
Tetrel Bond between 6-OTX3-Fulvene and NH3: Substituents and Aromaticity


 , and
, and
Abstract
1. Introduction
2. Theoretical Methods
3. Results and Discussion
3.1. MEPs of 6-OTX3-Fulvene
3.2. Geometries and Interaction Energy of Complexes
3.3. Substituents and AIM Analysis
3.4. NBO Analysis
3.5. Energy Decomposition Analysis
3.6. Aromaticity of the Fulvene Ring
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stone, K.J.; Little, R.D. An exceptionally simple and efficient method for the preparation of a wide variety of fulvenes. Cheminform 1984, 15, 1849–1853. [Google Scholar] [CrossRef]
- Peloquin, A.J.; Stone, R.L.; Avila, S.E. Synthesis of 1,3-diphenyl-6-alkyl/aryl-substituted fulvene chromophores: Observation of π-π interactions in a 6-pyrene-substituted 1,3-diphenylfulvene. J. Org. Chem. 2012, 77, 6371–6376. [Google Scholar] [CrossRef] [PubMed]
- Strohfeldt, K.; Tacke, M. Bioorganometallic fulvene-derived titanocene anti-cancer drugs. Chem. Soc. Rev. 2008, 37, 1174–1187. [Google Scholar] [CrossRef] [PubMed]
- Schaad, L.J.; Hess, B.A., Jr. Hueckel molecular orbital π resonance energies. Question of the σ structure. J. Am. Chem. Soc. 1972, 94, 3068–3074. [Google Scholar] [CrossRef]
- Cyrański, M.K. Energetic aspects of cyclic pi-electron delocalization: Evaluation of the methods of estimating aromatic stabilization energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef]
- Malar, E.J.P.; Neumann, F.; Jug, K. Investigation of aromaticity in the excited states of fulvene. J. Mol. Struct. Theochem. 1995, 336, 81–84. [Google Scholar] [CrossRef]
- Stepień, B.T.; Krygowski, T.M.; Cyrański, M.K. Aromaticity strongly affected by substituents in fulvene and heptafulvene as a new method of estimating the resonance effect. Chem. Phys. Lett. 2001, 350, 537–542. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Palusiak, M.; Oziminski, W.P. Aromaticity of substituted fulvene derivatives: Substituent-dependent ring currents. Phys. Chem. Chem. Phys. 2010, 12, 10740–10745. [Google Scholar] [CrossRef]
- Shakerzadeh, E.; Noorizadeh, S. Aromaticity study on tria-, penta-and hepta-fulvene derivatives. Comput. Theor. Chem. 2011, 964, 141–147. [Google Scholar]
- Krygowski, T.M.; Soncini, A.; Oziminski, W.P.; Fowler, P.W. Aromatization of fulvene by complexation with lithium. Org. Lett. 2010, 12, 4880–4883. [Google Scholar]
- Krygowski, T.M.; Zachara-horeglad, J.E.; Pelloni, S.; Lazzeretti, P.; Palusiak, M. Relation between π-electron localization/delocalization and H-bond strength in derivatives of omicron-hydroxy-schiff bases. J. Org. Chem. 2008, 73, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Vatanparast, M.; Nekoei, A.R. An intramolecular hydrogen bond study in some Schiff bases of fulvene: A challenge between the RAHB concept and the σ-skeleton influence. New J. Chem. 2014, 38, 5886–5891. [Google Scholar]
- Vianello, R.; Maksić, Z.B. Tailoring of strong neutral organic superacids: DFT-B3LYP calculations on some fulvene derivatives. New J. Chem. 2004, 28, 843–846. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yang, X.; Li, W.Z.; Cheng, J.; Li, Q.Z. A σ-hole interaction with radical species as electron donors: Does single-electron tetrel bonding exist? Phys. Chem. Chem. Phys. 2014, 16, 11617–11625. [Google Scholar]
- Guo, X.; Liu, Y.W.; Li, W.Z.; Cheng, J.B.; Li, Q.Z. Competition and cooperativity between tetrel bond and chalcogen bond in complexes involving F2CX (X=Se and Te). Chem. Phys. Lett. 2015, 620, 7–12. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel bonding interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Influence of ring size on the strength of carbon bonding complexes between anions and perfluorocycloalkanes. Phys. Chem. Chem. Phys. 2014, 16, 19192–19197. [Google Scholar] [CrossRef]
- Scheiner, S. Assembly of effective halide receptors from components. Comparing hydrogen, halogen, and tetrel Bonds. J. Phys. Chem. A 2017, 121, 3606–3615. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Servati, G.M.; Stilinović, V.; Bauzá, A.; Frontera, A.; McArdle, P.; Derveer, D.V.; Mahmoudi, G. Design of lead(II) metal-organic frameworks based on covalent and tetrel bonding. Chem. Eur. J. 2015, 21, 17951–17958. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A.; Garczarek, P.; Stilinović, V.; Kirillov, A.M.; Kennedy, A.; Ruiz-Pérez, C.; Mahmoudi, G. Metal–organic and supramolecular lead (ii) networks assembled from isomeric nicotinoylhydrazone blocks: The effects of ligand geometry and counter-ion on topology and supramolecular assembly. CrystEngComm 2016, 18, 5375–5385. [Google Scholar]
- Bauzá, A.; Frontera, A.; Mahmoudi, G. Concurrent agostic and tetrel bonding interactions in lead(ii) complexes with an isonicotinohydrazide based ligand and several anions. Dalton Trans. 2016, 45, 4965–4969. [Google Scholar]
- Grabowski, S.J. Tetrel bond-σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Cheng, J.; Li, H.B.; Li, Q.Z. Tetrel bond of pseudohalide anions with XH3F (X=C, Si, Ge, and Sn) and its role in SN2 reaction. J. Chem. Phys. 2016, 145, 224310. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Del Bene, J.E.; Alkorta, I. Carbon–carbon bonding between nitrogen heterocyclic carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar]
- Mani, D.; Arunan, E. Microwave spectroscopic and atoms in molecules theoretical investigations on the Ar···propargyl alcohol complex: Ar···H-O, Ar···π, and Ar···C interactions. ChemPhysChem 2013, 14, 754–763. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C···Y (X=O/F, Y=O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Thomas, S.P.; Pavan, M.S.; Row, T.N.G. Experimental evidence for ‘carbon bonding’ in the solid state from charge density analysis. Chem. Commun. 2014, 50, 49–51. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Non-covalent sp3 carbon bonding with ArCF3 is analogous to CH-π interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridine TF3 and furan TF3 (T=C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. RCH3···O interactions in biological systems: Are they trifurcated H-bonds or noncovalent carbon bonds? Crystals 2016, 6, 26. [Google Scholar] [CrossRef]
- Bučar, D.K.; Halasz, I.; Stilinović, V. V=O···C interactions in crystal structures of oxovanadium-coordination compounds. New J. Chem. 2013, 37, 619–623. [Google Scholar]
- Azofra, L.M.; Scheiner, S. Tetrel, chalcogen, and CH···O hydrogen bonds in complexes pairing carbonyl-containing molecules with 1, 2, and 3 molecules of CO2. J. Chem. Phys. 2015, 142, 034307. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Azines as electron-pair donors to CO2 for N···C tetrel bonds. J. Phys. Chem. A 2017, 121, 8017–8025. [Google Scholar] [CrossRef] [PubMed]
- Cormanich, R.A.; Rittner, R.; O’Hagan, D.; Buhl, M. Inter- and intramolecular CF···C=O interactions on aliphatic and cyclohexane carbonyl derivatives. J. Comput. Chem. 2016, 37, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Theoretical study on σ-and π-hole carbon···carbon bonding interactions: Implications in CFC chemistry. Phys. Chem. Chem. Phys. 2016, 18, 32155–32159. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Yang, X.; Cheng, J.; Li, Q. Comparison for σ-hole and π-hole tetrel-bonded complexes involving F2C=CFTF3 (T=C, Si, and Ge): Substitution, hybridization, and solvation effects. J. Fluorine Chem. 2018, 207, 38–44. [Google Scholar] [CrossRef]
- Wei, Y.; Scheiner, S.; Li, Q. The π-tetrel bond and its influence on hydrogen bonding and proton transfer. ChemPhysChem 2018, 19, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, J.; Yang, X.; Li, Q. Comparison of σ-hole and π-hole tetrel bonds formed by pyrazine and 1,4-dicyanobenzene: The interplay between anion-π and tetrel bonds. ChemPhysChem 2017, 18, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, D. Sigma-hole carbon-bonding interactions in carbon-carbon double bonds: An unnoticed contact. Phys. Chem. Chem. Phys. 2017, 19, 15530–15540. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.; Schulz, N.; Andersson, H.; Nekoueishahraki, B.; Carlsson, A.C.C.; Sarabi, D.; Valkonen, A.; Rissanen, K.; Gräfenstein, J.; Keller, S.; et al. Carbon’s three-center-four-electron tetrel bond, treated experimentally. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.; Konar, S.; Lama, P.; Kinsuk, D.; Bauzá, A.; Frontera, A.; Mukhopadhyay, S. On the importance of noncovalent carbon-bonding interactions in the stabilization of a 1D Co(II) polymeric chain as a precursor of a novel 2D coordination polymer. J. Phys. Chem. B 2016, 120, 6803–6811. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Bader, R.F.W. AIM2000 Program, V. 2.0; McMaster University: Hamilton, ON, Canada, 2000. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- ADF2008.01, SCM, Theoretical Chemistry, VrijeUniversiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 20 November 2018).
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Schleyer, P.R.; Maerker, C.; Dransfeld, A.; Jiao, H.J.; van Eikema Hommes, N.J.R. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of monomer deformation for tetrel and pnicogen bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef]
- Aronld, W.D.; Oldfield, E. The chemical nature of hydrogen bonding in proteins via NMR: J-couplings, chemical shifts, and AIM theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Solel, E.; Kozuch, S. On the power of geometry over tetrel bonds. Molecules 2018, 23, 2742. [Google Scholar] [CrossRef] [PubMed]






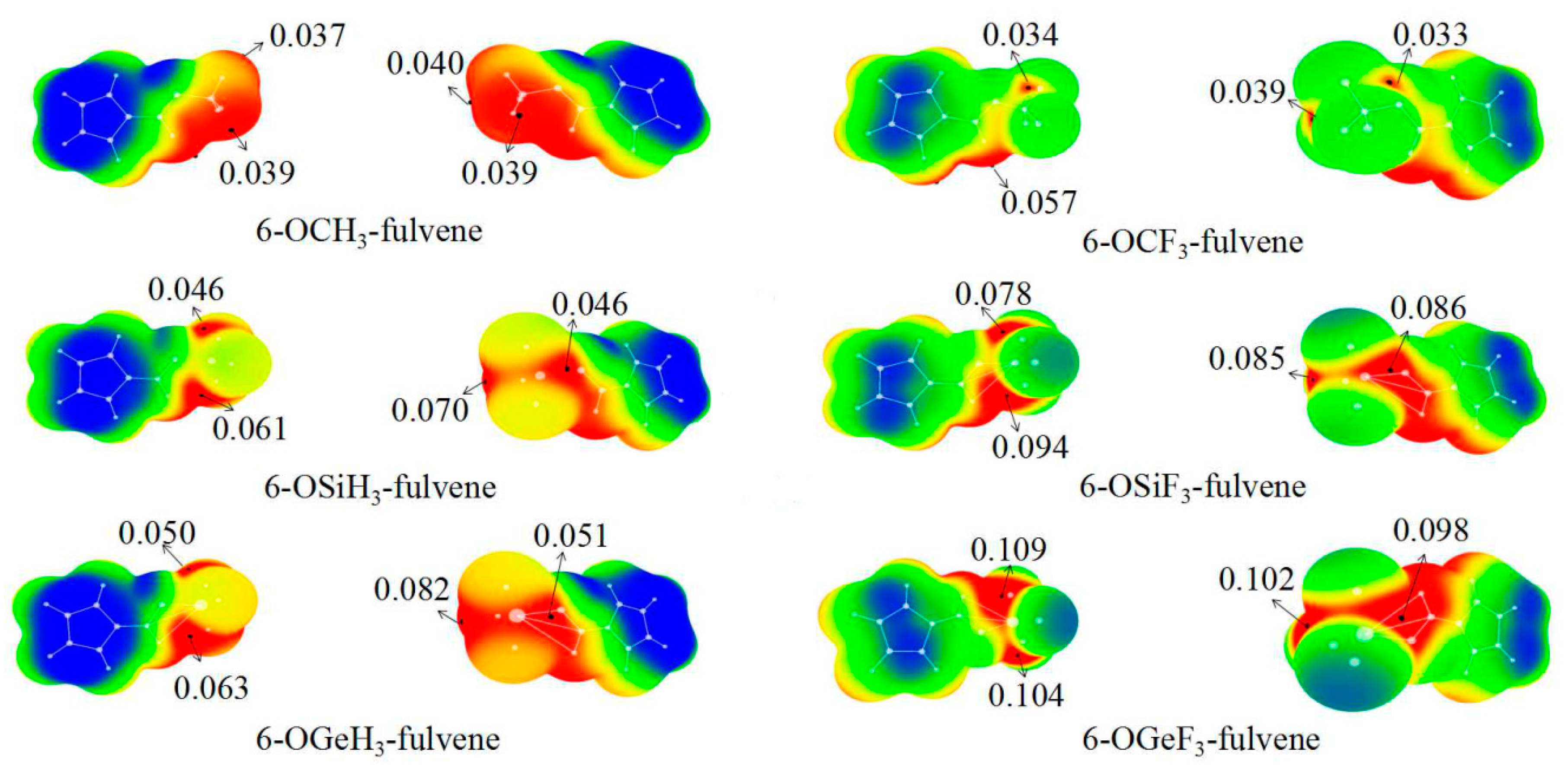{kind=link}
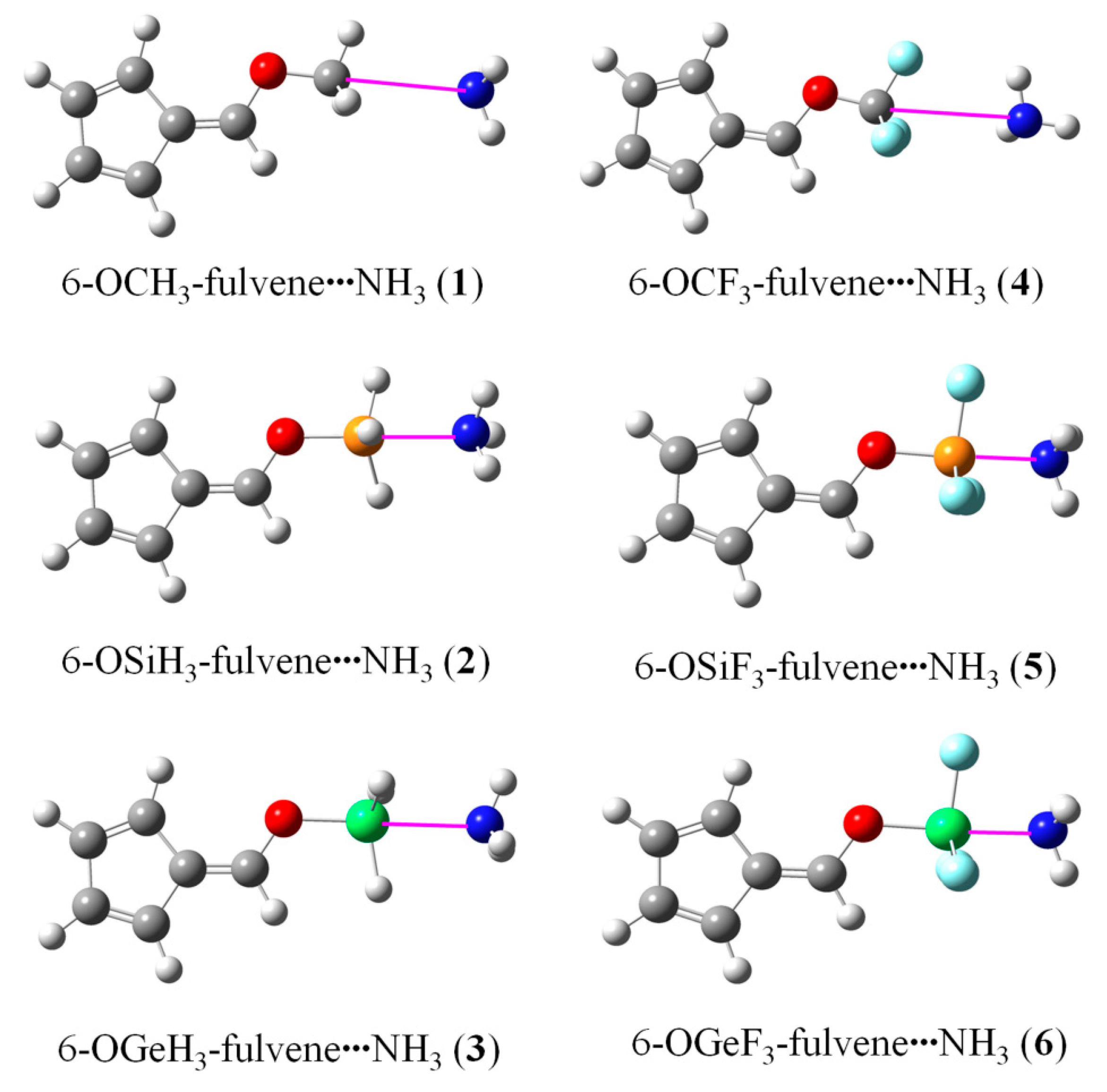{kind=link}
{kind=link}
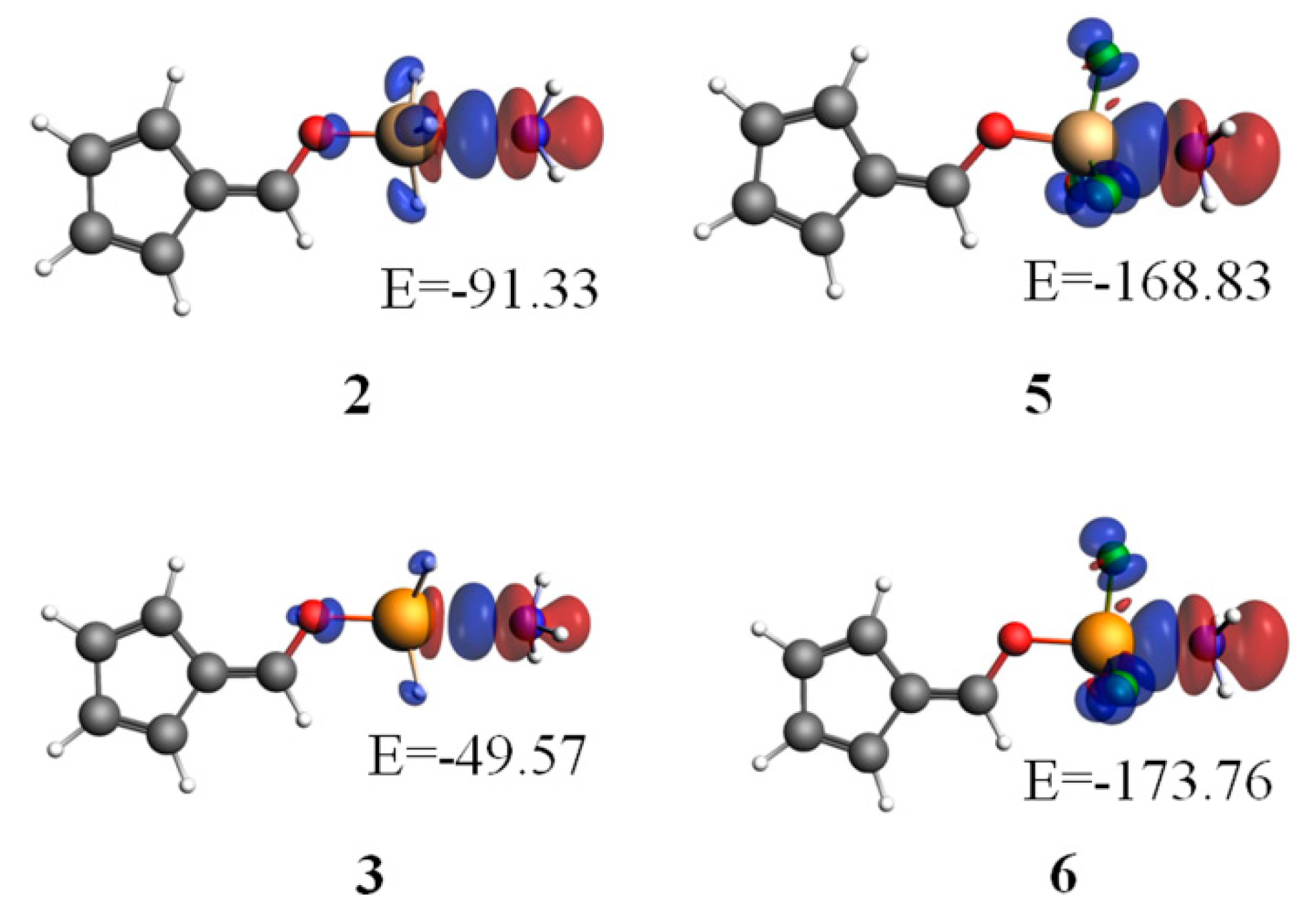{kind=link}
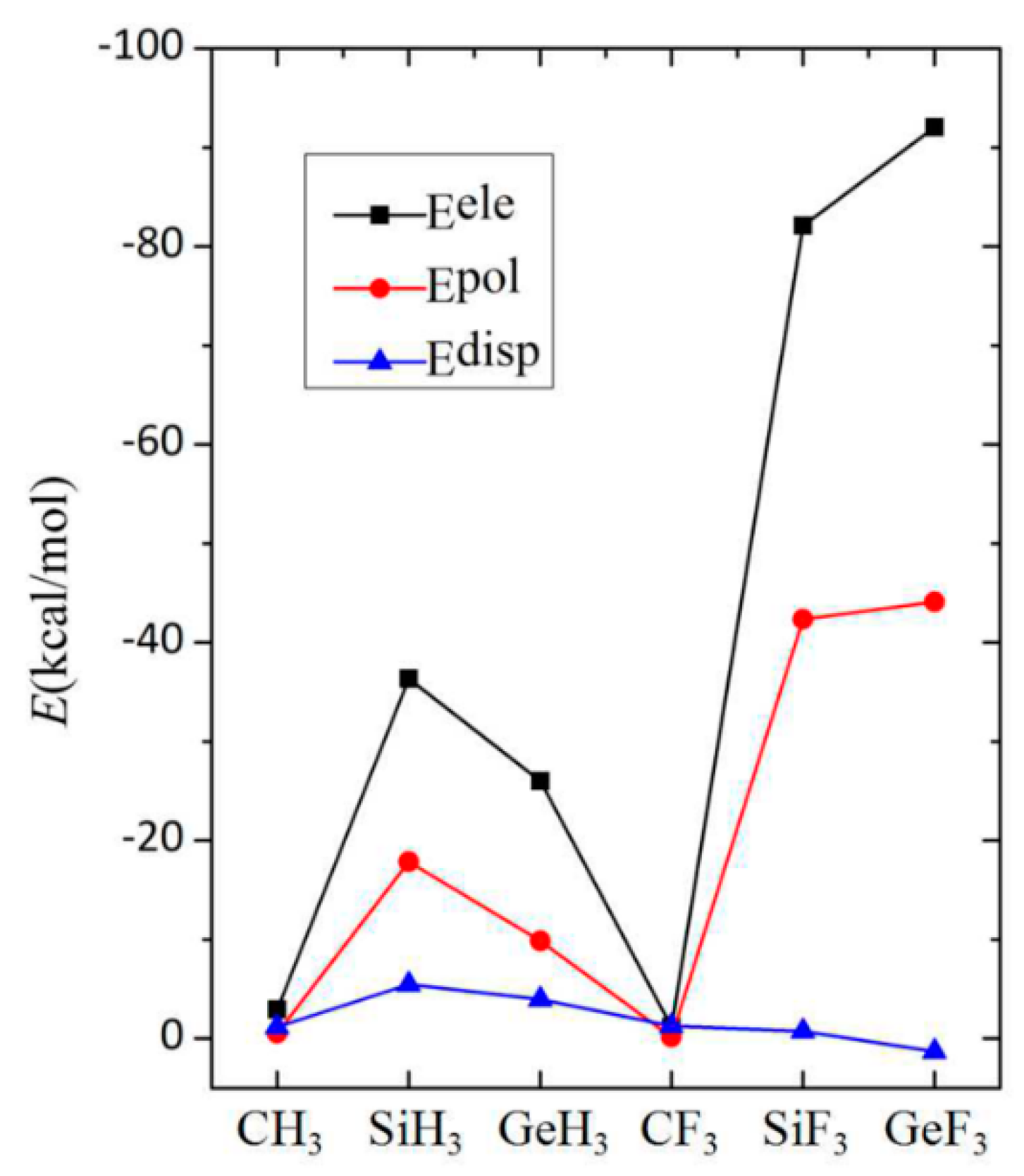{kind=link}
| Eint | Eb | DE | R | θ | ∆r | Vmax | |
|---|---|---|---|---|---|---|---|
| 6-OTX3-fulvene···NH3 | |||||||
| –CH3 | −9.93 | −9.90 | 0.02 | 3.228 | 178.3 | 0.006 | 0.040 |
| –SiH3 | −50.13 | −30.07 | 20.39 | 2.353 | 178.0 | 0.051 | 0.074 |
| –GeH3 | −39.30 | −29.73 | 9.56 | 2.565 | 180.0 | 0.044 | 0.084 |
| –CF3 | −4.12 | −3.97 | 0.15 | 3.498 | 169.3 | 0.003 | 0.039 |
| –SiF3 | −137.07 | −38.04 | 99.03 | 2.052 | 176.9 | 0.063 | 0.095 |
| –GeF3 | −151.31 | −70.36 | 80.94 | 2.074 | 176.7 | 0.046 | 0.113 |
| HOTX3···NH3 | |||||||
| –CH3 | −4.76 | −4.70 | 0.06 | 3.384 | 174.9 | 0.003 | 0.019 |
| –SiH3 | −18.24 | −14.89 | 3.35 | 2.792 | 175.9 | 0.017 | 0.051 |
| –GeH3 | −18.13 | −15.89 | 2.24 | 2.872 | 175.5 | 0.018 | 0.059 |
| –CF3 | −3.55 | −3.42 | 0.12 | 3.620 | 166.6 | 0.000 | 0.029 |
| –SiF3 | −101.03 | −14.96 | 86.07 | 2.120 | 176.2 | 0.041 | 0.078 |
| –GeF3 | −126.75 | −49.49 | 77.27 | 2.102 | 176.0 | 0.032 | 0.097 |
| PhOTX3···NH3 | |||||||
| –CH3 | −7.55 | −7.43 | 0.12 | 3.302 | 176.0 | 0.004 | 0.028 |
| –SiH3 | −30.78 | −20.56 | 10.22 | 2.539 | 177.6 | 0.032 | 0.057 |
| –GeH3 | −27.44 | −21.96 | 5.48 | 2.697 | 178.1 | 0.030 | 0.067 |
| –CF3 | −3.66 | −3.57 | 0.09 | 3.634 | 166.6 | 0.000 | 0.024 |
| –SiF3 | −115.29 | −22.83 | 92.46 | 2.088 | 176.9 | 0.050 | 0.080 |
| –GeF3 | −134.93 | −−56.92 | 79.17 | 2.094 | 175.8 | 0.038 | 0.096 |
| ρ | ∇2ρ | H | |
|---|---|---|---|
| 1 | 0.006 | 0.026 | 0.002 |
| 2 | 0.033 | 0.079 | −0.008 |
| 3 | 0.027 | 0.082 | −0.008 |
| 4 | 0.004 | 0.019 | 0.001 |
| 5 | 0.063 | 0.251 | −0.020 |
| 6 | 0.083 | 0.230 | −0.033 |
| CT | Orbitals | E(2) | Orbitals | E(2) | |
|---|---|---|---|---|---|
| 1 | 0.003 | Lp(N)→σ*C-O | 5.23 | ||
| 2 | 0.093 | Lp(N)→p*Si | 186.72 | Lp(N)→σ*Si-H | 60.69 |
| 3 | 0.062 | Lp(N)→p*Ge | 129.91 | Lp(N)→σ*Ge-H | 34.44 |
| 4 | −0.002 | Lp(F)→σ*N-H | 0.67 | ||
| 5 | 0.163 | Lp(N)→σ*Si-O | 102.66 | Lp(N)→σ*Si-F | 296.53 |
| 6 | 0.186 | Lp(N)→σ*Ge-O | 134.76 | Lp(N)→σ*Ge-F | 401.61 |
| NICS(1)zz | Δ | |
|---|---|---|
| 1 | −6.0781 (−5.8122) | −0.2659 |
| 2 | −6.7149 (−5.7074) | −1.0125 |
| 3 | −6.9462 (−6.0684) | −0.8778 |
| 4 | −5.2846 (−4.7814) | −0.5032 |
| 5 | −6.0377 (−5.7074) | −0.3303 |
| 6 | −6.2645 (−4.9984) | −1.2661 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, M.-C.; Yang, S.-B.; Li, Q.-Z.; Cheng, J.-B.; Li, H.-B.; Liu, S.-F. Tetrel Bond between 6-OTX3-Fulvene and NH3: Substituents and Aromaticity. Molecules 2019, 24, 10. https://doi.org/10.3390/molecules24010010
Hou M-C, Yang S-B, Li Q-Z, Cheng J-B, Li H-B, Liu S-F. Tetrel Bond between 6-OTX3-Fulvene and NH3: Substituents and Aromaticity. Molecules. 2019; 24(1):10. https://doi.org/10.3390/molecules24010010
Chicago/Turabian StyleHou, Ming-Chang, Shu-Bin Yang, Qing-Zhong Li, Jian-Bo Cheng, Hai-Bei Li, and Shu-Feng Liu. 2019. "Tetrel Bond between 6-OTX3-Fulvene and NH3: Substituents and Aromaticity" Molecules 24, no. 1: 10. https://doi.org/10.3390/molecules24010010
APA StyleHou, M.-C., Yang, S.-B., Li, Q.-Z., Cheng, J.-B., Li, H.-B., & Liu, S.-F. (2019). Tetrel Bond between 6-OTX3-Fulvene and NH3: Substituents and Aromaticity. Molecules, 24(1), 10. https://doi.org/10.3390/molecules24010010