
Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
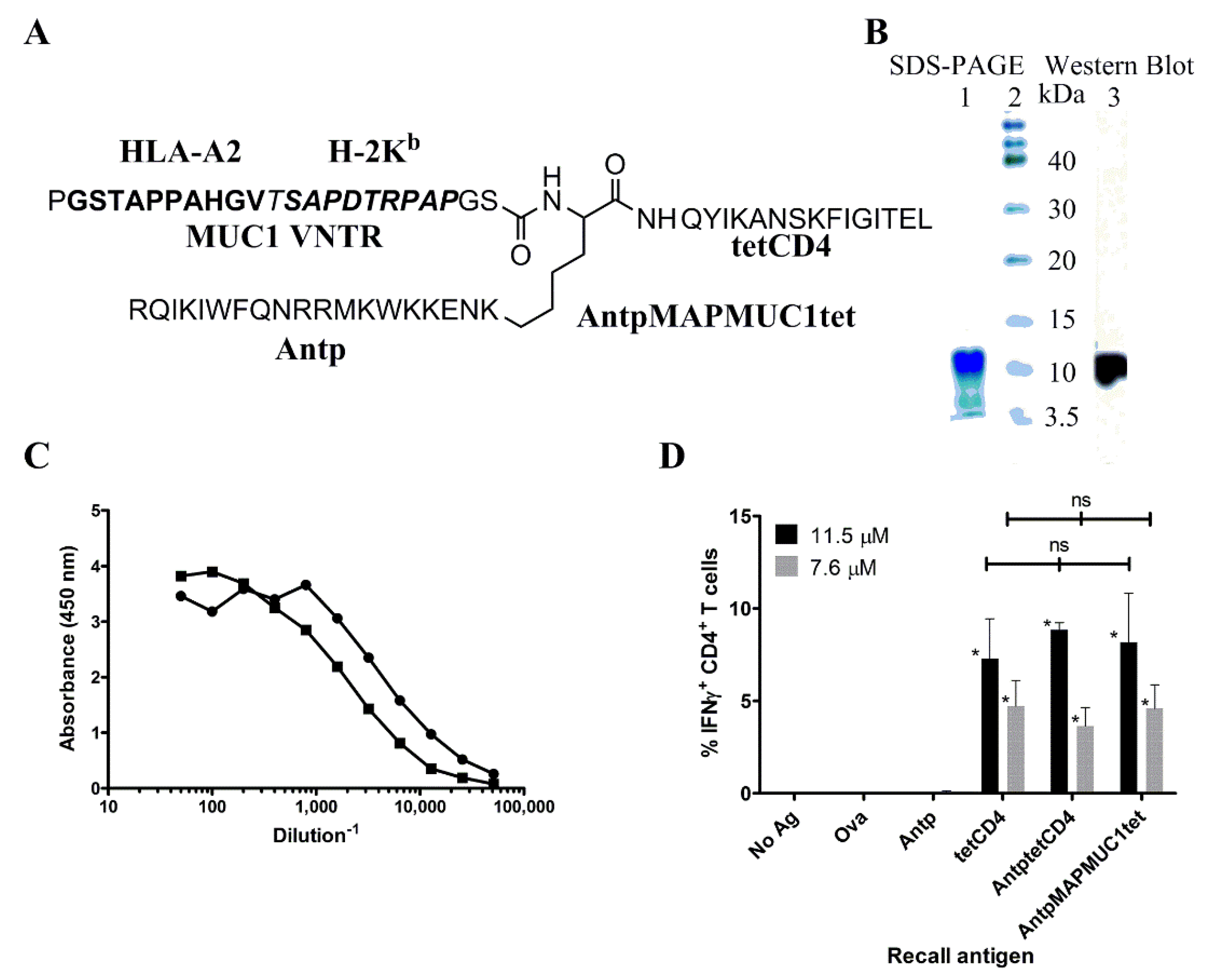
2.1. Biochemical and Immunochemical Characterisation of the Tripartite Peptide Containing Penetratin, MUC1 VNTR and Tetanus Toxin CD4 Peptide
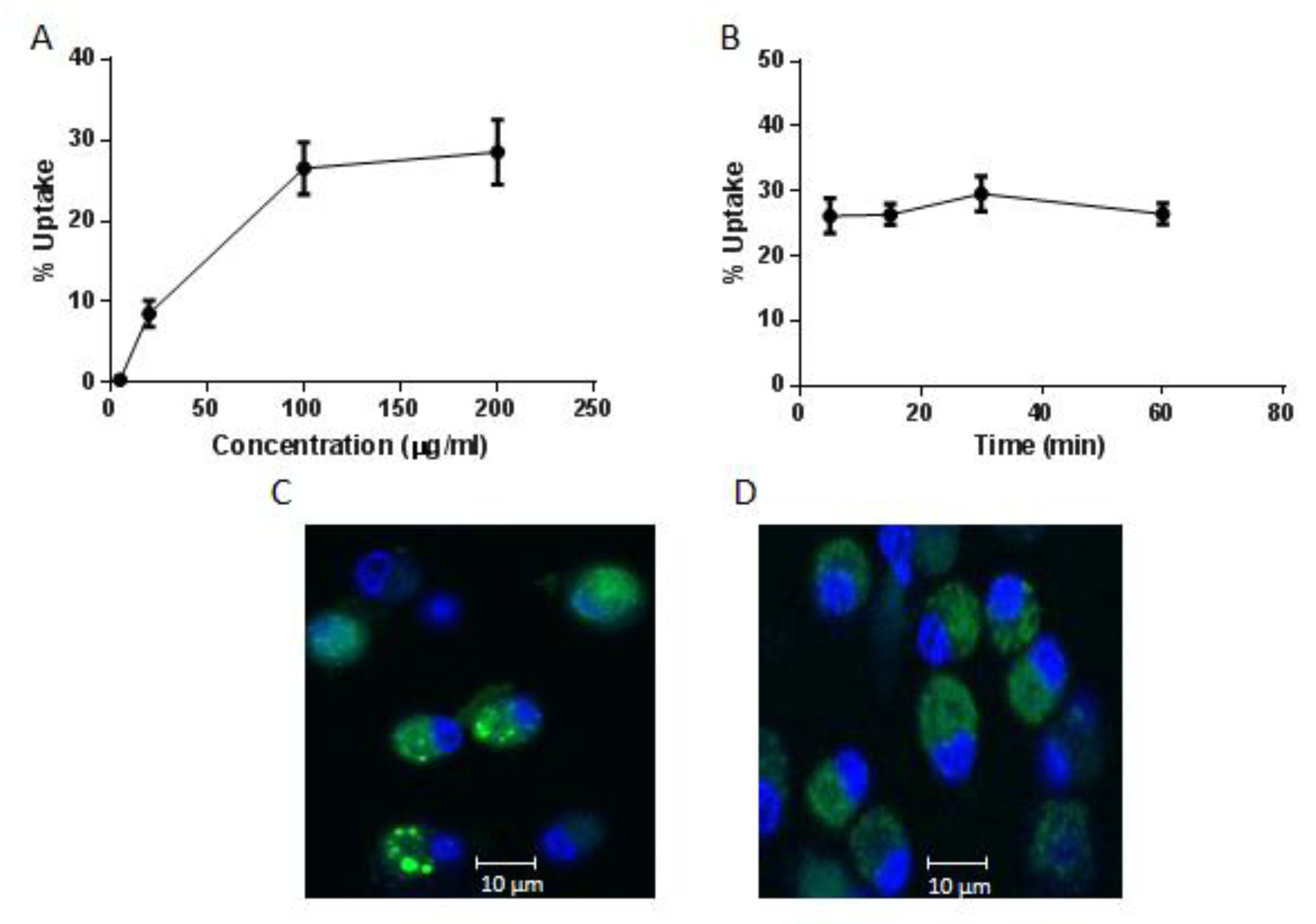
2.2. Internalisation of AntpMAPMUC1tet by Bone Marrow Derived Dendritic Cells (BMDC)
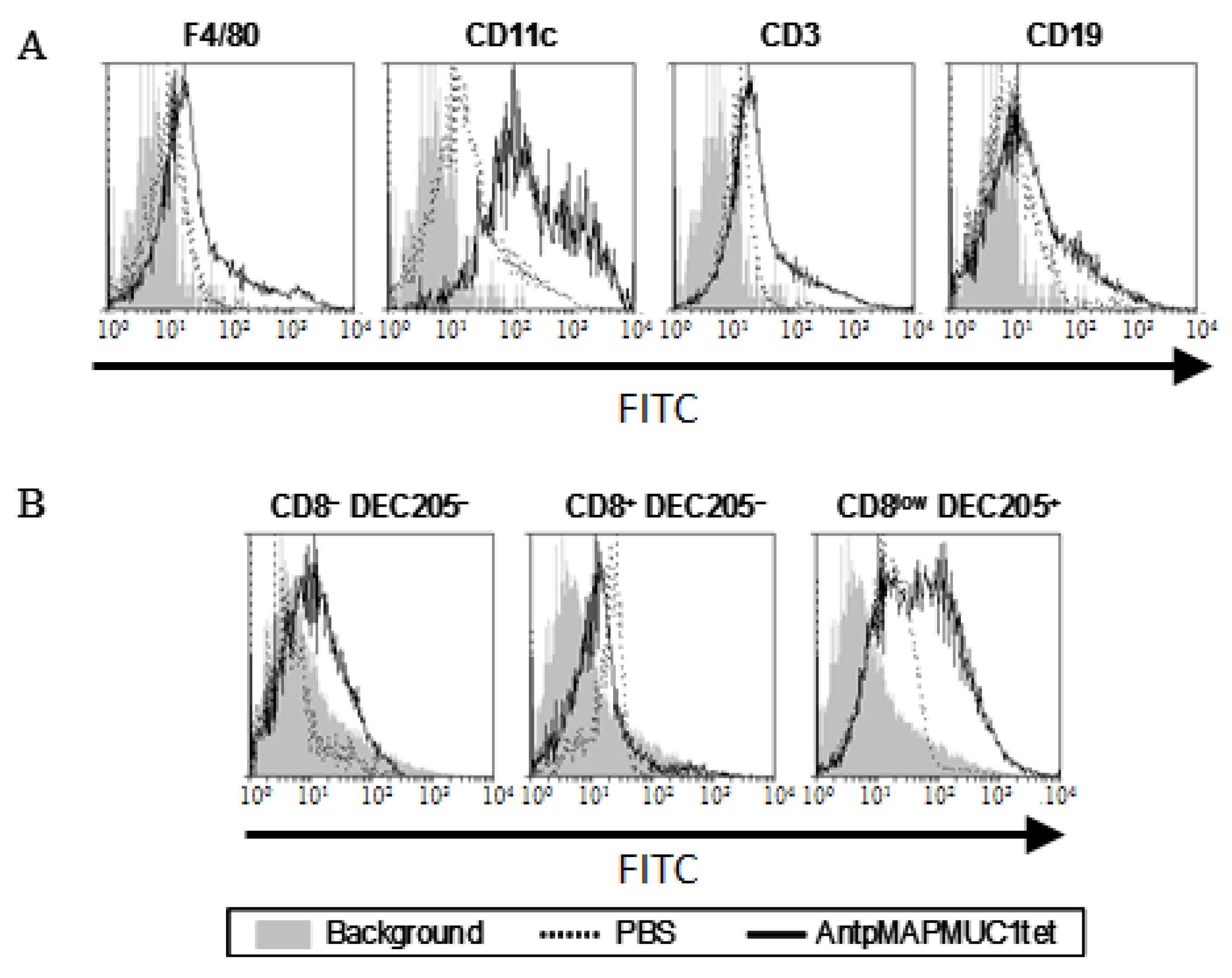
2.3. AntpMAPMUC1tet Targets Antigen Presenting Cells (APC) In Vivo
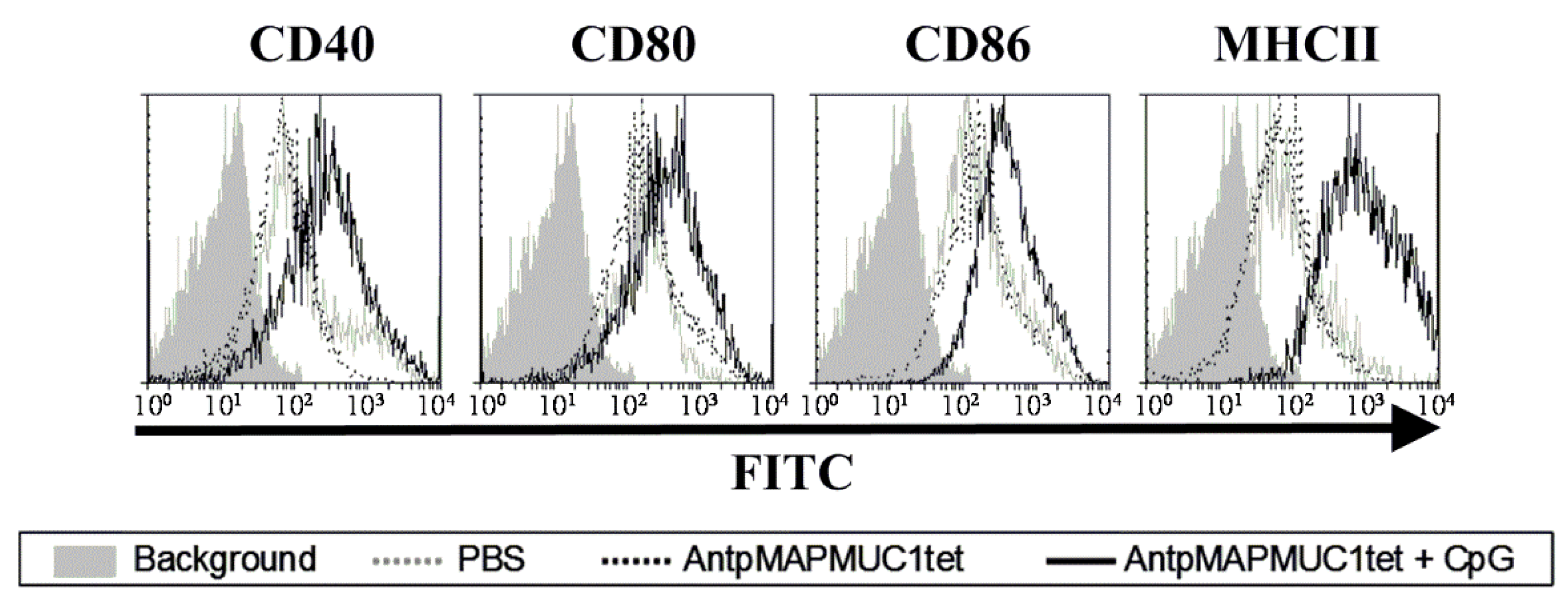
2.4. Addition of CpG to AntpMAPMUC1tet Induces Maturation of DCs In Vivo
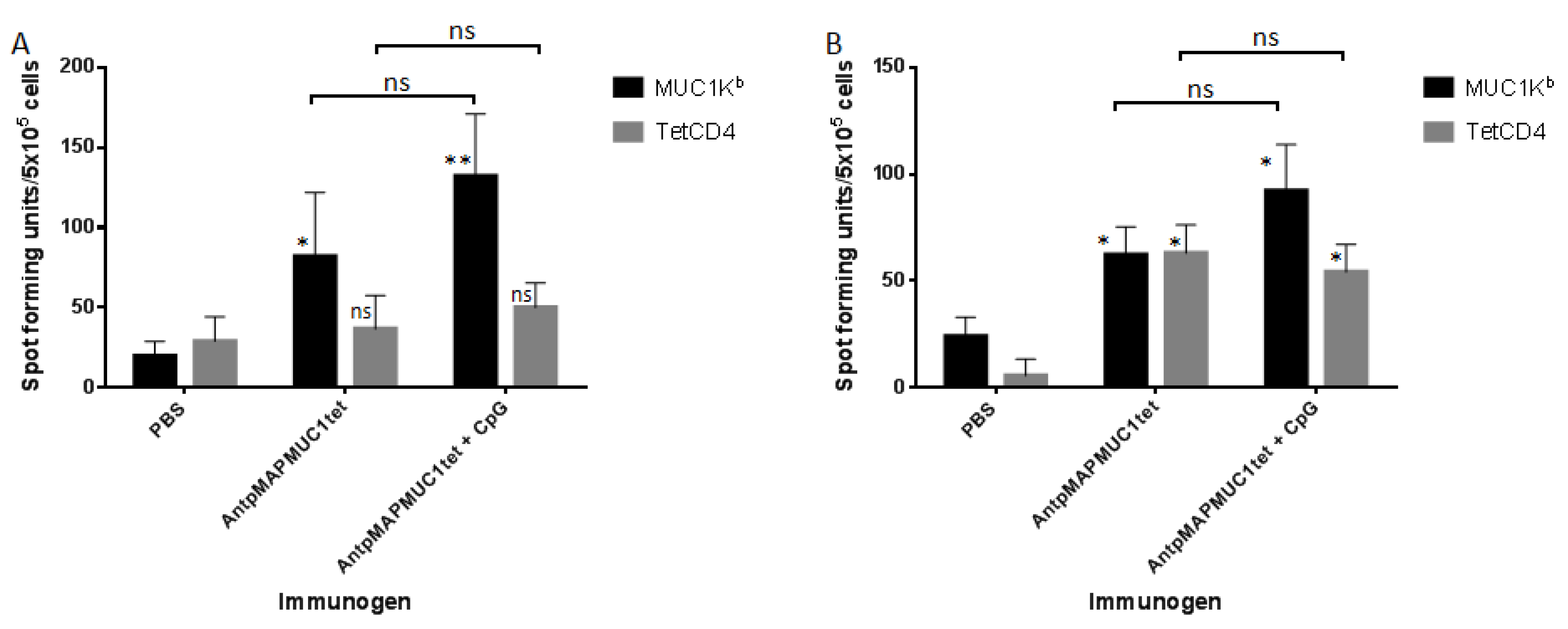
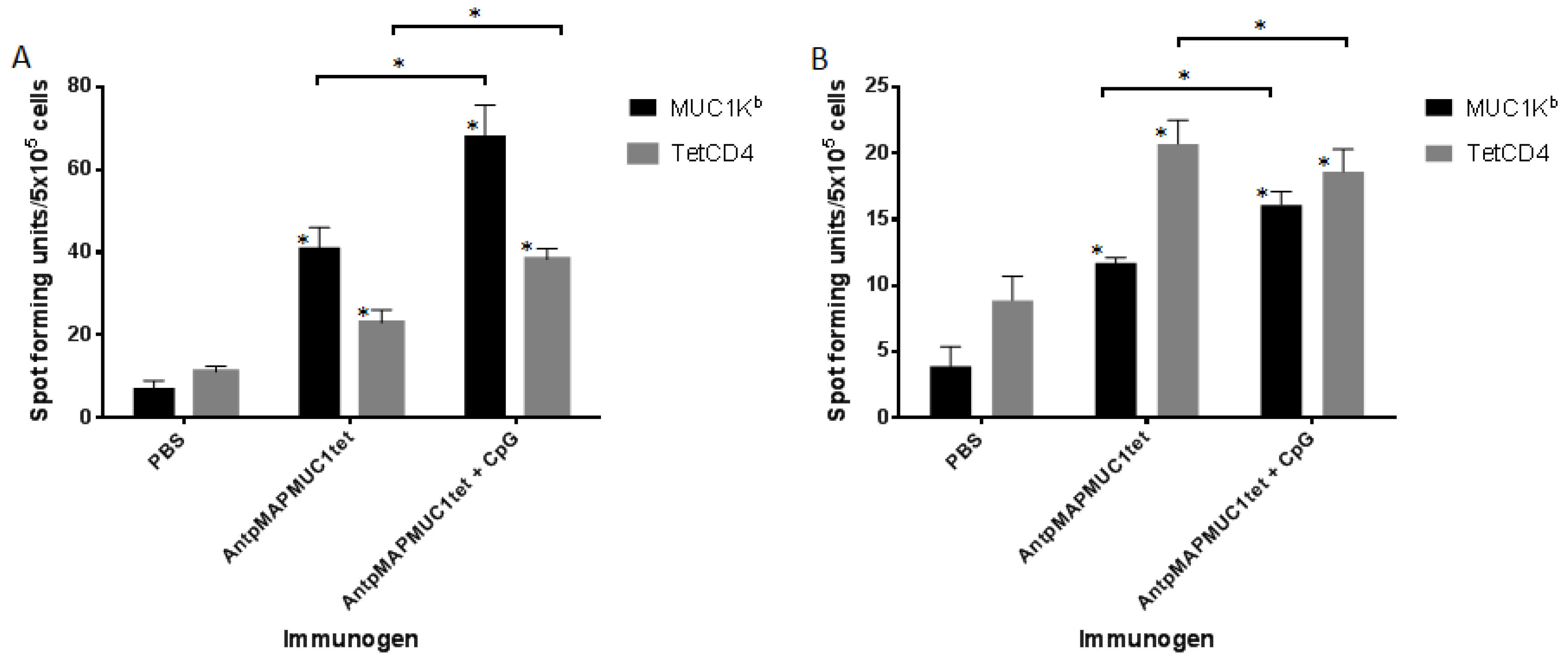
2.5. AntpMAPMUC1tet Induces MUC1-Specific T Cell Responses In Vivo in C57BL/6 Mice
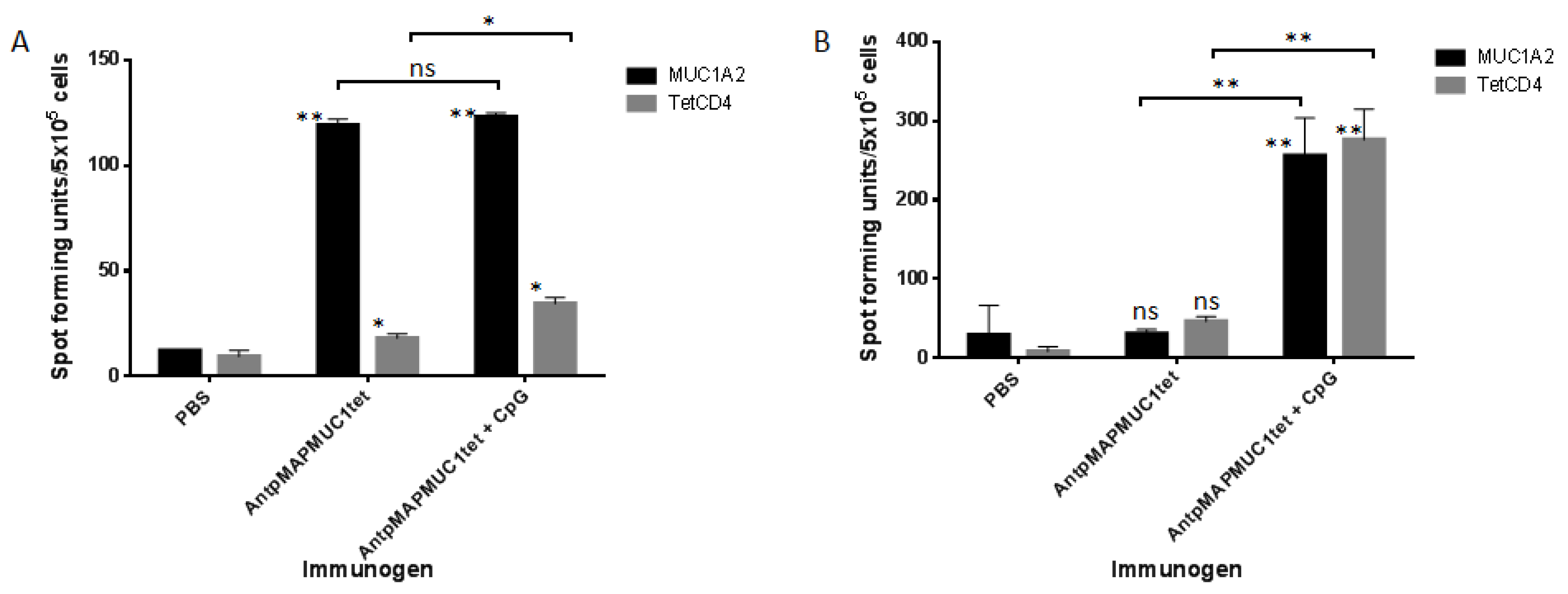
2.6. AntpMAPMUC1tet Immunisation Induces Specific T Cell Responses In Vivo in HLA-A2 Transgenic Mice
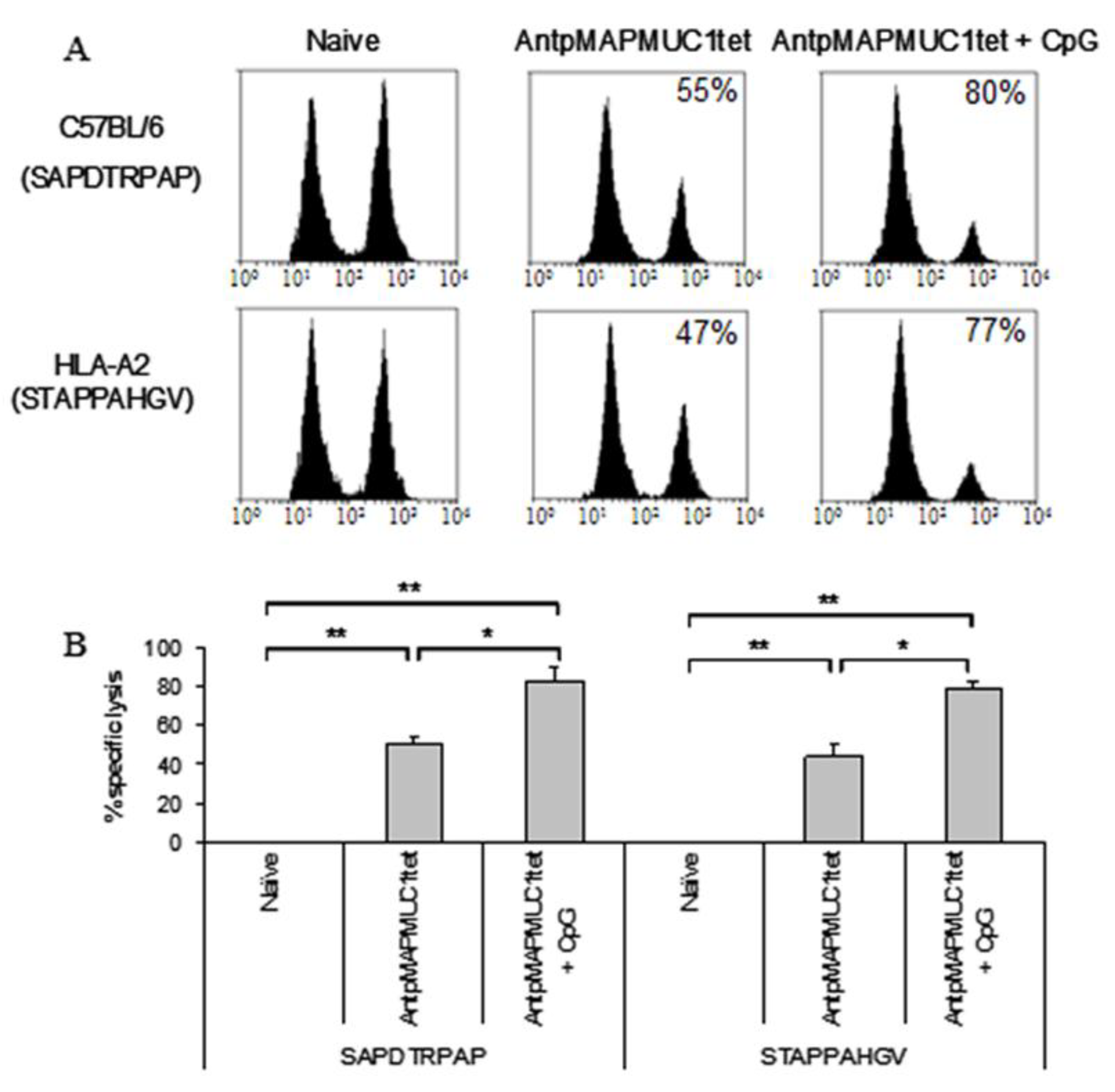
2.7. Combination of CpG with AntpMUC1tet Induces Enhanced In Vivo Antigen Specific Cytotoxic Killing
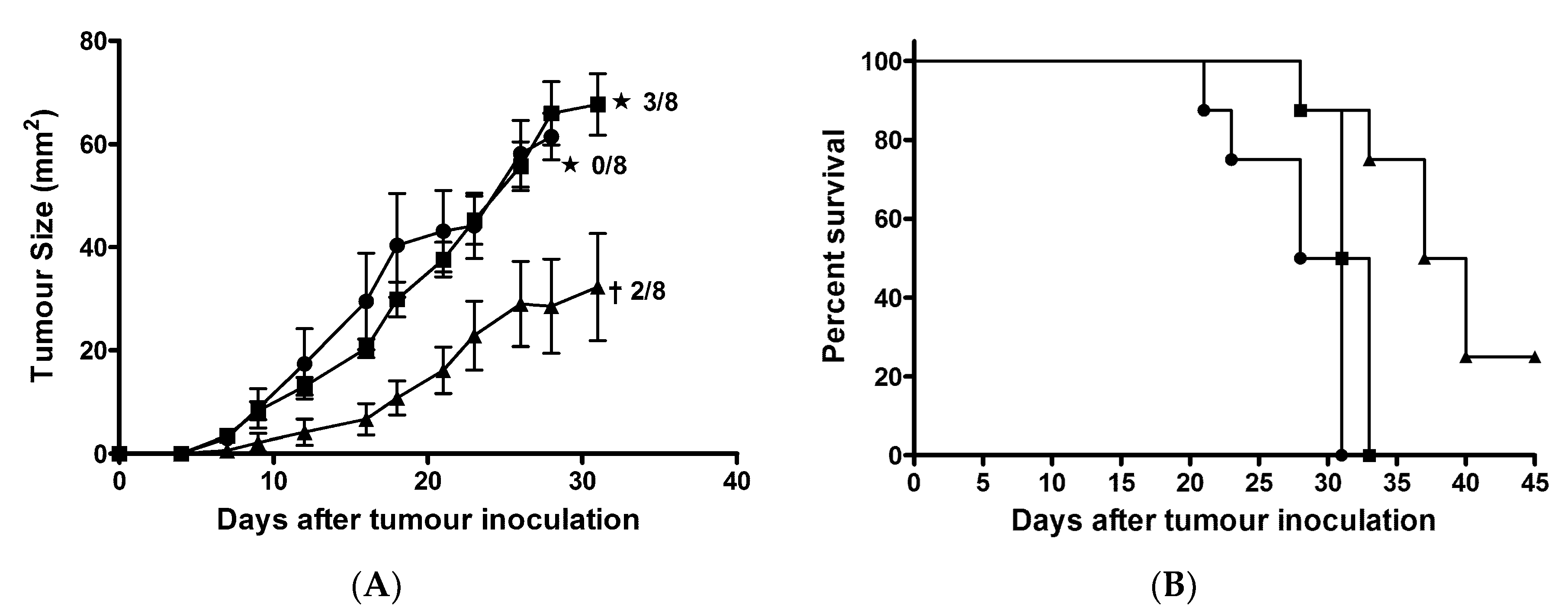
2.8. AntpMAPMUC1tet + CpG Immunisation Delayed Tumour Growth in Prophylactic Tumour Challenge
2.9. Induction of Long Term Immunity is Enhanced by CpG
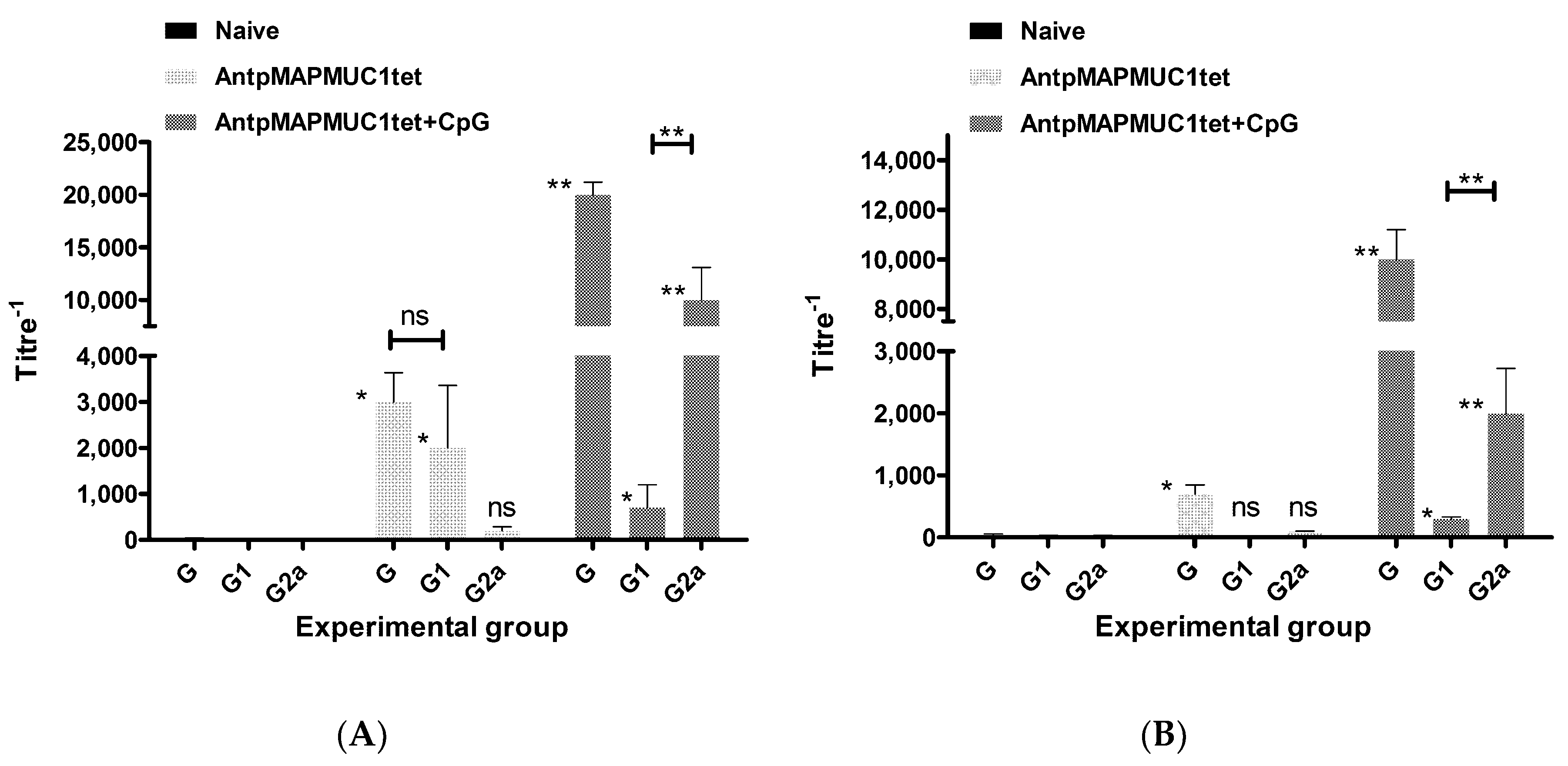
2.10. Generation of MUC1-Specific Antibodies
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Conjugation of AntpMAPMUC1tet to Fluorochromes
4.3. Mice and Immunisations
4.4. Adjuvants
4.5. ELISA
4.6. Generation of Bone Marrow Derived Dendritic Cells (BMDC)
4.7. Internalisation into BMDC
4.8. In Vitro Maturation
4.9. In Vivo Maturation
4.10. In Vivo Binding Specificity
4.11. Enzyme-Linked Immunosorbent Spot-Forming Cell Assay (ELISpot)
4.12. Antibody ELISA
4.13. Confocal Microscopy
4.14. Generation of Human Monocyte Derived Dendritic Cells (MoDC)
4.15. Generation of Tetanus Toxoid-Specific T Cell Lines
4.16. Analysis of Antigen-Specific T Cell Responses
4.17. Prophylactic Tumour Protection
4.18. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schlom, J.; Hodge, J.W.; Palena, C.; Tsang, K.Y.; Jochems, C.; Greiner, J.W.; Farsaci, B.; Madan, R.A.; Heery, C.R.; Gulley, J.L. Therapeutic cancer vaccines. Adv. Cancer Res. 2014, 121, 67–124. [Google Scholar] [PubMed]
- Guo, Y.; Lei, K.; Tang, L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol. 2018, 9, 1499. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting antigens to dendritic cell receptors for vaccine development. J. Drug Deliv. 2013, 2013, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.A.; Pouniotis, D.S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Cell-penetrating peptides: Application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.H.; Heger, L.; Heidkamp, G.F.; Baranska, A.; Luhr, J.J.; Hoffmann, A.; Dudziak, D. Direct delivery of antigens to dendritic cells via antibodies specific for endocytic receptors as a promising strategy for future therapies. Vaccines 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Sun, Z.Y.; Gao, Y.; Chen, P.G.; Liu, Y.F.; Chen, Y.X.; Li, Y.M. Strategy for designing a synthetic tumor vaccine: Multi-component, multivalency and antigen modification. Vaccines 2014, 2, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Baz, A.; Buttigieg, K.; Zeng, W.; Rizkalla, M.; Jackson, D.C.; Groves, P.; Kelso, A. Branched and linear lipopeptide vaccines have different effects on primary CD4+ and CD8+ T-cell activation but induce similar tumor-protective memory CD8+ T-cell responses. Vaccine 2008, 26, 2570–2579. [Google Scholar] [CrossRef] [PubMed]
- Justesen, S.; Buus, S.; Claesson, M.H.; Pedersen, A.E. Addition of tat protein transduction domain and GrpE to human p53 provides soluble fusion proteins that can be transduced into dendritic cells and elicit p53-specific T-cell responses in HLA-A*0201 transgenic mice. Immunology 2007, 122, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Peres Lde, P.; da Luz, F.A.; Pultz Bdos, A.; Brigido, P.C.; de Araujo, R.A.; Goulart, L.R.; Silva, M.J. Peptide vaccines in breast cancer: The immunological basis for clinical response. Biotechnol. Adv. 2015, 33, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T help in BALB/c mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.K.; Bright, J.D.; Byrne, J.A. Overexpressed oncogenic tumor-self antigens. Hum. Vaccines Immunother. 2014, 10, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Blanc, C.; Granier, C.; Saldmann, A.; Tanchot, C.; Tartour, E. Therapeutic Cancer Vaccine: Building the Future from Lessons of the Past. Seminars in Immunopathology; Springer Berlin Heidelberg: Heidelberg, Germany, 2018; pp. 1–17. [Google Scholar]
- Apostolopoulos, V.; Stojanovska, L.; Gargosky, S.E. MUC1 (CD227): A multi-tasked molecule. Cell. Mol. Life Sci. 2015, 72, 4475–4500. [Google Scholar] [CrossRef] [PubMed]
- Rivalland, G.; Loveland, B.; Mitchell, P. Update on Mucin-1 immunotherapy in cancer: A clinical perspective. Expert Opin. Biol. Ther. 2015, 15, 1773–1787. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Finn, O.J. Muc1 immunotherapy is here to stay. Expert Opin. Biol. Ther. 2013, 13, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Arriola, E.; Ottensmeier, C. Tg4010: A vaccine with a therapeutic role in cancer. Immunotherapy 2016, 8, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Scheid, E.; Major, P.; Bergeron, A.; Finn, O.J.; Salter, R.D.; Eady, R.; Yassine-Diab, B.; Favre, D.; Peretz, Y.; Landry, C.; et al. Tn-MUC1 dc vaccination of rhesus macaques and a phase I/II trial in patients with nonmetastatic castrate-resistant prostate cancer. Cancer Immunol. Res. 2016, 4, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, V.; Pouniotis, D.S.; van Maanen, P.J.; Andriessen, R.W.; Lodding, J.; Xing, P.X.; McKenzie, I.F.; Loveland, B.E.; Pietersz, G.A. Delivery of tumor associated antigens to antigen presenting cells using penetratin induces potent immune responses. Vaccine 2006, 24, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.A.; Pouniotis, D.S.; Sheng, K.C.; Apostolopoulos, V.; Pietersz, G.A. A membrane penetrating multiple antigen peptide (MAP) incorporating ovalbumin CD8 epitope induces potent immune responses in mice. Biochim. Biophys. Acta (BBA) Biomembr. 2010, 1798, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Chikh, G.G.; Kong, S.; Bally, M.B.; Meunier, J.C.; Schutze-Redelmeier, M.P. Efficient delivery of antennapedia homeodomain fused to CTL epitope with liposomes into dendritic cells results in the activation of CD8+ T cells. J. Immunol. 2001, 167, 6462–6470. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.T.; Mitchell, D.J.; Brockstedt, D.G.; Fong, L.; Nolan, G.P.; Fathman, C.G.; Engleman, E.G.; Rothbard, J.B. Introduction of soluble proteins into the MHC Class I pathway by conjugation to an HIV tat peptide. J. Immunol. 1997, 159, 1666–1668. [Google Scholar] [PubMed]
- Lu, J.; Higashimoto, Y.; Appella, E.; Celis, E. Multiepitope trojan antigen peptide vaccines for the induction of antitumor CTL and TH immune responses. J. Immunol. 2004, 172, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Pietersz, G.A.; Li, W.; Apostolopoulos, V. A 16-mer peptide (RQIKIWFQNRRMKWKK) from antennapedia preferentially targets the Class I pathway. Vaccine 2001, 19, 1397–1405. [Google Scholar] [CrossRef]
- Pouniotis, D.S.; Apostolopoulos, V.; Pietersz, G.A. Penetratin tandemly linked to a CTL peptide induces anti-tumour T-cell responses via a cross-presentation pathway. Immunology 2006, 117, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Pouniotis, D.S.; Esparon, S.; Apostolopoulos, V.; Pietersz, G.A. Whole protein and defined CD8+ and CD4+ peptides linked to penetratin targets both MHC class I and II antigen presentation pathways. Immunol. Cell Biol. 2011, 89, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Schutze-Redelmeier, M.P.; Gournier, H.; Garcia-Pons, F.; Moussa, M.; Joliot, A.H.; Volovitch, M.; Prochiantz, A.; Lemonnier, F.A. Introduction of exogenous antigens into the MHC class I processing and presentation pathway by Drosophila antennapedia homeodomain primes cytotoxic T cells in vivo. J. Immunol. 1996, 157, 650–655. [Google Scholar] [PubMed]
- Schutze-Redelmeier, M.P.; Kong, S.; Bally, M.B.; Dutz, J.P. Antennapedia transduction sequence promotes anti tumour immunity to epicutaneously administered CTL epitopes. Vaccine 2004, 22, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Fu, T.; Wang, G.; Zeng, G.; Perry-Lalley, D.M.; Yang, J.C.; Restifo, N.P.; Hwu, P.; Wang, R.F. Induction of CD4+ T cell-dependent antitumor immunity by tat-mediated tumor antigen delivery into dendritic cells. J. Clin. Investig. 2002, 109, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Cho, N.H.; Seong, S.Y. The tat-conjugated n-terminal region of mucin antigen 1 (MUC1) induces protective immunity against MUC1-expressing tumours. Clin. Exp. Immunol. 2009, 158, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, L.; Wang, H.; Shang, X.; Niu, W.; Li, J.; Wu, Y. A novel mimovirus vaccine containing survivin epitope with adjuvant IL-15 induces long-lasting cellular immunity and high antitumor efficiency. Mol. Immunol. 2008, 45, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Cell-penetrating peptides as delivery enhancers for vaccine. Curr. Pharm. Biotechnol. 2014, 15, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Belnoue, E.; Di Berardino-Besson, W.; Gaertner, H.; Carboni, S.; Dunand-Sauthier, I.; Cerini, F.; Suso-Inderberg, E.M.; Walchli, S.; Konig, S.; Salazar, A.M.; et al. Enhancing antitumor immune responses by optimized combinations of cell-penetrating peptide-based vaccines and adjuvants. Mol. Ther. 2016, 24, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, S.; Bolhassani, A. Comparison of six cell penetrating peptides with different properties for in vitro and in vivo delivery of HPV16 E7 antigen in therapeutic vaccines. Int. Immunopharmacol. 2018, 62, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Koo, J.H.; Choi, J.M. Use of cell-penetrating peptides in dendritic cell-based vaccination. Immune Netw. 2016, 16, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Voutsas, I.F.; Tsitsilonis, O.E. Toll-like receptor agonists: Current status and future perspective on their utility as adjuvants in improving anticancer vaccination strategies. Immunotherapy 2013, 5, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, H.S.; Park, H.M.; Kim, C.H.; Kim, T.G. Efficient induction of anti-tumor immunity by a tat-cea fusion protein vaccine with poly(I:C) in a murine colorectal tumor model. Vaccine 2011, 29, 8642–8648. [Google Scholar] [CrossRef] [PubMed]
- Pouniotis, D.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G. Vaccine delivery by penetratin: Mechanism of antigen presentation by dendritic cells. Immunol. Res. 2016, 64, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Brooks, N.; Esparon, S.; Pouniotis, D.; Pietersz, G.A. Comparative immunogenicity of a cytotoxic T cell epitope delivered by penetratin and tat cell penetrating peptides. Molecules 2015, 20, 14033–14050. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.K.; Sheng, K.C.; Esparon, S.E.; Proudfoot, O.; Apostolopoulos, V.; Pietersz, G.A. Molecular basis of improved immunogenicity in DNA vaccination mediated by a mannan based carrier. Biomaterials 2009, 30, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Anjuere, F.; Martin, P.; Ferrero, I.; Fraga, M.L.; del Hoyo, G.M.; Wright, N.; Ardavin, C. Definition of dendritic cell subpopulations present in the spleen, peyer’s patches, lymph nodes, and skin of the mouse. Blood 1999, 93, 590–598. [Google Scholar] [PubMed]
- Klinman, D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004, 4, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.C.; Kalkanidis, M.; Pouniotis, D.S.; Wright, M.D.; Pietersz, G.A.; Apostolopoulos, V. The adjuvanticity of a mannosylated antigen reveals TLR4 functionality essential for subset specialization and functional maturation of mouse dendritic cells. J. Immunol. 2008, 181, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.K.; Sheng, K.C.; Pouniotis, D.; Esparon, S.; Son, H.Y.; Kim, C.W.; Pietersz, G.A.; Apostolopoulos, V. Oxidized and reduced mannan mediated MUC1 DNA immunization induce effective anti-tumor responses. Vaccine 2008, 26, 3827–3834. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Delamarre, L. Dendritic cell-targeted vaccines. Front. Immunol. 2014, 5, 255. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sznol, M.; Zhao, B.; Wang, D.; Carvajal, R.D.; Keohan, M.L.; Chuang, E.; Sanborn, R.E.; Lutzky, J.; Powderly, J.; et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. 2014, 6, 232RA251. [Google Scholar] [CrossRef] [PubMed]
- Kastenmuller, W.; Kastenmuller, K.; Kurts, C.; Seder, R.A. Dendritic cell-targeted vaccines—Hope or hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Chapman, R.; Powderly, J.; Blackwell, K.; Keler, T.; Green, J.; Riggs, R.; He, L.Z.; Ramakrishna, V.; Vitale, L.; et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin. Cancer Res. 2011, 17, 4844–4853. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot phase III immunotherapy study in stage ii breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Karan, D.; Krieg, A.M.; Lubaroff, D.M. Paradoxical enhancement of CD8 T cell-dependent anti-tumor protection despite reduced CD8 T cell responses with addition of a TLR9 agonist to a tumor vaccine. Int. J. Cancer 2007, 121, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Celis, E. Toll-like receptor ligands energize peptide vaccines through multiple paths. Cancer Res. 2007, 67, 7945–7947. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, A.; Das, P.; Chakravortty, D. Engagement of TLR signaling as adjuvant: Towards smarter vaccine and beyond. Vaccine 2008, 26, 6777–6783. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.J.; Kim, C.H.; Park, M.Y.; Kim, H.S.; Sohn, H.J.; Park, J.S.; Kim, H.J.; Oh, S.T.; Kim, T.G. Co-administration of carcinoembryonic antigen and HIV TAT fusion protein with CpG-oligodeoxynucleotide induces potent antitumor immunity. Cancer Sci. 2008, 99, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Toes, R.E.; Ossendorp, F.; Offringa, R.; Melief, C.J. CD4 T cells and their role in antitumor immune responses. J. Exp. Med. 1999, 189, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.K.; Raz, E. Induction of Antigen Cross-Presentation by Toll-Like Receptors. Springer Seminars in Immunopathology; Springer-Verlag: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, P.M.; Kroesen, B.J.; Harmsen, M.C.; de Leij, L.F. Cancer immunotherapy: Insights from transgenic animal models. Crit. Rev. Oncol. 2001, 40, 53–76. [Google Scholar] [CrossRef]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Archer, G.E.; Tedder, T.F. Isolation and generation of human dendritic cells. Curr. Protoc. Immunol. 2012, 99, 7–32. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, N.; Hsu, J.; Esparon, S.; Pouniotis, D.; Pietersz, G.A. Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope. Molecules 2018, 23, 2233. https://doi.org/10.3390/molecules23092233
Brooks N, Hsu J, Esparon S, Pouniotis D, Pietersz GA. Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope. Molecules. 2018; 23(9):2233. https://doi.org/10.3390/molecules23092233
Chicago/Turabian StyleBrooks, Nicole, Jennifer Hsu, Sandra Esparon, Dodie Pouniotis, and Geoffrey A. Pietersz. 2018. "Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope" Molecules 23, no. 9: 2233. https://doi.org/10.3390/molecules23092233
APA StyleBrooks, N., Hsu, J., Esparon, S., Pouniotis, D., & Pietersz, G. A. (2018). Immunogenicity of a Tripartite Cell Penetrating Peptide Containing a MUC1 Variable Number of Tandem Repeat (VNTR) and A T Helper Epitope. Molecules, 23(9), 2233. https://doi.org/10.3390/molecules23092233