Esterification of Aryl/Alkyl Acids Catalysed by N-bromosuccinimide under Mild Reaction Conditions




Abstract
1. Introduction
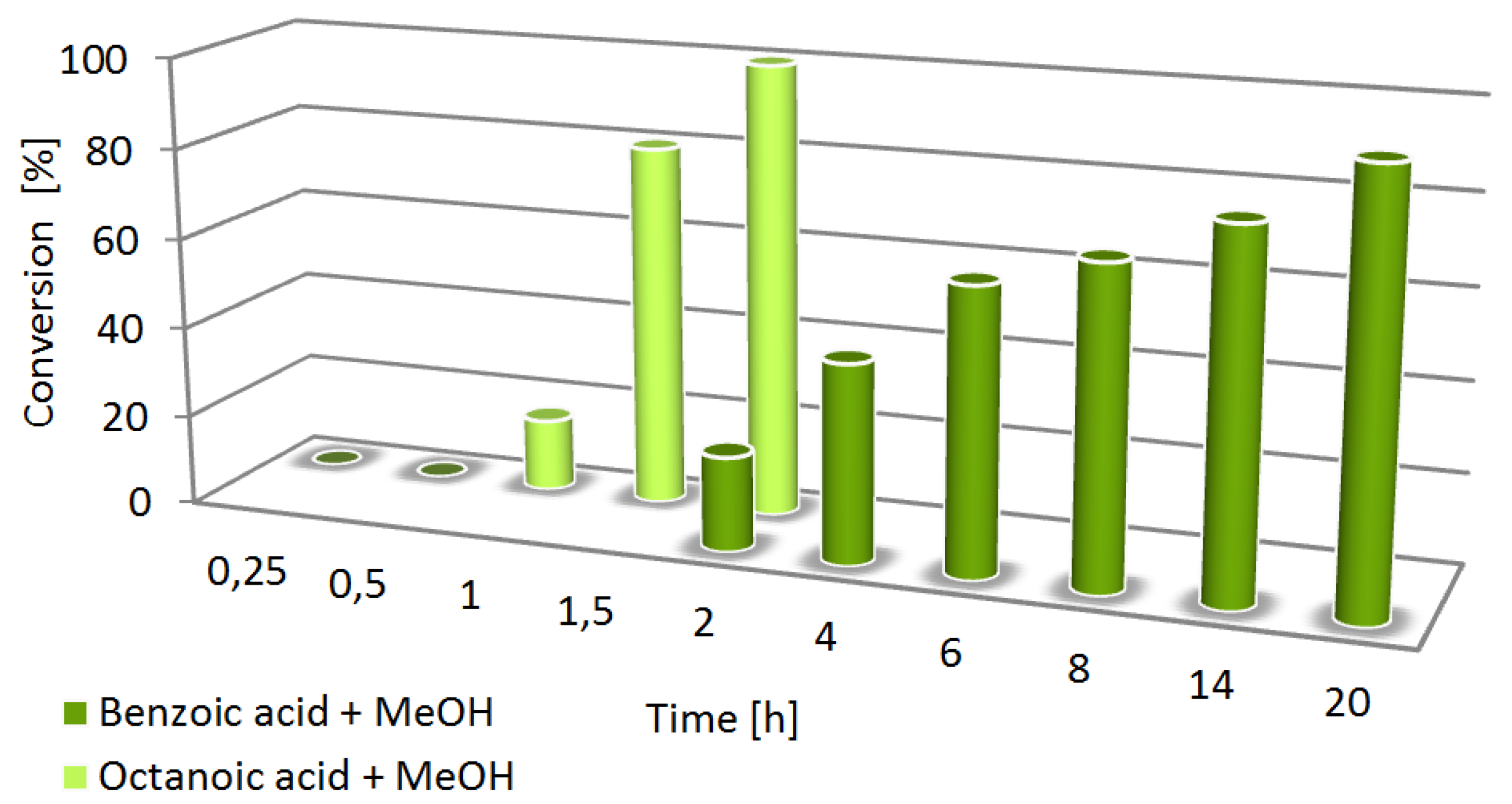
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental Procedures
3.2.1. General Procedure for the Esterification between Carboxylic Acids and Alcohols
3.2.2. Scale-Up Procedure for Preparation of Methyl Benzoate (1a) and Isolation of Succinimide
- Yield (methyl benzoate): 4.60 g, 85%.
- Yield (succinimide [57]): 272 mg, 98%.
- 1H NMR (300 MHz, CDCl3) δ 10.04 (s, 1H), 2.72 (s, 4H).
- 13C NMR (76 MHz, CDCl3) δ 178.8, 29.4.
3.2.3. Scale-Up Procedure for Preparation of Trimethyl Citrate (16a)
- Yield: 2.55 g, 99%.
3.2.4. Scale-Up Procedure for Preparation of Methyl Stearate (13a)
- Yield: 2.99 g, 100%.
3.2.5. Procedure for Recycling of N-bromosuccinimide (NBS) from Waste Succinimide
3.2.6. Detailed Procedures for the Preparation of Synthesized Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Otera, J. Esterification. Esterification; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; pp. 3–144. ISBN 9783527601844. [Google Scholar]
- Baskar, G.; Aiswarya, R. Trends in catalytic production of biodiesel from various feedstocks. Renew. Sustain. Energy Rev. 2016, 57, 496–504. [Google Scholar] [CrossRef]
- Talebian-Kiakalaieh, A.; Amin, N.A.S.; Mazaheri, H. A review on novel processes of biodiesel production from waste cooking oil. Appl. Energy 2013, 104, 683–710. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Sodagar, E. Esterification of free fatty acids (Biodiesel) using nano sulfated-titania as catalyst in solvent-free conditions. C R. Chim. 2013, 16, 229–238. [Google Scholar] [CrossRef]
- Mallesham, B.; Govinda Rao, B.; Reddy, B.M. Production of biofuel additives by esterification and acetalization of bioglycerol. C R. Chim. 2016, 19, 1194–1202. [Google Scholar] [CrossRef]
- Tsakos, M.; Schaffert, E.S.; Clement, L.L.; Villadsen, N.L.; Poulsen, T.B. Ester coupling reactions-An enduring challenge in the chemical synthesis of bioactive natural products. Nat. Prod. Rep. 2015, 32, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Larock, R.C.; Dubrovskiy, A.V.; Markina, N.A.; Pletnev, A.A.; Kesharwani, T.; Raminelli, C.; Yao, T.; Zeni, G.; Zhang, L.; Rozhkov, R. Comprehensive Organic Transformations, 4 Volume Set: A Guide to Functional Group Preparations, 3rd ed.; Larock, R.C., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2018; Volume 1, pp. 3753–3764. ISBN 9780470927953. [Google Scholar]
- Ishihara, K. Dehydrative condensation catalyses. Tetrahedron 2009, 65, 1085–1109. [Google Scholar] [CrossRef]
- Sakakura, A.; Koshikari, Y.; Ishihara, K. Open-air and solvent-free ester condensation catalyzed by sulfonic acids. Tetrahedron Lett. 2008, 49, 5017–5020. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Yang, T.; Cai, B.; Chang, X.; Pan, H. Highly efficient metal salt catalyst for the esterification of biomass derived levulinic acid under microwave irradiation. RSC Adv. 2016, 6, 2106–2111. [Google Scholar] [CrossRef]
- Jeschke, J.; Korb, M.; Rüffer, T.; Gäbler, C.; Lang, H. Atom economic ruthenium-catalyzed synthesis of bulky β-Oxo esters. Adv. Synth. Catal. 2015, 357, 4069–4081. [Google Scholar] [CrossRef]
- Da Silva, M.J.; Liberto, N.A.; De Andrade Leles, L.C.; Pereira, U.A. Fe4(SiW12O40)3-catalyzed glycerol acetylation: Synthesis of bioadditives by using highly active Lewis acid catalyst. J. Mol. Catal. A Chem. 2016, 422, 69–83. [Google Scholar] [CrossRef]
- Kato, C.N.; Ogasawara, T.; Kondo, A.; Kato, D. Heterogeneous esterification of fatty acids with methanol catalyzed by Lewis acidic organozirconium complexes with Keggin-type mono-aluminum-substituted polyoxotungstates. Catal. Commun. 2017, 96, 41–45. [Google Scholar] [CrossRef]
- Minakawa, M.; Baek, H.; Yamada, Y.M.A.; Han, J.W.; Uozumi, Y. Direct dehydrative esterification of alcohols and carboxylic acids with a macroporous polymeric acid catalyst. Org. Lett. 2013, 15, 5798–5801. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anna, M.M.; Capodiferro, V.F.; Mali, M.; Mastrorilli, P. Esterification, transesterification and hydrogenation reactions of polyunsaturated compounds catalyzed by a recyclable polymer supported palladium catalyst. J. Organomet. Chem. 2016, 818, 106–114. [Google Scholar] [CrossRef]
- Furuta, A.; Fukuyama, T.; Ryu, I. Efficient flow fischer esterification of carboxylic acids with alcohols using sulfonic acid-functionalized silica as supported catalyst. Bull. Chem. Soc. Jpn. 2017, 90, 607–612. [Google Scholar] [CrossRef]
- Chen, Z.; Wen, Y.; Fu, Y.; Chen, H.; Ye, M.; Luo, G. Graphene oxide: An efficient acid catalyst for the construction of esters from acids and alcohols. Synlett 2017, 28, 981–985. [Google Scholar] [CrossRef]
- Han, X.-X.; Du, H.; Hung, C.-T.; Liu, L.-L.; Wu, P.-H.; Ren, D.-H.; Huang, S.-J.; Liu, S.-B. Syntheses of novel halogen-free Bronsted-Lewis acidic ionic liquid catalysts and their applications for synthesis of methyl caprylate. Green Chem. 2015, 17, 499–508. [Google Scholar] [CrossRef]
- Dong, B.; Song, H.; Zhang, W.; He, A.; Yao, S. Ionic liquids as heterogeneous and homogeneous catalysts for condensation and esterification reactions. Curr. Org. Chem. 2016, 20, 2894–2910. [Google Scholar] [CrossRef]
- Phakhodee, W.; Duangkamol, C.; Pattarawarapan, M. Ph3P-I2 mediated aryl esterification with a mechanistic insight. Tetrahedron Lett. 2016, 57, 2087–2089. [Google Scholar] [CrossRef]
- Yeh, W.K.; Yang, H.C.; McCarthy, J.R. Enzyme Technologies: Metagenomics, Evolution, Biocatalysis and Biosynthesis; Wiley: Hoboken, NJ, USA, 2011; pp. 125–250. ISBN 9781118125038. [Google Scholar]
- Bezbradica, D.; Crovic, M.; Tanaskovic, S.J.; Lukovic, N.; Carevic, M.; Milivojevic, A.; Knezevic-Jugovic, Z. Enzymatic Syntheses of Esters-Green Chemistry for Valuable Food, Fuel and Fine Chemicals. Curr. Org. Chem. 2017, 21, 104–138. [Google Scholar] [CrossRef]
- Gokulakrishnan, N.; Pandurangan, A.; Sinha, P.K. Esterification of acetic acid with propanol isomers under autogeneous pressure: A catalytic activity study of Al-MCM-41 molecular sieves. J. Mol. Catal. A Chem. 2007, 263, 55–61. [Google Scholar] [CrossRef]
- Chung, K.-H.; Park, B.-G. Esterification of oleic acid in soybean oil on zeolite catalysts with different acidity. J. Ind. Eng. Chem. 2009, 15, 388–392. [Google Scholar] [CrossRef]
- Kolvari, E.; Ghorbani-Choghamarani, A.; Salehi, P.; Shirini, F.; Zolfigol, M.A. Application of N-halo reagents in organic synthesis. J. Iran. Chem. Soc. 2007, 4, 126–174. [Google Scholar] [CrossRef]
- Koval, I.V. N-Halo Reagents. N-Halosuccinimides in organic synthesis and in chemistry of natural compounds. Russ. J. Org. Chem. 2002, 38, 301–337. [Google Scholar] [CrossRef]
- Barton, D.; Ollis, W.D. Comprehensive Organic Chemistry: The Synthesis and Reactions of Organic Compounds, 1st ed.; Pergamon Press: Oxford, UK; New York, NY, USA, 1979; Volume 2. [Google Scholar]
- Kadam, S.T.; Kim, S.S. N-Iodosuccinimide (NIS) a novel and effective catalyst for the cyanosilylation of aldehydes under mild reaction conditions. Catal. Commun. 2008, 9, 1342–1345. [Google Scholar] [CrossRef]
- Nagarajappa Giridhar, B.; Pandey Krishna, K.; Shinde Aniket, S.; Vagdevi Hosadu, M. N-Bromosuccinimide (NBS)–An efficient catalyst for acetylation of wood. Holzforschung 2016, 70, 421–427. [Google Scholar] [CrossRef]
- Maleki, B.; Sedigh Ashrafi, S. N-Bromosuccinimide catalyzed three component one-pot efficient synthesis of 2,4,5-Triaryl-1H-imidazoles from aldehyde, ammonium acetate, and 1,2-Diketone or ±-Hydroxyketone. J. Mex. Chem. Soc. 2014, 58, 76–81. [Google Scholar] [CrossRef]
- Karimi, B.; Zamani, A.; Zareyee, D. N-Iodosuccinimide (NIS) as a mild and highly chemoselective catalyst for deprotection of tert-butyldimethylsilyl ethers. Tetrahedron Lett. 2004, 45, 9139–9141. [Google Scholar] [CrossRef]
- Saikia, I.; Borah, A.J.; Phukan, P. Use of bromine and bromo-organic compounds in organic synthesis. Chem. Rev. 2016, 116, 6837–7042. [Google Scholar] [CrossRef] [PubMed]
- Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Aerobic oxidative iodination of organic compounds with iodide catalyzed by sodium nitrite. Adv. Synth. Catal. 2008, 350, 2921–2929. [Google Scholar] [CrossRef]
- Stavber, G.; Iskra, J.; Zupan, M.; Stavber, S. Aerobic oxidative iodination of ketones catalysed by sodium nitrite "on water" or in a micelle-based aqueous system. Green Chem. 2009, 11, 1262–1267. [Google Scholar] [CrossRef]
- Stavber, G.; Stavber, S. Towards Greener Fluorine Organic Chemistry: Direct Electrophilic Fluorination of Carbonyl Compounds in Water and Under Solvent-Free Reaction Conditions. Adv. Synth. Catal. 2010, 352, 2838–2846. [Google Scholar] [CrossRef]
- Prebil, R.; Stavber, G.; Stavber, S. Aerobic oxidation of alcohols by using a completely metal-free catalytic system. Eur. J. Org. Chem. 2014, 2014, 395–402. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Direct halogenation of alcohols with halosilanes under catalyst- and organic solvent-free reaction conditions. Tetrahedron Lett. 2016, 57, 2430–2433. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Transformation of tertiary benzyl alcohols into the vicinal halo-substituted derivatives using N-Halosuccinimides. Molecules 2016, 21, 1325. [Google Scholar] [CrossRef] [PubMed]
- Vražič, D.; Jereb, M.; Laali, K.; Stavber, S. Brønsted acidic ionic liquid accelerated halogenation of organic compounds with N-Halosuccinimides (NXS). Molecules 2013, 18, 74–96. [Google Scholar] [CrossRef] [PubMed]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Environmentally benign electrophilic and radical bromination ‘on water’: H2O2–HBr system versus N-bromosuccinimide. Tetrahedron 2009, 65, 4429–4439. [Google Scholar] [CrossRef]
- Jereb, M.; Zupan, M.; Stavber, S. Visible-light-promoted wohl–ziegler functionalization of organic molecules with N-Bromosuccinimide under solvent-free reaction conditions. Helv. Chim. Acta 2009, 92, 555–566. [Google Scholar] [CrossRef]
- Pravst, I.; Zupan, M.; Stavber, S. Halogenation of ketones with N-halosuccinimides under solvent-free reaction conditions. Tetrahedron 2008, 64, 5191–5199. [Google Scholar] [CrossRef]
- Pravst, I.; Zupan, M.; Stavber, S. Directed regioselectivity of bromination of ketones with NBS: Solvent-free conditions versus water. Tetrahedron Lett. 2006, 47, 4707–4710. [Google Scholar] [CrossRef]
- Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Visible light induced ‘on water’ benzylic bromination with N-bromosuccinimide. Tetrahedron Lett. 2006, 47, 1097–1099. [Google Scholar] [CrossRef]
- Pravst, I.; Zupan, M.; Stavber, S. Solvent-free bromination of 1,3-diketones and β-keto esters with NBS. Green Chem. 2006, 8, 1001–1005. [Google Scholar] [CrossRef]
- Bandgar, B.P.; Uppalla, L.S.; Sadavarte, V.S. Chemoselective transesterification of β-Keto esters under neutral conditions using NBS as a catalyst. Synlett 2001, 2001, 1715–1718. [Google Scholar] [CrossRef]
- Karimi, B.; Seradj, H. N-Bromosuccinimide (NBS), a novel and highly effective catalyst for acetylation of alcohols under mild reaction conditions. Synlett 2001, 2001, 519–520. [Google Scholar] [CrossRef]
- Sucheta, K.; Reddy, G.S.R.; Ravi, D.; Rama Rao, N. A novel general route to the synthesis of carboxylic acid esters and thiolesters. Tetrahedron Lett. 1994, 35, 4415–4416. [Google Scholar] [CrossRef]
- Ramalinga, K.; Vijayalakshmi, P.; Kaimal, T.N.B. A mild and efficient method for esterification and transesterification catalyzed by iodine. Tetrahedron Lett. 2002, 43, 879–882. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Dual behavior of alcohols in iodine-catalyzed esterification under solvent-free reaction conditions. Tetrahedron Lett. 2009, 50, 2347–2352. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Iodine-catalyzed transformation of molecules containing oxygen functional groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Vairamani, M.; Rao, G.K.V. Use of bromine in methanol - preparation of methyl-esters. Indian J. Chem. Sect B 1985, 24, 691. [Google Scholar]
- Bowman, P.T.; Ko, E.I.; Sides, P.J. A potential hazard in preparing bromine-methanol solutions. J. Electrochem. Soc. 1990, 137, 1309–1311. [Google Scholar] [CrossRef]
- Virtanen, E.; Kolehmainen, E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004, 2004, 3385–3399. [Google Scholar] [CrossRef]
- Cravotto, G.; Binello, A.; Boffa, L.; Rosati, O.; Boccalini, M.; Chimichi, S. Regio- and stereoselective reductions of dehydrocholic acid. Steroids 2006, 71, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Mal, P. Electrophilic aryl-halogenation using N-halosuccinimides under ball-milling. Tetrahedron Lett. 2014, 55, 2154–2156. [Google Scholar] [CrossRef]
- Annese, C.; D'Accolti, L.; Fusco, C.; Licini, G.; Zonta, C. Heterolytic (2 e) vs Homolytic (1 e) Oxidation reactivity: N−H versus C−H switch in the oxidation of lactams by Dioxirans. Chem. Eur. J. 2017, 23, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.; Lyons, D.J.M. A novel aromatic carbocation-based coupling reagent for esterification and amidation reactions. Chem. Commun. 2015, 51, 3131–3134. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Zhao, L.; Liu, W.; Huang, X.; Song, H.; Zhang, T. Convenient, metal-free ipso-nitration of arylboronic acids using nitric acid and trifluoroacetic acid. Synth. Commun. 2017, 47, 10–14. [Google Scholar] [CrossRef]
- Jia, J.; Jiang, Q.; Zhao, A.; Xu, B.; Liu, Q.; Luo, W.-P.; Guo, C.-C. Copper-catalyzed O-methylation of carboxylic acids using DMSO as a methyl source. Synthesis 2016, 48, 421–428. [Google Scholar] [CrossRef]
- Powell, A.B.; Stahl, S.S. Aerobic Oxidation of diverse primary alcohols to methyl esters with a readily accessible heterogeneous Pd/Bi/Te catalyst. Org. Lett. 2013, 15, 5072–5075. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.W.; Buchwald, S.L. Mild and General palladium-catalyzed synthesis of methyl aryl ethers enabled by the use of a palladacycle precatalyst. Org. Lett. 2013, 15, 3998–4001. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.D.; Yodsanit, N.; Melander, C. Potentiation of the fosmidomycin analogue FR 900098 with substituted 2-oxazolines against Francisella novicida. MedChemComm 2016, 7, 1952–1956. [Google Scholar] [CrossRef] [PubMed]
- Offermann, D.A.; McKendrick, J.E.; Sejberg, J.J.P.; Mo, B.; Holdom, M.D.; Helm, B.A.; Leatherbarrow, R.J.; Beavil, A.J.; Sutton, B.J.; Spivey, A.C. Synthesis and incorporation into cyclic peptides of tolan amino acids and their hydrogenated congeners: construction of an array of A–B-loop mimetics of the Cε3 domain of human IgE. J. Org. Chem. 2012, 77, 3197–3214. [Google Scholar] [CrossRef] [PubMed]
- Strazzolini, P.; Gambi, A.G.; Giumanini, A.; Vancik, H. The reaction between ethanedioyl (oxalyl) dihalides and Ag2C2O4: A route to Staudinger's elusive ethanedioic (oxalic) acid anhydride. J. Chem. Soc. Perkin Trans. 1998, 1, 2553–2558. [Google Scholar] [CrossRef]
- Sun, H.-B.; Hua, R.; Yin, Y. ZrOCl2·8H2O: An efficient, cheap and reusable catalyst for the esterification of acrylic acid and other carboxylic acids with equimolar amounts of alcohols. Molecules 2006, 11, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Maslov, M.A.; Morozova, N.G.; Solomatina, T.V.; Shaforostova, N.G.; Serebrennikova, G.A. Synthesis of amino analogues of cholic acid. Russ. J. Bioorg. Chem. 2011, 37, 507–515. [Google Scholar] [CrossRef]
- Rohacova, J.; Marin, M.L.; Martinez-Romero, A.; O'Connor, J.-E.; Gomez-Lechon, M.J.; Donato, M.T.; Castell, J.V.; Miranda, M.A. Synthesis of new, UV-photoactive dansyl derivatives for flow cytometric studies on bile acid uptake. Org. Biomol. Chem. 2009, 7, 4973–4980. [Google Scholar] [CrossRef] [PubMed]
- Hutchby, M.; Houlden, C.E.; Haddow, M.F.; Tyler, S.N.G.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Switching pathways: room-temperature neutral solvolysis and substitution of amides. Angew. Chem. Int. Ed. 2012, 51, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zatolochnaya, O.V.; Wang, X.-J.; Rodríguez, S.; Qu, B.; Desrosiers, J.-N.; Mangunuru, H.P.R.; Biswas, S.; Rivalti, D.; Karyakarte, S.D.; Sieber, J.D.; Grinberg, N.; Wu, L.; Lee, H.; Haddad, N.; Fandrick, D.R.; Yee, N.K.; Song, J.J.; Senanayake, C.H. BABIPhos family of biaryl dihydrobenzooxaphosphole ligands for asymmetric hydrogenation. Org. Lett. 2018, 20, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Dethe, D.H.; Erande, R.D.; Ranjan, A. Biomimetic total syntheses of borreverine and flinderole alkaloids. J. Org. Chem. 2013, 78, 10106–10120. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Li, A.N.; Zhou, J.; Guo, Y.; Lin, L.; Chen, W.; Wang, R. Mg(OMe)2 promoted allylic isomerization of γ-hydroxy-α,β-alkenoic esters to synthesize γ-ketone esters. Org. Biomol. Chem. 2017, 15, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Suwada, M.; Fukuhara, T.; Hara, S. Selective mono-fluorination of diols via a cyclic acetal of N,N-diethyl-4-methoxybenzamide. J. Fluorine Chem. 2007, 128, 1121–1125. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhao, A.; Xu, B.; Jia, J.; Liu, X.; Guo, C. PIFA-Mediated Esterification Reaction of Alkynes with Alcohols via Oxidative Cleavage of Carbon Triple Bonds. J. Org. Chem. 2014, 79, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Fujita, T.; Sakamoto, M.; Kuramochi, T.; Kitazume, T. Reactions of monoesters of ethylene glycol with N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine. J. Fluorine Chem. 1987, 36, 361–372. [Google Scholar] [CrossRef]
- Umeda, R.; Nishimura, T.; Kaiba, K.; Tanaka, T.; Takahashi, Y.; Nishiyama, Y. Rhenium complex-catalyzed acylative cleavage of ethers with acyl chlorides. Tetrahedron 2011, 67, 7217–7221. [Google Scholar] [CrossRef]
- Whittaker, A.M.; Dong, V.M. Nickel-catalyzed dehydrogenative cross-coupling: direct transformation of aldehydes into esters and amides. Angew. Chem. Int. Ed. 2015, 54, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Gianetti, T.L.; Annen, S.P.; Santiso-Quinones, G.; Reiher, M.; Driess, M.; Grützmacher, H. Nitrous Oxide as a Hydrogen Acceptor for the Dehydrogenative Coupling of Alcohols. Angew. Chem. Int. Ed. 2016, 55, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Kumar, A.; Sain, B.; Gupta, A.K. A simple and convenient one-pot synthesis of fatty acid esters from hindered alcohols using N,N-dimethylchloro-sulfitemethaniminium chloride as dehydrating agent. Synth. Commun. 2002, 32, 2885–2891. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
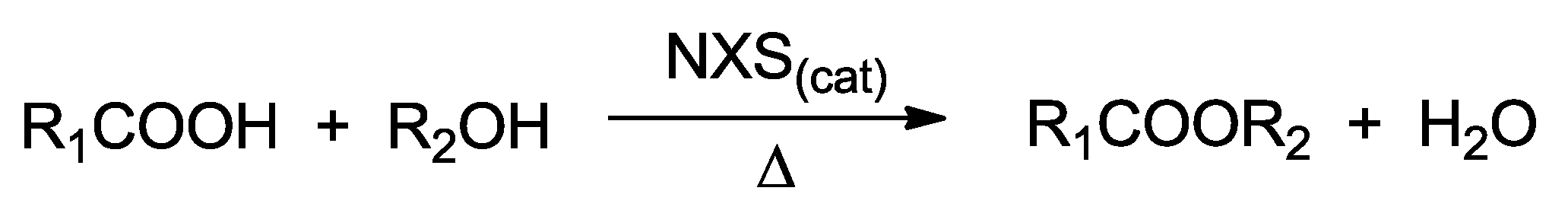{kind=link}

| Entry | NXS | Loading [mol%] | Temperature [°C] | Conversion of Benzoic Acid to Methyl Benzoate [%] 2 |
|---|---|---|---|---|
| 1 | / | / | 70 | 0 |
| 2 | NCS | 15 | 70 | 17 |
| 3 | NBS | 15 | 70 | 94 |
| 4 | NIS | 15 | 70 | 30 |
| 5 | NBS | 10 | 70 | 94 |
| 6 | NBS | 7 | 70 | 94 |
| 7 | NBS | 3 | 70 | 76 |
| 8 3 | NBS | 7 | 70 | <10 |
| 9 | NBS | 7 | 30 | 0 |
| 10 | NBS | 7 | 50 | 75 |
| 11 | NBS | 7 | 100 | 94 |
| 12 | HCl | 7 | 70 | 93 |
| 13 | HBr | 7 | 70 | 84 |
| 14 | HI | 7 | 70 | 68 |
| 15 | Br2 | 7 | 70 | 94 |

| R2OH | R1 = Ph | R1 = Heptyl |
|---|---|---|
 | ||

| Methyl Benzoates |
 |
 |
| Methyl Alkyl Esters and Methyl Esters of Cholic Acid Derivatives |
 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čebular, K.; Božić, B.Đ.; Stavber, S. Esterification of Aryl/Alkyl Acids Catalysed by N-bromosuccinimide under Mild Reaction Conditions. Molecules 2018, 23, 2235. https://doi.org/10.3390/molecules23092235
Čebular K, Božić BĐ, Stavber S. Esterification of Aryl/Alkyl Acids Catalysed by N-bromosuccinimide under Mild Reaction Conditions. Molecules. 2018; 23(9):2235. https://doi.org/10.3390/molecules23092235
Chicago/Turabian StyleČebular, Klara, Bojan Đ. Božić, and Stojan Stavber. 2018. "Esterification of Aryl/Alkyl Acids Catalysed by N-bromosuccinimide under Mild Reaction Conditions" Molecules 23, no. 9: 2235. https://doi.org/10.3390/molecules23092235
APA StyleČebular, K., Božić, B. Đ., & Stavber, S. (2018). Esterification of Aryl/Alkyl Acids Catalysed by N-bromosuccinimide under Mild Reaction Conditions. Molecules, 23(9), 2235. https://doi.org/10.3390/molecules23092235