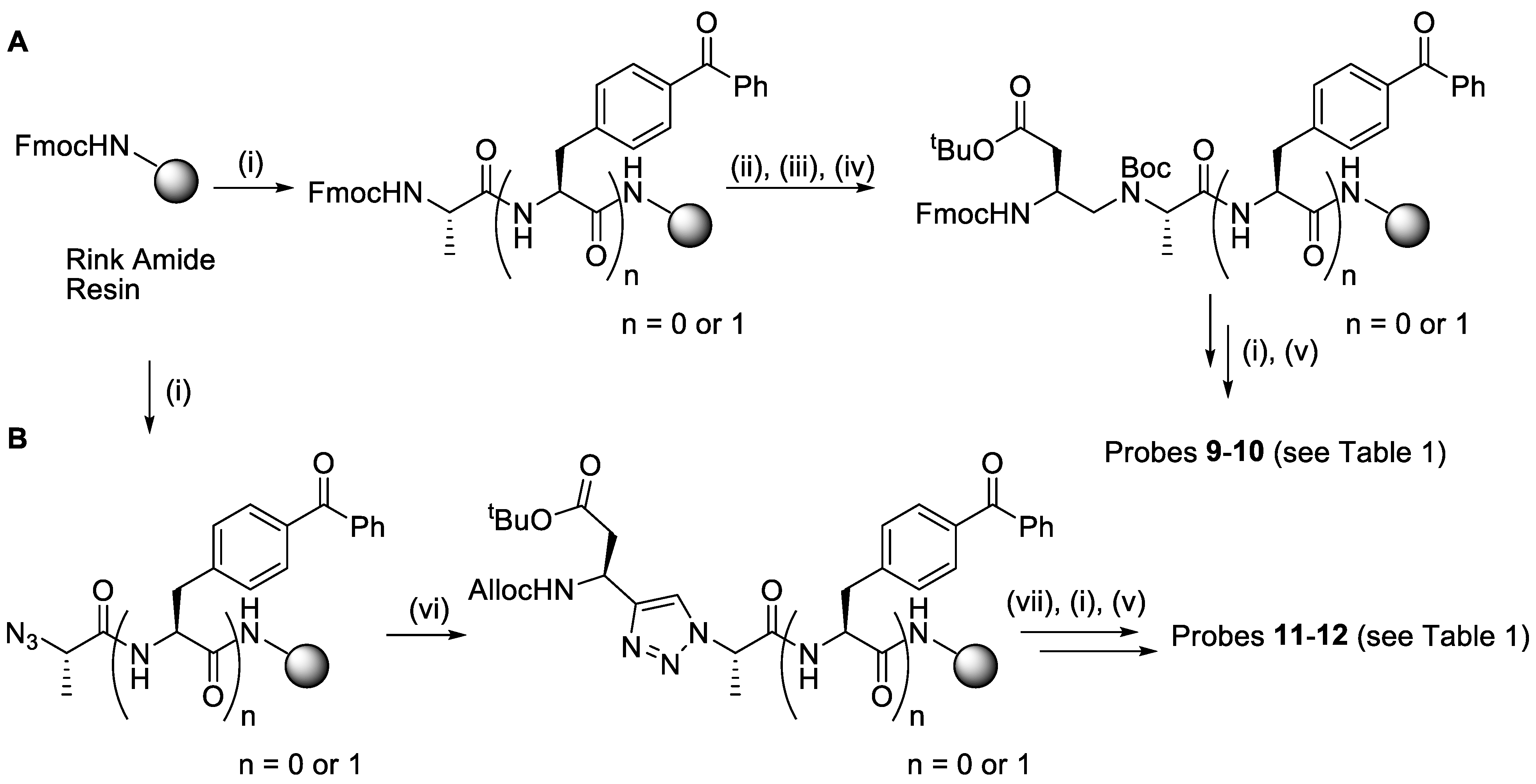
2.1. Synthesis
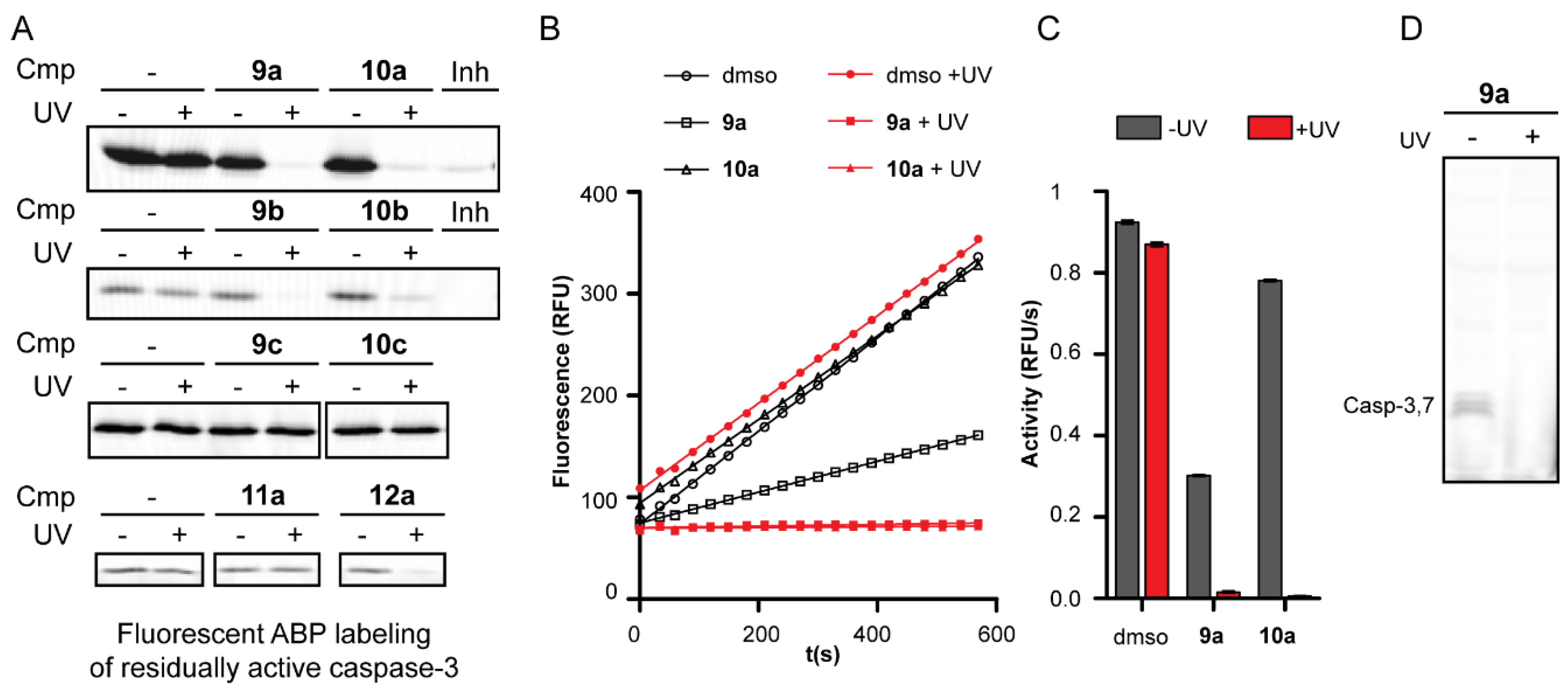
2.2. Inhibition Studies
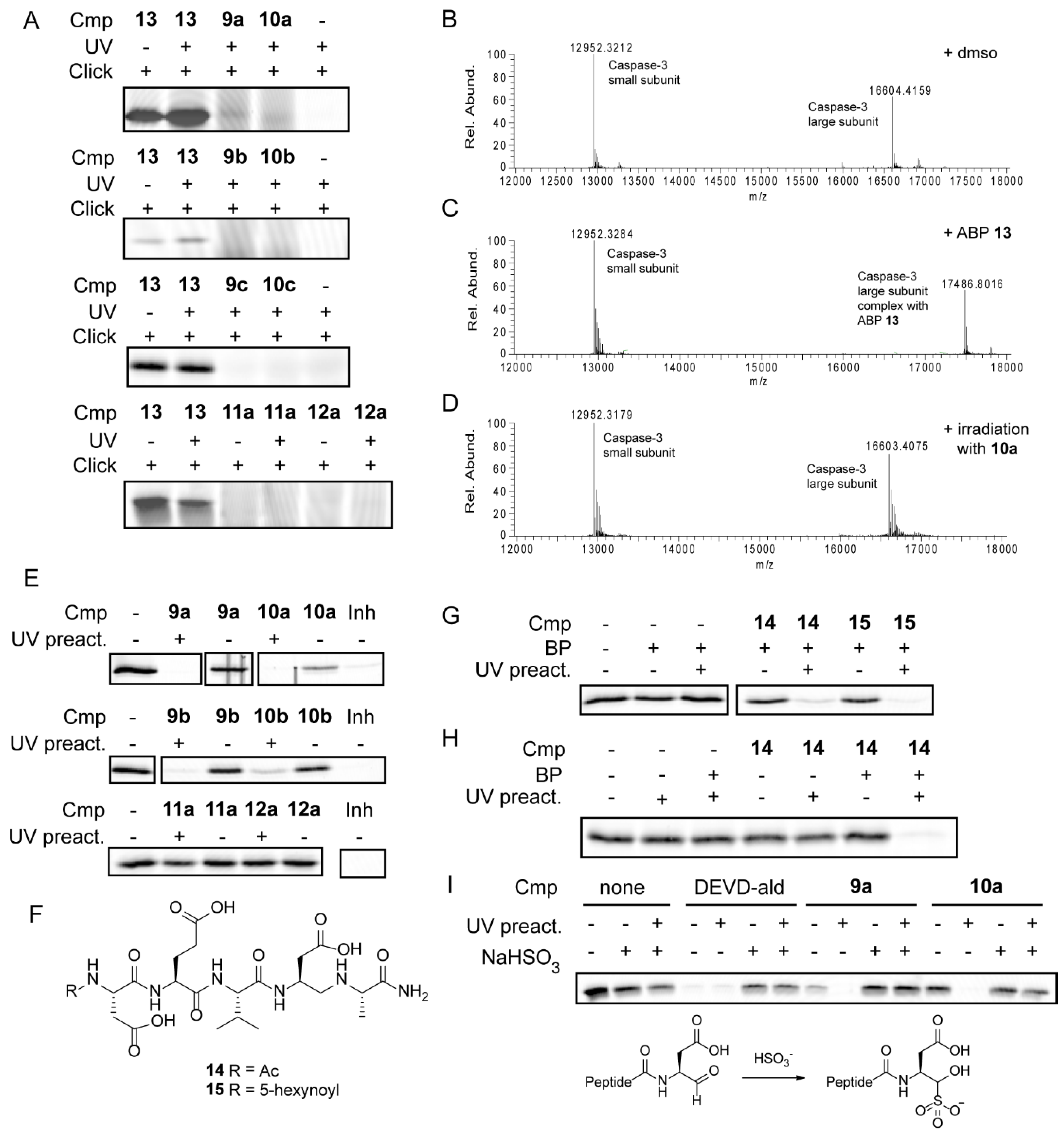
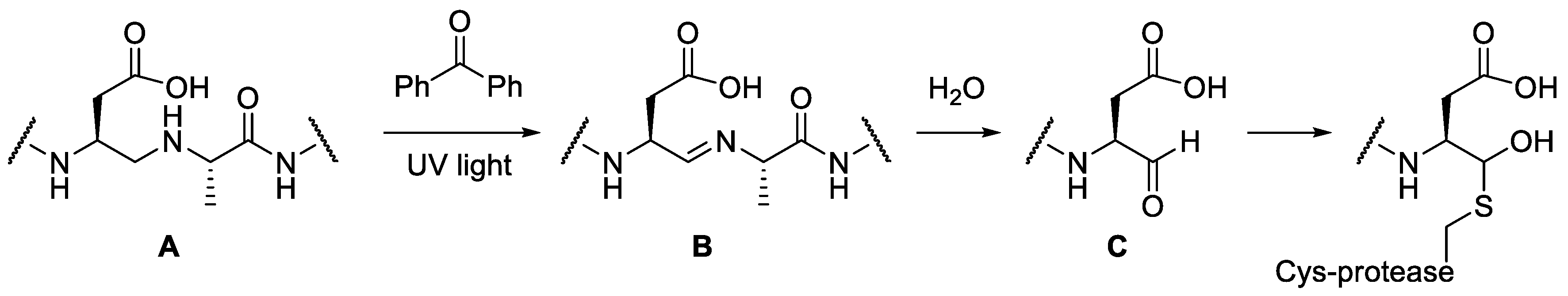
2.3. Mechanism of Action
3. Discussion
4. Materials and Methods
4.1. Materials and Equipment
4.2. General Procedures
4.2.1. Kaiser Test
4.2.2. Chloranil Test
4.2.3. General SPPS Elongation Procedure
4.2.4. On Resin Reductive Amination
4.2.5. On Resin Click Chemistry
4.2.6. On Resin Alloc Deprotection
4.2.7. Resin Cleavage and Purification
4.3. Synthesis
4.4. Biochemistry
4.4.1. Gel-Based Competitive ABPP
4.4.2. Protease Kinetics Experiments
4.4.3. Fluorescent Labeling Experiments
4.4.4. ESI-MS on Caspase-3
4.4.5. MALDI-MS on Caspase-3
4.4.6. Competitive ABPP with Bisulfite Aldehyde Quenching
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Riddel, J.P., Jr.; Aouizerat, B.E.; Miaskowski, C.; Lillicrap, D.P. Theories of blood coagulation. J. Pediatr. Oncol. Nurs. 2007, 24, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S. Caspases and apoptosis. Essays Biochem. 2002, 38, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Verdoes, M.; Verhelst, S.H. Detection of protease activity in cells and animals. Biochim. Biophys. Acta 2016, 1864, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, P.; Verhelst, S.H.L. Chemical Probes Targeting Proteases for Imaging and Diagnostics in Cancer, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 351–375. [Google Scholar]
- Sanman, L.E.; Bogyo, M. Activity-based profiling of proteases. Annu. Rev. Biochem. 2014, 83, 249–273. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Kahler, J.P.; van de Plassche, M.A.T.; Vanhoutte, R.; Verhelst, S.H.L. Recent Advances in Activity-Based Protein Profiling of Proteases. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Serim, S.; Haedke, U.; Verhelst, S.H.L. Activity-based probes for the study of proteases: Recent advances and developments. ChemMedChem 2012, 7, 1146–1159. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Groborz, K.; Drag, M. Emerging challenges in the design of selective substrates, inhibitors and activity-based probes for indistinguishable proteases. Febs J. 2017, 284, 1518–1539. [Google Scholar] [CrossRef]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef]
- Saghatelian, A.; Jessani, N.; Joseph, A.; Humphrey, M.; Cravatt, B.F. Activity-based probes for the proteomic profiling of metalloproteases. Proc. Natl. Acad. Sci. USA 2004, 101, 10000–10005. [Google Scholar] [CrossRef] [PubMed]
- Sieber, S.A.; Niessen, S.; Hoover, H.S.; Cravatt, B.F. Proteomic profiling of metalloprotease activities with cocktails of active-site probes. Nat. Chem. Biol. 2006, 2, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, M.A.; Geurink, P.P.; Klein, T.; Kauffman, H.F.; van der Marel, G.A.; Bischoff, R.; Overkleeft, H.S. Solid-phase synthesis of succinylhydroxamate peptides: Functionalized matrix metalloproteinase inhibitors. Org. Lett. 2006, 8, 1705–1708. [Google Scholar] [CrossRef]
- Rut, W.; Kasperkiewicz, P.; Byzia, A.; Poreba, M.; Groborz, K.; Drag, M. Recent advances and concepts in substrate specificity determination of proteases using tailored libraries of fluorogenic substrates with unnatural amino acids. Biol. Chem. 2015, 396, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Geurink, P.P.; Prely, L.M.; van der Marel, G.A.; Bischoff, R.; Overkleeft, H.S. Photoaffinity Labeling in Activity-Based Protein Profiling. Top. Curr. Chem. 2012, 324, 85–113. [Google Scholar] [PubMed]
- Kleiner, P.; Heydenreuter, W.; Stahl, M.; Korotkov, V.S.; Sieber, S.A. A Whole Proteome Inventory of Background Photocrosslinker Binding. Angew. Chem. Int. Ed. Engl. 2017, 56, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Angell, Y.L.; Burgess, K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. [Google Scholar] [CrossRef]
- Szelke, M.; Leckie, B.; Hallett, A.; Jones, D.M.; Sueiras, J.; Atrash, B.; Lever, A.F. Potent new inhibitors of human renin. Nature 1982, 299, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Pav, S.; Pargellis, C.; Do, F.; Lamarre, D.; Anderson, P.C. Crystal structure of human immunodeficiency virus (HIV) type 2 protease in complex with a reduced amide inhibitor and comparison with HIV-1 protease structures. Proc. Natl. Acad. Sci. USA 1993, 90, 8387–8391. [Google Scholar] [CrossRef]
- Seyferth, D.; Marmor, R.S.; Hilbert, P. Reactions of Dimethylphosphono-Substituted Diazoalkanes—(Meo)2P(O)CR Transfer to Olefins and 1,3-Dipolar Additions of (Meo)2P(O)C(N2)R. J. Org. Chem. 1971, 36, 1379–1386. [Google Scholar] [CrossRef]
- Gilbert, J.C.; Weerasooriya, U. Diazoethenes—Their Attempted Synthesis from Aldehydes and Aromatic Ketones by Way of the Horner-Emmons Modification of the Wittig Reaction—A Facile Synthesis of Alkynes. J. Org. Chem. 1982, 47, 1837–1845. [Google Scholar] [CrossRef]
- Jepsen, T.H.; Kristensen, J.L. In Situ Generation of the Ohira-Bestmann Reagent from Stable Sulfonyl Azide: Scalable Synthesis of Alkynes from Aldehydes. J. Org. Chem. 2014, 79, 9423–9426. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [PubMed]
- Schilling, O.; Overall, C.M. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 2008, 26, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Timmer, J.C.; Zhu, W.; Pop, C.; Regan, T.; Snipas, S.J.; Eroshkin, A.M.; Riedl, S.J.; Salvesen, G.S. Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 2009, 16, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.N.; Shema, G.; Zahedi, R.P.; Verhelst, S.H.L. Protease Specificity Profiling in a Pipet Tip Using “Charge-Synchronized” Proteome-Derived Peptide Libraries. J. Proteome Res. 2018, 17, 1923–1933. [Google Scholar] [CrossRef]
- Punna, S.; Finn, M.G. A convenient colorimetric test for aliphatic azides. Synlett 2004, 1, 99–100. [Google Scholar]
- Cravatt, B.F.; Wright, A.T.; Kozarich, J.W. Activity-based protein profiling: From enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008, 77, 383–414. [Google Scholar] [CrossRef]
- Berger, A.B.; Witte, M.D.; Denault, J.B.; Sadaghiani, A.M.; Sexton, K.M.; Salvesen, G.S.; Bogyo, M. Identification of early intermediates of caspase activation using selective inhibitors and activity-based probes. Mol. Cell 2006, 23, 509–521. [Google Scholar] [CrossRef]
- Galibert, M.; Wartenberg, M.; Lecaille, F.; Saidi, A.; Mavel, S.; Joulin-Giet, A.; Korkmaz, B.; Bromme, D.; Aucagne, V.; Delmas, A.F.; et al. Substrate-derived triazolo- and azapeptides as inhibitors of cathepsins K and S. Eur. J. Med. Chem. 2018, 144, 201–210. [Google Scholar] [CrossRef]
- Bartholomew, R.F.; Davidson, R.S. Photosensitised Oxidation of Amines.1. Use of Benzophenone as a Sensitiser. J. Chem. Soc. C-Org. 1971, 12, 2342–2346. [Google Scholar] [CrossRef]
- Stennicke, H.R.; Salvesen, G.S. Caspases: Preparation and characterization. Methods 1999, 17, 313–319. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 9–15 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure 1 | Type |
|---|---|---|
| 9a–c |  | ψ(CH2NH) |
| 10a–c |  | ψ(CH2NH) |
| 11a |  | triazolo |
| 12a |  | triazolo |
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Kersavond, T.; Konopatzki, R.; Chakrabarty, S.; Blank-Landeshammer, B.; Sickmann, A.; Verhelst, S.H.L. Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors. Molecules 2019, 24, 206. https://doi.org/10.3390/molecules24010206
Van Kersavond T, Konopatzki R, Chakrabarty S, Blank-Landeshammer B, Sickmann A, Verhelst SHL. Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors. Molecules. 2019; 24(1):206. https://doi.org/10.3390/molecules24010206
Chicago/Turabian StyleVan Kersavond, Tim, Raphael Konopatzki, Suravi Chakrabarty, Bernhard Blank-Landeshammer, Albert Sickmann, and Steven H. L. Verhelst. 2019. "Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors" Molecules 24, no. 1: 206. https://doi.org/10.3390/molecules24010206
APA StyleVan Kersavond, T., Konopatzki, R., Chakrabarty, S., Blank-Landeshammer, B., Sickmann, A., & Verhelst, S. H. L. (2019). Short Peptides with Uncleavable Peptide Bond Mimetics as Photoactivatable Caspase-3 Inhibitors. Molecules, 24(1), 206. https://doi.org/10.3390/molecules24010206