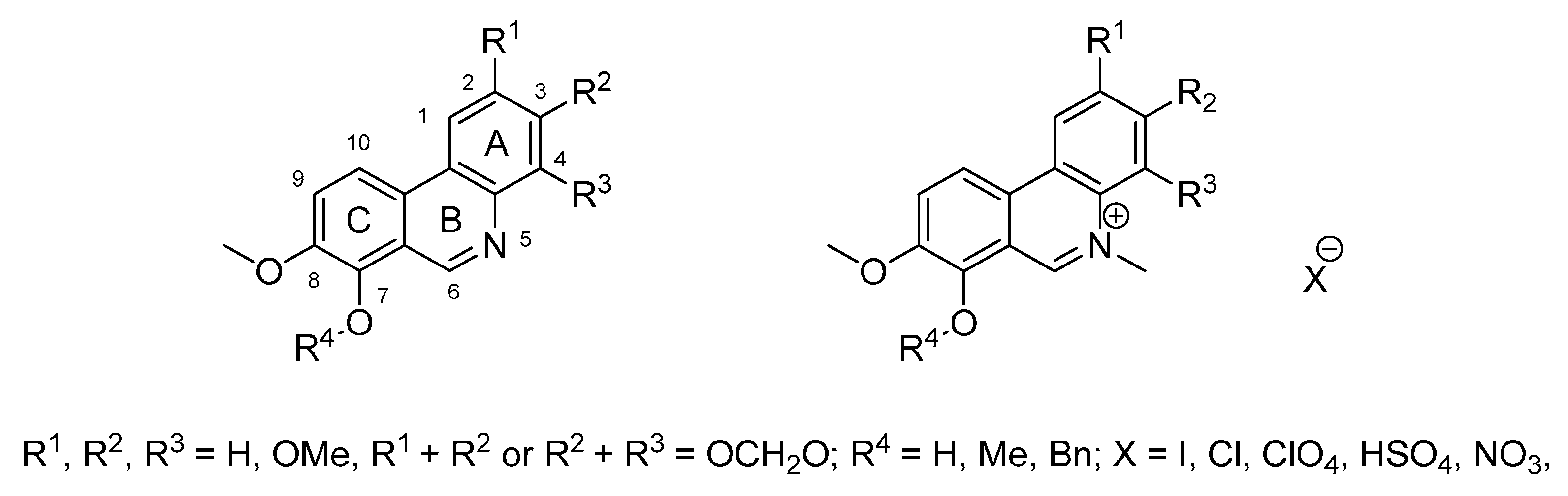
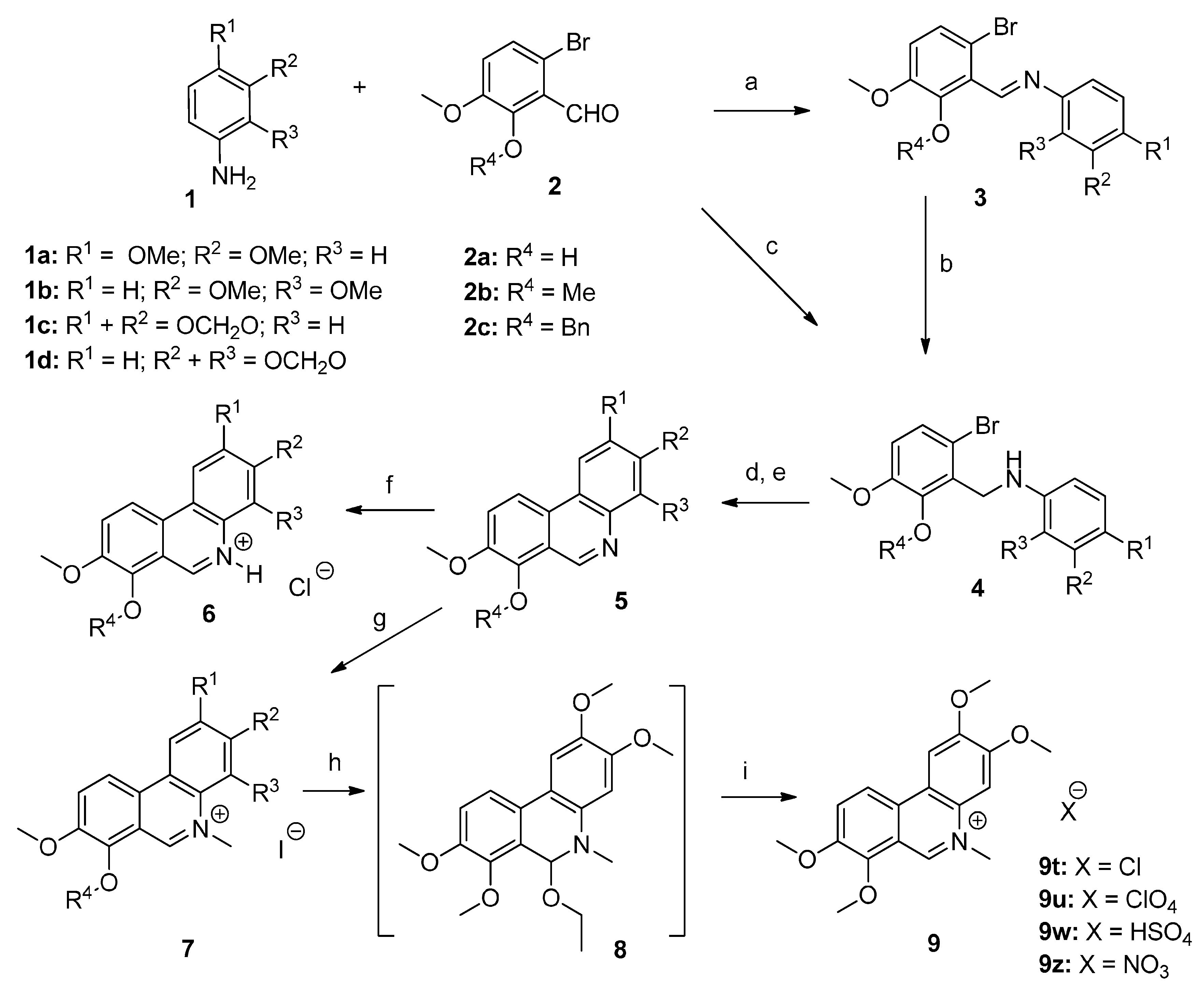
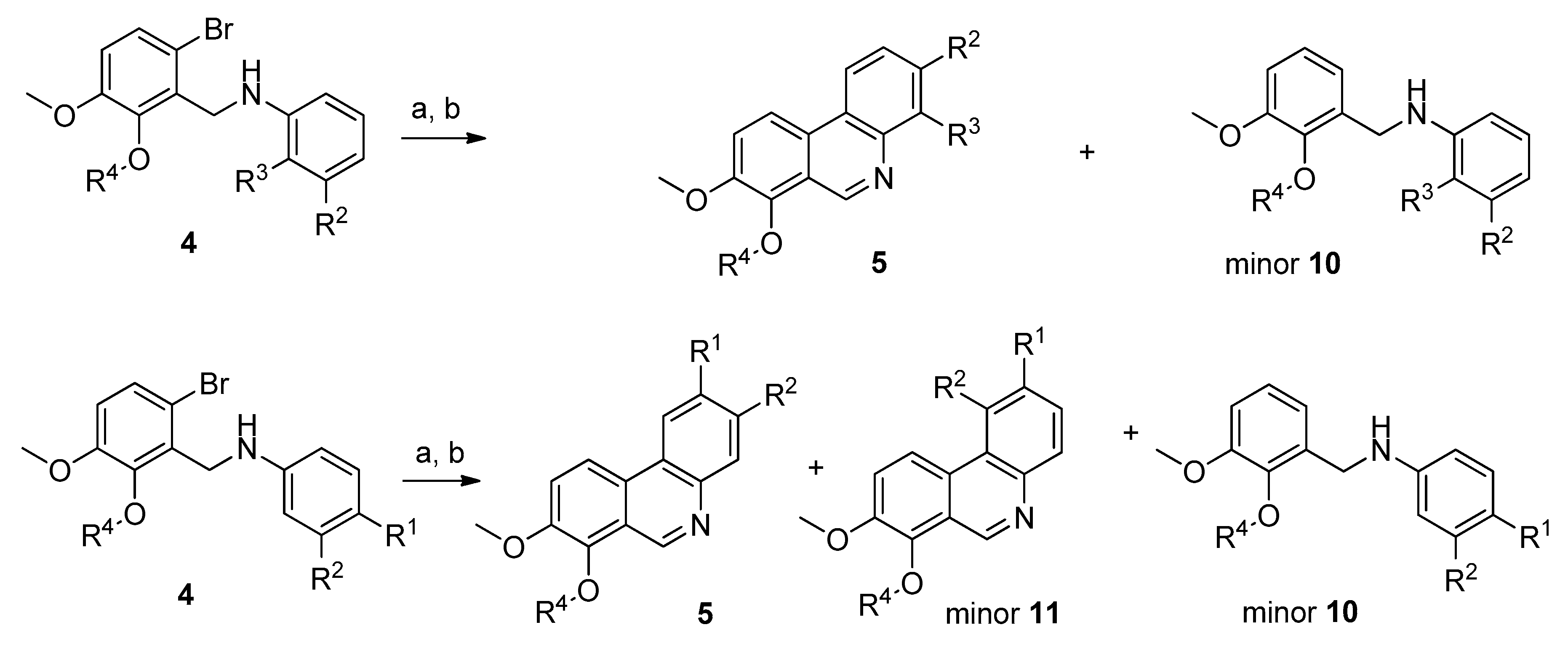
3.1.2. Synthesis of Compounds 3–9
3-Bromo-2-{[(3,4-dimethoxyphenyl)imino]methyl}-6-methoxyphenol (3a). To a solution of aniline 1a (410 mg; 2.67 mmol) in anhydrous EtOH (15 mL) was added benzaldehyde 2a (600 mg; 2.60 mmol). The reaction mixture was stirred at reflux for 1 h and then slowly cooled to 0–5 °C. Resultant precipitate was filtered off, washed with ice-cold EtOH (2 mL) and dried. Compound 3a (921 mg, 97%) was obtained as an orange microcrystalline solid. Mp. = 157–159 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.27 (s, 1H), 9.05 (s, 1H), 7.02 (d, J = 8.8 Hz, 1H), 6.98–6.89 (m, 3H), 6.78 (d, J = 8.8 Hz, 1H), 3.94 (s, 3H), 3.92 (s, 3H), 3.90 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 160.4, 154.6, 149.6, 148.8, 148.7, 140.0, 121.8, 116.4, 115.7, 114.7, 113.1, 111.4, 105.1, 56.2, 56.1, 56.0; HRMS (HESI, m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0335.
2-[(1,3-Benzodioxole-5-ylimino)methyl]-3-bromo-6-methoxyphenol (3c). Schiff base 3c was obtained in a similar manner to compound 3a using benzaldehyde 2a (750 mg; 3.25 mmol), aniline 1c (445 mg; 3.25 mmol) and anhydrous EtOH (20 mL). Compound 3c (1.09 g; 92%) was obtained as a dark red microcrystalline solid. Mp. = 176–178 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.07 (br. s, 1H), 9.02 (s, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.92–6.89 (m, 1H), 6.88–6.85 (m 2H), 6.78 (d, J = 8.7 Hz, 1H), 6.03 (s, 2H), 3.91 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 160.6, 154.4, 148.8, 148.7, 147.4, 141.4, 122.0, 116.5, 116.0, 115.9, 114.9, 108.7, 101.8, 101.5, 56.3; HRMS (HESI, m/z): [M + H]+ calcd. for C15H13BrNO4, 350.0028; found 350.0023.
2-[(1,3-Benzodioxole-4-ylimino)methyl]-3-bromo-6-methoxyphenol (3d). Schiff base 3d was obtained in a similar manner to compound 3a using benzaldehyde 2a (572 mg; 2.48 mmol), aniline 1d (350 mg; 2.55 mmol) and anhydrous EtOH (15 mL). Compound 3d (737 mg; 85%) was obtained as a pale orange microcrystalline solid. Mp. = 195–196 °C; 1H-NMR (400 MHz, CDCl3) δ = 15.18 (s, 1H), 9.40 (s, 1H), 7.05 (d, J = 8.6 Hz, 1H), 6.96–6.88 (m, 2H), 6.82–6.77 (m, 2H), 6.09 (s, 2H), 3.92 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 164.3, 154.7, 148.9, 148.7, 140.3, 129.3, 122.2, 121.9, 116.7, 116.3, 116.0, 115.0, 107.7, 101.7, 56.2; HRMS (HESI, m/z): [M + H]+ calcd. for C15H13BrNO4, 350.0028; found 350.0022.
3-Bromo-2-{[(3,4-dimethoxyphenyl)amino]methyl}-6-methoxyphenol (4a). To a suspension of the Schiff base 3a (921 mg, 2.51 mmol) in anhydrous EtOH (25 mL) was added NaBH4 (95 mg; 2.51 mmol). After 2 h of stirring at room temperature the reaction mixture was neutralized by adding an aqueous solution of 10% AcOH (1 mL). The suspension dissolved into solution, which was concentrated under reduced pressure. The residue was extracted with a mixture of EtOAc (20 mL) and water (20 mL). The aqueous phase was washed with EtOAc (20 mL). The combined EtOAc phases were dried over Na2SO4, filtered and concentrated under reduced pressure to provide a yellow crude oil (737 mg, 80%). TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.05 (d, J = 9.3 Hz, 1H), 6.74 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 8.3 Hz, 1H), 6.47 (d, J = 2.1 Hz, 1H), 6.37 (dd, J = 2.1, 8.3, Hz, 1H), 5.89 (brs, 2H), 4.53 (s, 2H), 3.85 (s, 3H), 3.83 (s, 3H), 3.80 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 149.7, 146.6, 146.2, 142.7, 141.7, 123.4, 123.2, 115.1, 112.7, 111.2, 106.1, 100.5, 56.5, 56.1, 55.7, 45.8; HRMS (HESI, m/z): [M + H]+ calcd. for C16H19BrNO4, 368.0492; found 368.0498.
2-[(1,3-Benzodioxole-5-ylamino)methyl]-3-bromo-6-methoxyphenol (4c). The secondary amine 4c was obtained in a similar manner as compound 4a using Schiff base 3c (1.0 g; 2.9 mmol), NaBH4 (109 mg; 2.9 mmol) and anhydrous EtOH (25 mL). The resulting crude product was a pale yellow oil (1.0 g, 99%). A sample for analysis was prepared by crystallization from toluene (100 mg/3 mL) and standing this solution at −20 °C. Mp. = 95–98 °C as a colorless crystalline compound; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.34 (s, 1H), 7.02 (d, J = 8.7 Hz, 1H), 6.86 (d, J = 8.7 Hz, 1H), 6.64 (d, J = 8.2 Hz, 1H), 6.42 (d, J = 1.8 Hz, 1H), 6.12 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.21 (t, J = 5.2 Hz, 1H), 4.19 (d, J = 5.2 Hz, 2H), 3.79 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 147.6, 147.1, 146.2, 144.6, 138.1, 124.7, 122.2, 115.6, 112.2, 108.3, 103.4, 99.9, 95.4, 56.0, 42.9; HRMS (HESI, m/z): [M + H]+ calcd. for C15H15BrNO4, 352.0179; found 352.0182.
2-[(1,3-Benzodioxole-4-ylamino)methyl]-3-bromo-6-methoxyphenol (4d). The secondary amine 4d was obtained in a similar manner as compound 4a using Schiff base 3d (500 mg; 1.43 mmol), NaBH4 (54 mg; 1.43 mmol) and anhydrous EtOH (13 mL). The resulting crude product was a pale yellow oil (500 mg, 99%). A sample for analysis was prepared by crystallization from toluene (100 mg/3 mL) and standing this solution at −20 °C. Mp. = 89–90 °C as a colorless crystalline compound; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.07 (d, J = 8.7 Hz, 1H), 6.75 (t, J = 8.0 Hz, 1H), 6.68 (d, J = 8.7 Hz, 1H), 6.60 (d, J = 8.2 Hz, 1H), 6.36 (d, J = 7.8 Hz, 1H), 5.90 (s, 2H), 4.59 (s, 2H), 3.87 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 147.2, 146.2, 145.6, 134.6, 132.2, 123.9, 123.3, 122.2, 115.7, 111.1, 107.8, 100.6, 99.8, 56.1, 43.7; HRMS (HESI, m/z): [M + H]+ calcd. for C15H15BrNO4, 352.0179; found 352.0183.
N-(6-Bromo-2,3-dimethoxybenzyl)-3,4-dimethoxyaniline (4e). To a solution of bromobenzaldehyde 2b (1.0 g; 4.08 mmol) and aniline 1a (644 mg; 4.20 mmol) in anhydrous toluene (50 mL) was added NaBH(OAc)3 (2.6 g; 12.2 mmol). The resulting suspension was stirred at room temperature for 1 h, and then washed with water (2 × 35 mL) and brine (1 × 40 mL). The aqueous phase was extracted with toluene (1 × 40 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. To induce crystallization of the product, MeOH (8 mL) was added to the residue. The resulting solid was filtered off, washed with cold MeOH (1 mL) and dried. Compound 4e was obtained as a slightly pink crystalline solid (1.19 g; 76%). Mp. = 79–80 °C; TLC Rf = 0.2 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, CDCl3) δ = 7.26 (d, J = 8.8 Hz, 1H), 6.79–6.72 (m, 2H), 6.43 (d, J = 2.6 Hz, 1H), 6.33 (dd, J = 8.5, 2.6 Hz, 1H), 4.42 (s, 2H), 4.02 (br. s, 1H), 3.87 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.80 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 152.3, 149.8, 148.7, 142.7, 141.7, 132.6, 128.0, 115.2, 113.0, 112.9, 104.6, 99.5, 61.5, 56.6, 55.9, 55.7, 43.9; HRMS (HESI, m/z): [M + H]+ calcd. for C17H21BrNO4, 382.0654; found 382.0648.
N-(6-Bromo-2,3-dimethoxybenzyl)-2,3-dimethoxyaniline (4f). The secondary amine 4f was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (400 mg; 1.63 mmol), aniline 1b (258 mg; 1.68 mmol) and NaBH(OAc)3 (1.04 g; 4.9 mmol) in anhydrous toluene (20 mL). The residue was crystallized from EtOH/water to provide compound 4f (212 mg; 34%) as a colorless microcrystalline solid. Mp. = 52–53 °C; TLC Rf = 0.5 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.34 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 1H), 6.85 (t, J = 8.2 Hz, 1H), 6.52 (dd, J = 8.2, 1.2 Hz, 1H), 6.33 (dd, J = 8.2, 1.2 Hz, 1H), 4.85 (t, J = 6.2 Hz, 1H), 4.33 (d, J = 6.2 Hz, 2H), 3.80 (s, 3H), 3.77 (s, 3H), 3.72 (s, 3H), 3.60 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.4, 141.6, 135.0, 131.9, 127.9, 124.2, 119.5, 114.4, 113.8, 104.7, 101.6, 61.2, 59.5, 55.9, 55.5, 42.2; HRMS (HESI m/z): [M + H]+ calcd. for C17H21BrNO4, 382.0654; found 382.0648.
N-(6-Bromo-2,3-dimethoxybenzyl)-1,3-benzodioxole-5-amine (4g). The secondary amine 4g was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (400 mg; 1.63 mmol), aniline 1c (231 mg; 1.68 mmol) and NaBH(OAc)3 (1.04 g; 4.9 mmol) in anhydrous toluene (20 mL). The residue was crystallized from EtOH/water to provide compound 4g (275 mg; 46%) as a pale yellow microcrystalline solid. Mp. = 74–75 °C; TLC Rf = 0.4 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.34 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 6.65 (d, J = 8.3 Hz, 1H), 6.41 (d, J = 2.3 Hz, 1H), 6.12 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.26 (t, J = 5.3 Hz, 1H), 4.16 (d, J = 5.2 Hz, 2H), 3.81 (s, 3H), 3.75 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.5, 147.7, 144.6, 138.1, 132.0, 127.7, 115.0, 113.7, 108.4, 103.2, 99.9, 95.2, 61.1, 55.9, 43.0; HRMS (HESI m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0337.
N-(6-Bromo-2,3-dimethoxybenzyl)-1,3-benzodioxole-4-amine (4h). The secondary amine 4h was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2b (253 mg; 1.03 mmol), aniline 1d (142 mg; 1.04 mmol) and NaBH(OAc)3 (660 mg; 3.1 mmol) in anhydrous toluene (12 mL). The residue was crystallized from MeOH to provide compound 4h (302 mg; 80%) as a colorless microcrystalline solid. Mp. = 90–92 °C; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.33 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 9.2 Hz, 1H), 6.65 (t, J = 7.8 Hz, 1H), 6.46 (d, J = 8.2 Hz, 1H), 6.27 (dd, J = 7.8, 0.9 Hz, 1H), 5.90 (s, 2H), 4.82 (t, J = 6.0 Hz, 1H), 4.40 (d, J = 6.0 Hz, 2H), 3.80 (s, 3H), 3.77 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.1, 148.4, 147.0, 133.2, 132.7, 131.8, 127.7, 122.1, 114.6, 113.7, 107.2. 100.0, 98.3, 61.0, 55.9, 42.6; HRMS (HESI m/z): [M + H]+ calcd. for C16H17BrNO4, 366.0341; found 366.0337.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-3,4-dimethoxyaniline (4i). The secondary amine 4i was prepared in a similar manner as amine 4e using bromobenzaldehyde 2c (1.0 g; 3.11 mmol), aniline 1a (491 mg; 3.21 mmol) and NaBH(OAc)3 (1.98 g; 9.4 mmol) in anhydrous toluene (50 mL). The residue was crystallized from MeOH to provide compound 4i (1.16 g; 81%) as a light pink microcrystalline solid. Mp.= 79–81 °C; TLC Rf = 0.3 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.43–7.29 (m, 6H), 7.03 (d, J = 8.8 Hz, 1H), 6.69 (d, J = 8.8 Hz, 1H), 6.39 (d, J = 8.6 Hz, 1H), 6.15 (dd, J = 8.6, 2.6 Hz, 1H), 5.10 (t, J = 5.6 Hz, 1H), 4.99 (s, 2H), 4.17 (d, J = 5.4 Hz, 2H), 3.86 (s, 3H), 3.61 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 149.8, 147.1, 143.7, 140.3, 137.1, 132.4, 128.4, 128.3, 128.1, 127.9, 115.0, 114.4, 113.6, 103.0, 98.7, 75.1, 56.6, 56.0, 55.1, 42.9; HRMS (HESI m/z): [M + H]+ calcd. for C23H25BrNO4, 458.0967; found 458.0961.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-2,3-dimethoxyaniline (4j). The secondary amine 4j was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (900 mg; 2.80 mmol), aniline 1b (442 mg; 2.89 mmol) and NaBH(OAc)3 (1.78 g; 8.4 mmol) in anhydrous toluene (45 mL). The residue was crystallized from EtOH to provide compound 4j (758 mg; 75%) as a pale yellow microcrystalline solid. Mp. = 86–87 °C; TLC Rf = 0.5 (hexane/EtOAc 10/3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.46–7.30 (m, 6H), 7.02 (d, J = 8.8 Hz, 1H), 6.81 (t, J = 8.2 Hz, 1H), 6.46 (d, J = 8.0 Hz, 1H), 6.32 (d, J = 8.3 Hz, 1H), 5.01 (s, 2H), 4.85 (t, J = 6.2 Hz, 1H), 4.27 (d, J = 4.9 Hz, 2H), 3.85 (s, 3H), 3.71 (s, 3H), 3.55 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 152.1, 147.0, 141.5, 137.1, 135.1, 132.2, 128.3, 128.2, 128.1, 128.0, 124.1, 114.4, 113.7, 104.6, 101.6, 74.9, 59.4, 56.0, 55.5, 42.4; HRMS (HESI m/z): [M + H]+ calcd. for C23H25BrNO4, 458.0967; found 458.0961.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-1,3-benzodioxole-5-amine (4k). The secondary amine 4k was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (600 mg; 1.87 mmol), aniline 1c (263 mg; 1.93 mmol) and NaBH(OAc)3 (1.19 g; 5.6 mmol) in anhydrous toluene (33 mL). The residue was crystallized from MeOH/acetone/water mixture to provide compound 4k (791 mg; 96%) as a pale brown microcrystalline solid. Mp. = 77–80 °C; TLC Rf = 0.5 (hexane/EtOAc 10:3 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 7.41–7.27 (m, 6H), 7.04 (d, J = 9.1 Hz, 1H), 6.64 (d, J = 8.3 Hz, 1H), 6.38 (d, J = 2.3 Hz, 1H), 6.08 (dd, J = 8.3, 2.3 Hz, 1H), 5.82 (s, 2H), 5.23 (t, J = 5.2 Hz, 1H), 4.98 (s, 2H), 4.12 (d, J = 5.4 Hz, 2H), 3.86 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 147.7, 147.2, 144.6, 138.1, 137.1, 132.2, 128.3, 128.3, 128.1, 127.9, 115.1, 113.7, 108.4, 103.2, 99.9, 95.3, 75.0, 56.1, 43.2; HRMS (HESI m/z): [M + H]+ calcd. for C22H21BrNO4, 442.0654; found 442.0648.
N-[2-(Benzyloxy)-6-bromo-3-methoxybenzyl]-1,3-benzodioxole-4-amine (4l). The secondary amine 4l was prepared in a similar manner as the amine 4e using bromobenzaldehyde 2c (1.0 g; 3.11 mmol), aniline 1d (439 mg; 3.20 mmol) and NaBH(OAc)3 (1.98 g; 9.3 mmol) in anhydrous toluene (45 mL). The residue was crystallized from EtOH to provide compound 4l (1.24 g; 90%) as a white microcrystalline solid. Mp. = 94–96 °C; TLC Rf = 0.4 (toluene); 1H-NMR (400 MHz, DMSO-d6) δ = 7.41–7.31 (m, 6H), 7.03 (d, J = 9.6 Hz, 1H), 6.62 (t, J = 8.3 Hz, 1H), 6.41 (d, J = 7.6 Hz, 1H), 6.26 (d, J = 7.6 Hz, 1H), 5.86 (s, 2H), 5.01 (s, 2H), 4.75 (t, J = 6.0 Hz, 1H), 4.33 (d, J = 6.0 Hz, 2H), 3.85 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.2, 147.1, 147.0, 137.1, 133.2, 132.7, 132.1, 128.3 (2×), 128.1, 127.9, 122.1, 114.7, 113.8, 107.3, 100.0, 98.4, 74.9, 56.1, 42.7; HRMS (HESI m/z): [M + H]+ calcd. for C22H21BrNO4, 442.0654; found 442.0652.
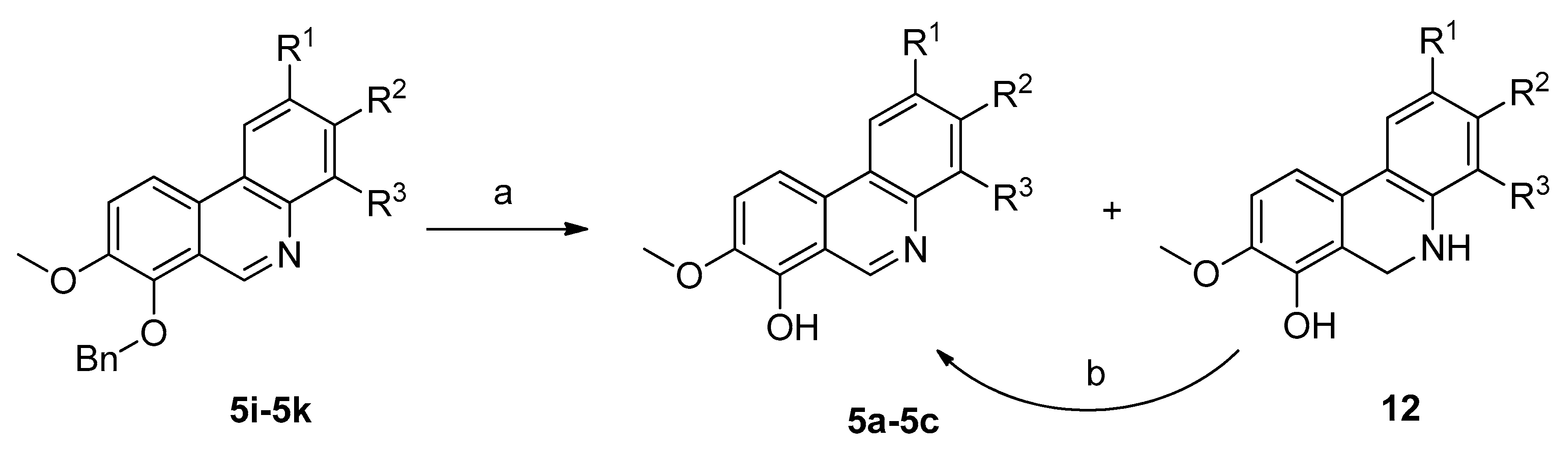
2,3,8-Trimethoxyphenanthridin-7-ol (5a). To a suspension of benzylated phenanthridine 5i (52 mg; 0.14 mmol) in MeOH (8 mL) was added 10% Pd/C (5 mg) and the mixture was vigorously stirred under atmospheric pressure of hydrogen for 4 h. It was then filtered and concentrated under reduced pressure to afford compound 5a (28 mg; 71%) as an orange microcrystalline solid. Mp. = 212–215 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.88 (br. s., 1H), 9.40 (s, 1H), 8.18 (d, J = 9.1 Hz, 1H), 7.98 (s, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.47 (s, 1H), 4.00 (s, 3H), 3.95 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 149.9, 149.3, 145.5, 143.7, 142.7, 138.7, 126.4, 118.0, 117.7, 116.4, 112.8, 109.7, 102.7, 56.9, 55.9, 55.5; HRMS (HESI m/z): [M + H]+ calcd. for C16H16NO4 286.1079; found 286.1074.
3,4,8-Trimethoxyphenanthridin-7-ol (5b). Compound 5b was prepared in a similar manner as the base 5a using benzylated phenanthridine 5j (20 mg; 0.09 mmol), 10% Pd/C (2 mg) and MeOH (4 mL). After stirring for 4 h under atmospheric pressure of hydrogen, the reaction mixture was filtered. The filtrate contained both fully aromatized debenzylated product 5b and partially reduced debenzylated form 12b as a side product. Therefore, the mixture was vigorously stirred in air until only fully aromatized product was present (ca. 2 h). The solution was allowed to stand overnight at −20 °C and the resulting precipitate was filtered off to afford 5b (10 mg; 66%) as an orange crystalline solid. Mp.= 199–202 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/2 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.88 (s, 1H), 9.51 (d, J = 0.5 Hz, 1H), 8.35 (d, J = 9.3 Hz, 1H), 8.11 (d, J = 9.1 Hz, 1H), 7.63 (d, J = 9.1 Hz, 1H), 7.48 (d, J = 9.3 Hz, 1H), 3.96 (s, 3H), 3.95 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.0, 147.9, 144.2, 144.0, 143.1, 138.0, 126.6, 118.6, 118.2, 117.7, 115.8, 114.3, 112.5, 61.2, 56.7, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C16H16NO4 286.1079; found 286.1074.
3-Methoxy-[
1,
3]
dioxolo[4,5-b]phenanthridin-4-ol (
5c). Compound
5k (100 mg; 0.28 mmol) was added to ethanol (4 mL) containing aqueous 35% HCl (3 mL). This reaction mixture was heated in a sealed vial at 100 °C for 2.5 h. After removal of volatiles, the residue was diluted with water (5 mL) and alkalized with aqueous NH
3 solution to pH = 8–9. The precipitated solid was filtered off, washed with water and dried to provide compound
5c (57 mg; 76%) as an orange crystalline solid. A sample for analysis was prepared by crystallization from larger amount of THF. Mp. = 260–270 °C (decomp.); R
f = 0.2 (CHCl
3/MeOH 40/2
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.85 (s, 1H), 9.39 (s, 1H), 8.14 (s, 1H), 8.11 (d,
J = 8.7 Hz, 1H), 7.63 (d,
J = 9.0 Hz, 1H), 7.43 (s, 1H), 6.21 (s, 2H), 3.96 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 148.2, 147.84, 145.6, 143.8, 142.5, 140.04, 126.9, 119.5, 118.2, 116.56, 113.0, 106.9, 101.8, 100.0, 56.8; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
15H
12NO
4, 270.0766; found 270.0760.
7-Methoxy-[
1,
3]
dioxolo[4,5-c]phenanthridin-6-ol (
5d). To a stirred solution of the secondary amine
4d (300 mg; 0.85 mmol) and Bu
3SnH (460 μL; 1.70 mmol) in anhydrous toluene (15 mL) at 70 °C was added AIBN (210 mg, 1.28 mmol) and temperature was raised and maintained at 104–108 °C for a period of 3 h. Due to no further conversion of starting material (
4d), another portion of Bu
3SnH (160 μL; 0.60 mmol) and AIBN (50 mg, 0.30 mmol) were added to the reaction mixture and heating continued for a further 3 h. The reaction mixture was then cooled to room temperature, activated MnO
2 [
44] (222 mg, 2.56 mmol) was added and stirring continued overnight. The suspension was filtered and the filtrate concentrated under reduced pressure. Addition of cyclohexane (30 mL) to the residue resulted in the exclusion of a red precipitate, which was filtered off and further purified by silica gel gradient column chromatography (hexane/EtOAc 5/1–5/3
v/
v) to afford compound
5d (30 mg; 13%) as an orange microcrystalline solid. Mp. = 246–248 °C; R
f = 0.2 (CHCl
3/MeOH 40/2
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.99 (s, 1H), 9.44 (s, 1H), 8.16 (d,
J = 8.8 Hz, 1H), 8.08 (d,
J = 8.8 Hz, 1H), 7.63 (d,
J = 8.8 Hz, 1H), 7.35 (d,
J = 8.8 Hz, 1H), 6.25 (s, 2H), 3.95 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 148.6, 146.1, 144.5, 143.3, 142.3, 129.3, 126.5, 119.5, 118.4, 115.8, 115.7, 112.7, 109.4, 102.0, 56.7; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
15H
12NO
4, 270.0766; found 270.0761.
2,3,7,8-Tetramethoxyphenanthridin (5e). To a stirred solution of the secondary amine 4e (600 mg, 1.57 mmol) and Bu3SnH (845 μL; 3.14 mmol) in anhydrous toluene (30 mL) at 70 °C was added AIBN (387 mg, 2.36 mmol) and temperature of the reaction mixture was raised and maintained at 104–108 °C for a period of 3 h. The reaction was carried out under an inert atmosphere of argon. After 3 h the reaction mixture was slowly cooled to room temperature and activated MnO2 (409 mg; 4.71 mmol) was added. The resulting mixture was stirred overnight, then filtered and concentrated under reduced pressure. Addition of cyclohexane (8 mL) to the residue resulted in the exclusion of a brown precipitate. The suspension was left to stand overnight at 5 °C, and the next day it was filtered, washed with cyclohexane (2 mL) and recrystallized from CHCl3/hexane. Compound 5e (185 mg, 39%) was obtained as a beige crystalline solid. Mp. = 154–158 °C (CHCl3/hexane); Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.35 (s, 1H), 8.51 (d, J = 8.8 Hz, 1H), 8.03 (s, 1H), 7.74 (d, J = 9.1 Hz, 1H), 7.51 (s, 1H), 4.02 (s, 3H), 4.00 (s, 6H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 150.1, 149.6, 148.9, 145.0, 144.1, 139.0, 126.4, 120.2, 118.9, 118.5, 117.6, 109.8, 102.6, 61.4, 56.6, 56.0, 55.6; HRMS (HESI m/z): [M + H]+ calcd. for C17H18NO4, 300.1236; found 300.1230.
3,4,7,8-Tetramethoxyphenanthridine (5f). Phenanthridine 5f was prepared in a similar manner as the compound 5e using amine 4f (467 mg; 1.22 mmol), Bu3SnH (660 μL; 2.44 mmol), AIBN (301 mg, 1.83 mmol) and MnO2 (318 mg; 3.66 mmol) in anhydrous toluene (23 mL). The recrystallization from CHCl3/hexane afforded compound 5f (150 mg; 41%) as a beige crystalline solid. Mp. = 142–144 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.47 (s, 1H), 8.46 (d, J = 9.1 Hz, 1H), 8.42 (d, J = 9.1 Hz, 1H), 7.75 (d, J = 9.1 Hz, 1H), 7.52 (d, J = 9.1 Hz, 1H), 4.01 (s, 3H), 3.99 (s, 3H), 3.96 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.2, 149.3, 147.5, 144.5, 144.0, 138.0, 126.7, 119.7, 119.3, 118.4, 118.2, 117.8, 114.6, 61.5, 61.3, 56.5, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C17H18NO4, 300.1236; found 300.1230.
3,4-Dimethoxy-[
1,
3]
dioxolo[4,5-b]phenanthridine (
5g). Phenanthridine
5g was prepared in a similar manner as compound
5e using secondary amine
4g (265 mg; 0.72 mmol), Bu
3SnH (390 μL; 1.45 mmol), AIBN (178 mg, 1.09 mmol), MnO
2 (189 mg; 2.17 mmol) in anhydrous toluene (13 mL). Compound
5g (55 mg, 27%) was obtained as a beige microcrystalline solid. Mp. = 172–174 °C; TLC R
f = 0.2 (CHCl
3/MeOH 40/1
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.33 (d,
J = 0.5 Hz, 1H), 8.44 (d,
J = 9.0 Hz, 1H), 8.20 (s, 1H), 7.73 (d,
J = 9.3 Hz, 1H), 7.46 (s, 1H), 6.23 (s, 2H), 3.99 (s, 3H), 3.98 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 149.0, 148.4, 148.1, 145.2, 143.9, 140.2, 126.9, 119.5, 119.5, 119.1, 118.7, 107.0, 102.0, 100.0, 61.4, 56.6; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
16H
14NO
4, 284.0923; found 284.0917.
6,7-Dimethoxy-[
1,
3]
dioxolo[4,5-c]phenanthridine (
5h). Phenanthridine
5h was prepared in a similar manner as the product
5e using amine
4h (478 mg; 1.31 mmol), Bu
3SnH (700 μL; 2.61 mmol), AIBN (322 mg; 1.96 mmol) and MnO
2 (340 mg; 3.92 mmol) in anhydrous toluene (23 mL). The recrystallization from CHCl
3/hexane afforded compound
5h (135 mg; 37%) as a brown crystalline solid. Mp. = 163–166 °C; TLC R
f = 0.2 (CHCl
3/MeOH 40/1
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.40 (s, 1H), 8.42 (d,
J = 9.1 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 7.75 (d,
J = 9.0 Hz, 1H), 7.39 (d,
J = 8.6 Hz, 1H), 6.27 (s, 2H), 4.00 (s, 3H), 3.99 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 149.5, 148.1, 146.2, 144.7, 142.3, 129.1, 126.4, 122.3, 119.8, 119.5, 118.3, 115.7, 109.7, 102.1, 61.5, 56.5; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
16H
14NO
4, 284.0923; found 284.0917.
7-(Benzyloxy)-2,3,8-trimethoxyphenanthridine (5i). Phenanthridine 5i was prepared in a similar manner as phenanthridine 5e using secondary amine 4i (1.27 g, 2.78 mmol), Bu3SnH (1.5 mL; 5.55 mmol), AIBN (684 mg; 4.16 mmol) and MnO2 (724 mg, 8.33 mmol) in anhydrous toluene (50 mL). The recrystallization from CHCl3/hexane afforded compound 5i (240 mg; 23%) as a beige crystalline solid. Mp. = 155–156 °C; TLC Rf = 0.2 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.25 (s, 1H), 8.51 (d, J = 9.1 Hz, 1H), 8.01 (s, 1H), 7.76 (d, J = 9.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.46 (s, 1H), 7.41–7.30 (m, 3H), 5.25 (s, 2H), 4.04 (s, 3H), 4.01 (s, 3H), 3.92 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 150.0, 149.5, 149.0, 145.2, 142.6, 138.9, 137.1, 128.6, 128.4, 128.2, 126.3, 120.5, 118.7, 118.5, 117.5, 109.8, 102.6, 75.0, 56.7, 56.0, 55.5; HRMS (HESI m/z): [M + H]+ calcd. for C23H22NO4, 376.1549; found 376.1543.
7-(Benzyloxy)-3,4,8-trimethoxyphenanthridine (5j). Phenanthridine 5j was prepared in a similar manner as phenanthridine 5e using secondary amine 4j (950 mg, 2.07 mmol), Bu3SnH (1115 μL; 4.15 mmol), AIBN (511 mg, 3.11 mmol) and MnO2 (541 mg, 6.22 mmol) in anhydrous toluene (37 mL). The recrystallization from CHCl3/hexane afforded compound 5j (233 mg; 30%) as a slightly gray microcrystalline solid. Mp. = 153–155 °C; TLC Rf = 0.3 (CHCl3/MeOH 40/1 v/v); 1H-NMR (400 MHz, DMSO-d6) δ = 9.38 (s, 1H), 8.45 (d, J = 8.8 Hz, 1H), 8.40 (d, J = 9.1 Hz, 1H), 7.77 (d, J = 9.1 Hz, 1H), 7.55–7.48 (m, 3H), 7.42–7.30 (m, 3H), 5.26 (s, 2H), 4.04 (s, 3H), 3.96 (s, 3H), 3.93 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 151.2, 149.4, 147.6, 143.9, 143.0, 137.9, 137.0, 128.7, 128.4, 128.2, 126.6, 120.1, 119.1, 118.3, 118.2, 117.8, 114.5, 75.1, 61.3, 56.6, 56.4; HRMS (HESI m/z): [M + H]+ calcd. for C23H22NO4, 376.1549; found 376.1543.
4-(Benzyloxy)-3-methoxy-[
1,
3]
dioxolo[4,5-b]phenanthridine (
5k). Phenanthridine
5k was prepared in a similar manner as phenanthridine
5e using secondary amine
4k (730 mg; 1.65 mmol), Bu
3SnH (890 μL; 3.30 mmol), AIBN (407 mg, 2.48 mmol) and MnO
2 (430 mg; 4.95 mmol) in anhydrous toluene (29 mL). The recrystallization from CHCl
3/hexane afforded compound
5k (280 mg; 47%) as a brown crystalline solid. Mp.= 129–131 °C; TLC R
f = 0.3 (CHCl
3/MeOH 40/1
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.25 (s, 1H), 8.43 (d,
J = 9.1 Hz, 1H), 8.18 (s, 1H), 7.76 (d,
J = 9.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.43–7.31 (m, 4H), 6.21 (s, 2H), 5.24 (s, 2H), 4.03 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 149.1, 148.4, 148.0, 145.4, 142.4, 140.0, 137.0, 128.6, 128.4, 128.2, 126.7, 120.6, 119.3, 118.9, 118.6, 106.9, 101.9, 99.9, 75.0, 56.6; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
22H
18NO
4, 360.1236; found 360.1230.
6-(Benzyloxy)-7-methoxy-[
1,
3]
dioxolo[4,5-c]phenanthridine (
5l). Phenanthridine
5l was prepared in a similar manner as phenanthridine
5e using secondary amine
4l (1.1 g; 2.48 mmol), Bu
3SnH (1.34 mL; 4.95 mmol), AIBN (610 mg, 3.72 mmol) and MnO
2 (645 mg; 7.42 mmol) in anhydrous toluene (40 mL). The recrystallization from CHCl
3/hexane afforded compound
5l (555 mg; 62%) as a beige crystalline solid. Mp.= 163–164 °C; TLC R
f = 0.3 (CHCl
3/MeOH 40/1
v/
v);
1H-NMR (400 MHz, DMSO-
d6) δ = 9.27 (s, 1H), 8.42 (d,
J = 9.1 Hz, 1H), 8.21 (d,
J = 8.7 Hz, 1H), 7.78 (d,
J = 9.1 Hz, 1H), 7.50 (d,
J = 6.9 Hz, 2H), 7.41 – 7.30 (m, 4H), 6.25 (s, 2H), 5.26 (s, 2H), 4.04 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 149.62, 148.32, 146.25, 143.13, 142.34, 136.87, 129.06, 128.77, 128.40, 128.28, 126.38, 120.24, 119.31, 119.10, 118.41, 115.68, 109.71, 102.07, 75.07, 56.56; HRMS (HESI
m/
z): [M + H]
+ calcd. for C
22H
18NO
4, 360.1236; found 360.1230.
3.1.3. General Procedure for the Preparation of Phenanthridine Hydrochlorides 6a–6l
The above prepared phenanthridine derivatives 5 (ca. 20 mg) were dissolved in dioxane (2–3 mL). To the resulting solution a 4 M HCl in dioxane solution (1.5 mL) was added resulting in formation of a yellow-orange precipitate. To the suspension was added ether (4 mL). The precipitate was filtered off, washed with ether (2 mL) and dried under vacuum over KOH. The yields of hydrochlorides 6 were within 80–95%. All compounds were proved by elemental analysis; the recorded values were within +/−0.4%.
7-Hydroxy-2,3,8-trimethoxyphenanthridin-5-ium chloride (6a). Elemental analysis, calcd. for C16H16ClNO4 (321.8), C, 59.73; H, 5.01; N, 4.35; found C, 59.83; H, 5.11; N, 4.08.
7-Hydroxy-3,4,8-trimethoxyphenanthridin-5-ium chloride (6b). Elemental analysis, calcd. for C16H16ClNO4 (321.8), C, 59.73; H, 5.01; N, 4.35; found C, 59.74; H, 4.90; N, 4.18.
4-Hydroxy-3-methoxy-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium chloride (
6c). Elemental analysis, calcd. for C
15H
12ClNO
4 (305.7), C, 58.93; H, 3.96; N, 4.58; found C, 58.73; H, 3.77; N, 4.88.
6-Hydroxy-7-methoxy-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium chloride (
6d). Elemental analysis, calcd. for C
15H
12ClNO
4 (305.7), C, 58.93; H, 3.96; N, 4.58; found C, 58.65; H, 3.98; N, 4.73.
2,3,7,8-Tetramethoxyphenanthridin-5-ium chloride (6e). Elemental analysis, calcd. for C17H18ClNO4 (335.8), C, 60.81; H, 5.40; N, 4.17; found C, 61.15; H, 5.63; N, 4.26.
3,4,7,8-Tetramethoxyphenanthridin-5-ium chloride (6f). Elemental analysis, calcd. for C17H18ClNO4 (335.8), C, 60.81; H, 5.40; N, 4.17; found C, 61.01; H, 5.31; N, 4.02.
3,4-Dimethoxy-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium chloride (
6g). Elemental analysis, calcd. for C
16H
14ClNO
4 (319.7), C, 60.10; H, 4.41; N, 4.38; found C, 60.01; H, 4.71; N, 4.22.
6,7-Dimethoxy-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium chloride (
6h). Elemental analysis, calcd. for C
16H
14ClNO
4 (319.7), C, 60.10; H, 4.41; N, 4.38; found C, 60.24; H, 4.69; N, 4.48.
7-(Benzyloxy)-2,3,8-trimethoxyphenanthridin-5-ium chloride (6i). Elemental analysis, calcd. for C23H22ClNO4 (411.9), C, 67.07; H, 5.38; N, 3.40; found C, 67.17; H, 5.31; N, 3.46.
7-(Benzyloxy)-3,4,8-trimethoxyphenanthridin-5-ium chloride (6j). Elemental analysis, calcd. for C23H22ClNO4 (411.9), C, 67.07; H, 5.38; N, 3.40; found C, 67.22; H, 5.46; N, 3.20.
4-(Benzyloxy)-3-methoxy-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium chloride (
6k). Elemental analysis, calcd. for C
22H
18ClNO
4 (395.8), C, 66.75; H, 4.58; N, 3.54; found C, 66.66; H, 4.75; N, 3.44.
6-(Benzyloxy)-7-methoxy-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium chloride (
6l). Elemental analysis, calcd. for C
22H
18ClNO
4 (395.8), C, 66.75; H, 4.58; N, 3.54; found C, 66.58; H, 4.80; N, 3.32.
4-Hydroxy-3-methoxy-6-methyl-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium iodide (
7c). The free base
5c (20 mg; 0.08 mmol) was dissolved in ACN (10 mL) and MeI (19 μL; 0.3 mmol) was added. The reaction mixture was allowed to stand at r.t. in the dark place without stirring. After 7 days, the precipitated crystalline solid was filtered and washed with cold ACN (1 mL) to obtain poorly soluble substance
7c (18 mg; 61%) as a dark orange crystalline solid. Mp. = 250–255 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 11.35 (br. s, 1H), 9.95 (s, 1H), 8.50 (s, 1H), 8.35 (d,
J = 9.0 Hz, 1H), 8.05 (d,
J = 9.1 Hz, 1H), 7.97 (s, 1H), 6.41 (s, 2H), 4.56 (s, 3H), 4.05 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 150.7, 150.1, 147.42, 145.7, 145.2, 130.22, 127.6, 124.0, 122.7, 114.6, 113.5, 103.71, 101.4, 98.6, 57.0, 46.1; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
16H
14NO
4, 284.0923; found 284.0917; elemental analysis, calcd. for C
16H
14INO
4 (411.2), C, 46.74; H, 3.43; N, 3.41; found C, 46.70; H, 3.66; N, 3.35.
6-Hydroxy-7-methoxy-4-methyl-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium iodide (
7d). The quaternary salt
7d was prepared in a similar manner as compound
7c using base
5d (10 mg; 0.04 mmol), MeI (9 μL; 0.15 mmol) and ACN (3 mL). Compound
7d (9 mg; 61%) was obtained as a dark orange crystalline solid. Mp. = 236–239 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 11.52 (br. s, 1H), 9.90 (s, 1H), 8.49 (d,
J = 8.6 Hz, 1H), 8.26 (d,
J = 8.8 Hz, 1H), 8.00 (d,
J = 8.8 Hz, 1H), 7.69 (dd,
J = 1.0, 8.8 Hz, 1H), 6.38 (s, 2H), 4.65 (s, 3H), 4.04 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 152.0, 148.8, 147.4, 145.7, 138.1, 126.9, 124.2, 120.9, 120.0, 118.4, 113.7, 113.0, 112.4, 103.1, 56.8, 48.2; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
16H
14NO
4, 284.0923; found 284.0917; elemental analysis, calcd. for C
16H
14INO
4 (411.2), C, 46.74; H, 3.43; N, 3.41; found C, 46.88; H, 3.54; N, 3.32.
2,3,7,8-Tetramethoxy-5-methylphenanthridinium iodide (7e). The quaternary salt 7e was prepared in a similar manner as compound 7d using base 5e (280 mg, 0.94 mmol), MeI (230 μL; 3.74 mmol) and ACN (14 mL). Compound 7e (372 mg; 90%) was obtained as a dark orange needle-like crystalline solid. Mp. = 263–266 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.96 (s, 1H), 8.84 (d, J = 9.3 Hz, 1H), 8.33 (s, 1H), 8.20 (d, J = 9.3 Hz, 1H), 7.73 (s, 1H), 4.70 (s, 3H), 4.13 (s, 6H), 4.10 (s, 6H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 149.9, 146.9, 145.6, 129.1, 127.7, 125.3, 120.8, 119.2, 118.7, 103.9, 100.7, 62.1, 57.0, 56.8, 56.6, 46.0; HRMS (HESI m/z): [M − I]+ calcd. for C18H20NO4, 314.1392; found 314.1387; elemental analysis, calcd. for C18H20INO4 (441.3), C, 48.99; H, 4.57 N, 3.17; found C, 48.92; H, 4.15; N, 3.12.
3,4,7,8-Tetramethoxy-5-methylphenanthridinium iodide (7f). The quaternary salt 7f was prepared in a similar manner as compound 7d using base 5f (60 mg, 0.20 mmol), MeI (50 μL, 0.80 mmol) and ACN (5 mL). Compound 7f (42 mg; 47%) was obtained as an orange crystalline solid. Mp. = 186–188 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.95 (s, 1H), 8.81 (d, J = 9.3 Hz, 1H), 8.70 (d, J = 9.3 Hz, 1H), 8.19 (d, J = 9.3 Hz, 1H), 7.89 (d, J = 9.3 Hz, 1H), 4.81 (s, 3H), 4.13 (s, 3H), 4.09 (s, 3H), 4.08 (s, 3H), 3.99 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 154.4, 153.6, 150.0, 146.4, 140.1, 128.3, 128.1, 126.2, 120.5, 120.2, 118.5, 118.0, 116.9, 62.2, 61.8, 56.9, 51.1, 39.5; HRMS (HESI m/z): [M − I]+ calcd. for C18H20NO4, 314.1392; found 314.1387.
3,4-Dimethoxy-6-methyl-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium iodide (
7g). The quaternary salt
7g was prepared in a similar manner as substance
7d using base
5g (30 mg, 0.11 mmol), MeI (25 μL; 0.42 mmol) and ACN (6 mL). Compound
7g (40 mg; 89%) was obtained as a bright yellow crystalline solid. Mp. > 300 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 9.96 (s, 1H), 8.71 (d,
J = 9.3 Hz, 1H), 8.58 (s, 1H), 8.19 (d,
J = 9.3 Hz, 1H), 8.04 (s, 1H), 6.44 (s, 2H), 4.62 (s, 3H), 4.11 (s, 3H), 4.08 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 151.1, 150.4, 150.0, 147.2, 145.5, 130.6, 128.1, 125.5, 122.8, 119.1, 118.7, 103.9, 101.2, 98.5, 62.1, 57.0, 46.4; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
17H
16NO
4, 298.1079; found 298.1074.
6,7-Dimethoxy-4-methyl-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium iodide (
7h). The quaternary salt
7h was prepared in a similar manner as the substance
7d using free base
5h (90 mg; 0.32 mmol), MeI (80 μL; 1.27 mmol) and ACN (5 mL). Compound
7h (26 mg; 19%) was obtained as a blood-red crystalline solid. Mp. = 223–224 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 9.95 (s, 1H), 8.64 (d,
J = 9.1 Hz, 1H), 8.59 (d,
J = 8.8 Hz, 1H), 8.16 (d,
J = 9.3 Hz, 1H), 7.76 (d,
J = 8.8 Hz, 1H), 6.42 (s, 2H), 4.71 (s, 3 H), 4.11 (s, 3H), 4.07 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 152.1, 150.2, 149.1, 146.7, 138.2, 127.6, 126.0, 120.9, 120.2, 118.7, 118.6, 118.0, 112.8, 103.3, 62.2, 56.9, 48.7; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
17H
16NO
4, 298.1079; found 298.1074.
7-(Benzyloxy)-2,3,8-trimethoxy-5-methylphenanthridinium iodide (7i). The quaternary salt 7i was prepared in a similar manner as compound 7d using free base 5i (91 mg; 0.24 mmol), MeI (60 μL; 0.97 mmol) and ACN (9 mL). Compound 7i (77 mg; 61%) was obtained as a bright yellow crystalline solid. Mp. = 207–210 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.79 (s, 1H), 8.83 (d, J = 9.3 Hz, 1H), 8.30 (s, 1H), 8.20 (d, J = 9.1 Hz, 1H), 7.70 (s, 1H), 7.58 (d, J = 7.5 Hz, 2H), 7.43–7.30 (m, 3H), 5.40 (s, 2H), 4.68 (s, 3H), 4.12 (s, 6H), 4.09 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 152.0, 151.5, 150.2, 146.7, 143.9, 136.3, 128.9, 128.9, 128.4, 128.3, 127.5, 125.0, 120.7, 119.4, 119.1, 103.9, 100.7, 75.4, 57.0, 56.7, 56.6, 46.2; HRMS (HESI m/z): [M − I]+ calcd. for C24H24NO4, 390.1705; found 390.1700.
7-(Benzyloxy)-3,4,8-trimethoxy-5-methylphenanthridinium iodide (7j). The quaternary salt 7j was prepared in a similar manner as compound 7d using free base 5j (150 mg; 0.40 mmol), MeI (100 μL; 1.6 mmol) and ACN (7 mL). Compound 7j (123 mg; 60%) was obtained as a light orange microcrystalline solid. Mp. = 169–173 °C; 1H-NMR (400 MHz, DMSO-d6) δ = 9.79 (s, 1H), 8.80 (d, J = 9.3 Hz, 1H), 8.70 (d, J = 9.1 Hz, 1H), 8.20 (d, J = 9.1 Hz, 1H), 7.89 (d, J = 9.3 Hz, 1H), 7.59 (d, J = 7.0 Hz, 2 H), 7.48–7.26 (m, 3H), 5.41 (s, 2H), 4.79 (s, 3H), 4.10 (s, 3H), 4.08 (s, 3H), 3.98 (s, 3H); 13C-NMR (101 MHz, DMSO-d6) δ = 154.4, 153.5, 150.3, 144.6, 140.1, 136.3, 128.9, 128.4, 128.3, 128.2, 127.9, 126.0, 120.4, 120.2, 118.8, 118.5, 117.0, 75.6, 61.8, 57.0, 56.9, 51.3; HRMS (HESI m/z): [M − I]+ calcd. for C24H24NO4, 390.1705; found 390.1700.
4-(Benzyloxy)-3-methoxy-6-methyl-[
1,
3]
dioxolo[4,5-b]phenanthridin-6-ium iodide (
7k). The quaternary salt
7k was prepared in a similar manner as compound
7d using free base
5k (91 mg, 0.25 mmol), MeI (60 μL, 1.0 mmol) and ACN (8 mL). The reaction was conducted at 50 °C for 36 h. Compound
7k (77 mg; 61%) was obtained as a pale yellow crystalline solid. Mp. = 198–201 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 9.81 (s, 1H), 8.69 (d,
J = 9.1 Hz, 1H), 8.55 (s, 1H), 8.19 (d,
J = 9.3 Hz, 1H), 8.01 (s, 1H), 7.61–7.56 (m, 2H), 7.42–7.31 (m, 3H), 6.44 (s, 2H), 5.38 (s, 2H), 4.60 (s, 3H), 4.10 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 151.1, 150.4, 150.2, 147.0, 143.7, 136.3, 130.5, 128.9, 128.4, 128.3, 128.0, 125.3, 122.8, 119.2, 119.2, 103.9, 101.3, 98.5, 75.4, 56.9, 46.7; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
23 H
20NO
4, 374.1392; found 374.1387.
6-(Benzyloxy)-7-methoxy-4-methyl-[
1,
3]
dioxolo[4,5-c]phenanthridin-4-ium iodide (
7l). The quaternary salt
7l was prepared in a similar manner as compound
7d with using free base
5l (36 mg, 0.1 mmol), MeI (24 μL, 0.4 mmol) and ACN (4 mL). The reaction was conducted at 50 °C for 36 h. Compound
7l (28 mg; 55%) was obtained as a light orange solid. Mp. = 169–172 °C;
1H-NMR (400 MHz, DMSO-
d6) δ = 9.80 (s, 1H), 8.64 (d,
J = 9.2 Hz, 1H), 8.58 (d,
J = 8.7 Hz, 1H), 8.17 (d,
J = 9.2 Hz, 1H), 7.75 (d,
J = 8.7 Hz, 1H), 7.64–7.52 (m, 2H), 7.44–7.28 (m, 3H), 6.40 (s, 2H), 5.39 (s, 2H), 4.69 (s, 3H), 4.08 (s, 3H);
13C-NMR (101 MHz, DMSO-
d6) δ = 152.0, 150.2, 149.2, 145.0, 138.2, 136.2, 128.9, 128.5, 128.4, 127.6, 125.8, 120.8, 120.2, 119.0, 118.6, 118.5, 112.9, 103.3, 75.6, 56.9, 48.9; HRMS (HESI
m/
z): [M − I]
+ calcd. for C
23 H
20NO
4, 374.1392; found 374.1387.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}