Structure-Based Discovery and Synthesis of Potential Transketolase Inhibitors

 ,
,
Abstract
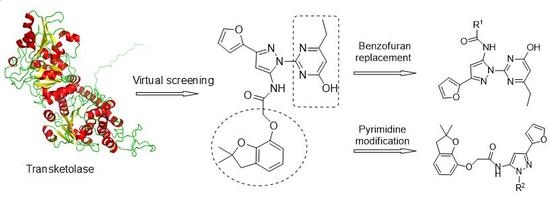
1. Introduction
2. Results and Discussion
2.1. Homology Modeling of AtTKL1
2.2. Virtual Screening and Biological Activity
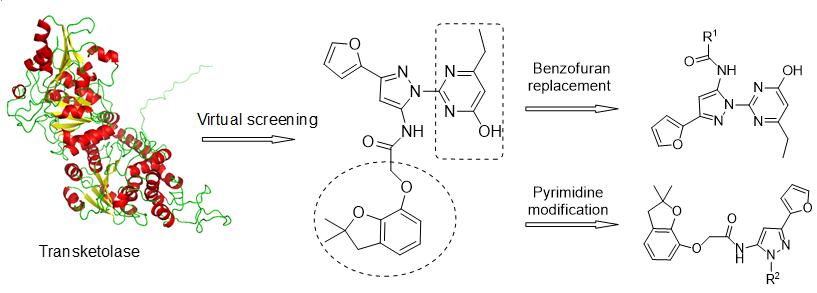
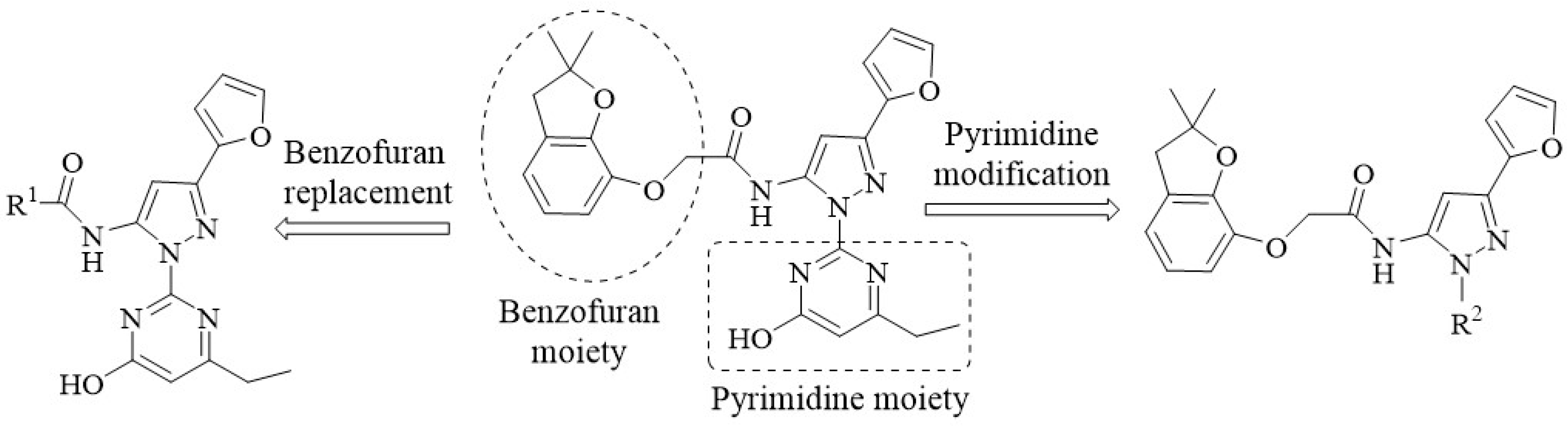
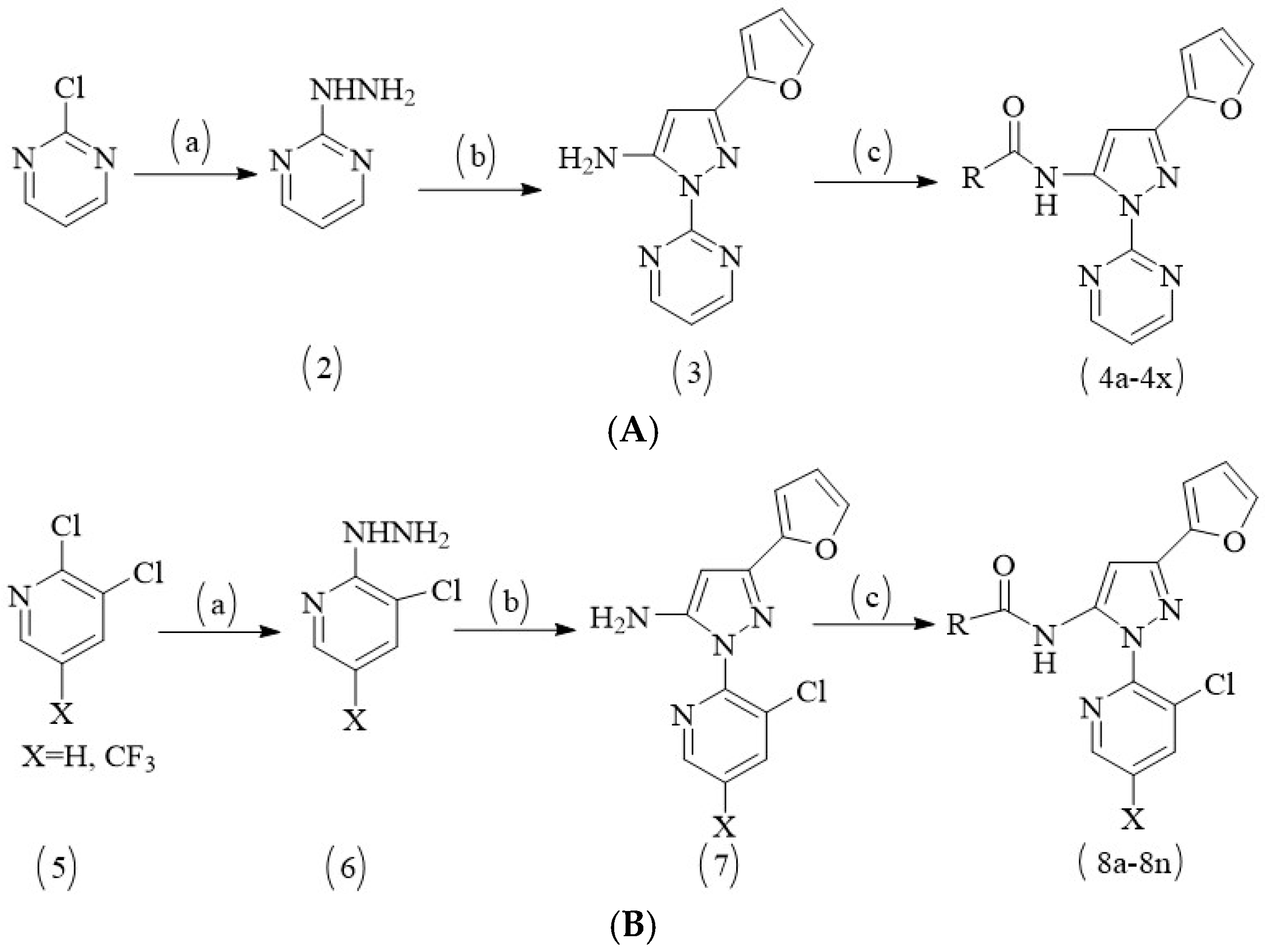
2.3. The Syntheses and Biological Activities of the New Compounds
2.4. The Biological Activities of the New Compounds 4a–4x and 8a–8n
2.5. Molecular Docking of Compounds 4u and 8h with Transketolase
3. Materials and Methods
3.1. Homology Modeling of A. thaliana Transketolase 1 (AtTKL1)
3.2. Virtual Screening
3.3. Biological Activities of Compounds
3.3.1. Herbicidal Bioassay
3.3.2. Fungicide Bioassay
3.4. Designing and Synthesizing New Compounds as Potent Herbicide Candidates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fan, Z.J.; Ai, Y.W.; Qian, C.F.; Li, Z.M. Herbicide activity of monosulfuron and its mode of action. J. Environ. Sci. 2005, 17, 399–403. [Google Scholar]
- Song, Y. Insight into the mode of action of 2,4-dichlorophenoxyacetic acid (2,4-D) as an herbicide. J. Integr. Plant Biol. 2014, 56, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Henkes, S.; Sonnewald, U.; Badur, R.; Flachmann, R.; Stitt, M. A small decrease of plastid transketolase activity in antisense tobacco transformants has dramatic effects on photosynthesis and phenylpropanoid metabolism. Plant. Cell 2001, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Kamada, N.; Takano, Y.; Nakano, T. Molecular analysis of the Corynebacterium glutamicum transketolase gene. Biosci. Biotechnol. Biochem. 1999, 63, 1806–1810. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Kondo, E.; Makino, A. Effects of co-overexpression of the genes of rubisco and transketolase on photosynthesis in rice. Photosynth. Res. 2017, 131, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Linnemann, T.V.; Schreiber, L.; Bartels, D. The role of transketolase and octulose in the resurrection plant Craterostigma plantagineum. J. Exp. Bot. 2016, 67, 3551–3559. [Google Scholar] [CrossRef] [PubMed]
- Bagherzadeh, K.; Shirgahi Talari, F.; Sharifi, A.; Ganjali, M.R.; Saboury, A.A.; Amanlou, M. A new insight into mushroom tyrosinase inhibitors: Docking, pharmacophore-based virtual screening, and molecular modeling studies. J. Biomol. Struct. Dyn. 2015, 33, 487–501. [Google Scholar] [CrossRef] [PubMed]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, S.; Echt, S.; Busch, M.; Freigang, J.; Auerbach, G.; Bader, G.; Martin, W.F.; Bacher, A.; Huber, R.; Fischer, M. Structure and properties of an engineered transketolase from maize. Plant Physiol. 2003, 132, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Huo, J.; Liu, N.; Zhang, J.; Dong, J. Transketolase is identified as a target of herbicidal substance alpha-terthienyl by proteomics. Toxins 2018, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using pymol. J. Comput.-Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with pymol and autodock/vina. J. Comput.-Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Han, R.; Cao, Q.; Yu, J.; Mao, J.; Zhang, T.; Wang, S.; Niu, Y.; Liu, D. Pharmacophore-based virtual screening of novel inhibitors and docking analysis for CYP51a from Penicillium italicum. Mar. Drugs 2017, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Wadood, A.; Ali, M.; Zia, U.; Ul-Haq, Z.; Lodhi, M.A.; Khan, M.; Perveen, S.; Choudhary, M.I. Identification of potent urease inhibitors via ligand- and structure-based virtual screening and in vitro assays. J. Mol. Graph. Model. 2010, 28, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-L.; Zhang, L.; Hu, X.-P.; Yin, B.; Liang, P.; Yang, X.-L. Target-based design, synthesis and biological activity of new pyrazole amide derivatives. Chin. Chem. Lett. 2016, 27, 251–255. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, P.; Bi, Y.; Luo, X. Analysis of a drug target-based classification system using molecular descriptors. Comb. Chem. High Throughput Screen. 2016, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, A.P.; Boyd, S.; Walse, B. Fragment-based drug discovery and protein-protein interactions. Res. Rep. Biochem. 2014, 4, 13–26. [Google Scholar] [CrossRef]
- Moreno, M.A.; Alonso, A.; Alcolea, P.J.; Abramov, A.; De Lacoba, M.G.A.; Abendroth, J.; Zhang, S.; Edwards, T.; Lorimer, D.; Myler, P.J. Tyrosine aminotransferase from leishmania infantum: A new drug target candidate. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Anderson, G.; Dean, O.; Berk, M.; Galecki, P.; Martin-Subero, M.; Maes, M. The glutathione system: A new drug target in neuroimmune disorders. Mol. Neurobiol. 2014, 50, 1059–1084. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Hontecillas, R.; Casandra, W.P.; Bassaganya-Riera, J. Lanthionine synthetase component c-like protein 2: A new drug target for inflammatory diseases and diabetes. Curr. Drug Targets 2014, 15, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Duke, S.O. Natural compounds as next-generation herbicides. Plant Physiol. 2014, 166, 1090–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.H.; Zhan, Y.Z.; Zhang, L.Y.; Li, Z.M.; Li, Y.H.; Zhang, X.; Wang, B.L. Synthesis and biological activities of novel 5-substituted-1,3,4-oxadiazole mannich bases and bis-mannich bases as ketol-acid reductoisomerase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4661–4665. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Zhang, L.Y.; Liu, X.H.; Ma, Y.; Zhang, Y.; Li, Z.M.; Zhang, X. Synthesis, biological activities and sar studies of new 3-substitutedphenyl-4-substitutedbenzylideneamino-1,2,4-triazole mannich bases and bis-mannich bases as ketol-acid reductoisomerase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 5457–5462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Y.; Cui, Z.; Beattie, D.; Gu, Y.; Wang, Q. Design, synthesis, and biological activities of arylmethylamine substituted chlorotriazine and methylthiotriazine compounds. J. Agric. Food Chem. 2011, 59, 11711–11717. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shang, Y.; Xu, D. Multi-dimensional scaling and modeller-based evolutionary algorithms for protein model refinement. In Proceedings of the 2014 IEEE Congress on Evolutionary computation, Beijing, China, 6–11 July 2014; pp. 1038–1045. [Google Scholar]
- Parida, B.K.; Panda, P.K.; Misra, N.; Mishra, B.K. MaxMod: A hidden markov model based novel interface to modeller for improved prediction of protein 3D models. J. Mol. Model. 2015, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.Y.; Sali, A. Protein structure modeling with modeller. Methods Mol. Biol. 2008, 426, 145–159. [Google Scholar] [PubMed]
- Kassler, K.; Meier, J.; Eichler, J.; Sticht, H. Structural basis for species selectivity in the HIV-1 gp120-cd4 interaction: Restoring affinity to gp120 in murine cd4 mimetic peptides. Adv. Bioinform. 2011, 2011, 736593. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. A Publ. Protein Soc. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the all target compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | ZINC ID and Chemical Name | Affinity (kcal/mol) | xlogP | Molecular Weight (g/mol) | H-Bond Donors/Acceptors | H-Bond Binding Site/Bond Distance | Structural Formula |
|---|---|---|---|---|---|---|---|
| a | ZINC12007063 2-[(2,2-dimethyl-3H-benzofuran-7-yl)oxy]-N-[2-(4-ethyl-6-oxo-1H-pyrimidin-2-yl)-5-(2-furyl)pyrazol-3 | −9.1 | 3.81 | 474.497 | 1/10 | Ser266/3.3Å |  |
| Ser140/3.0Å | |||||||
| Gly232/3.4Å | |||||||
| His340/3.3Å | |||||||
| Leu194/2.9Å | |||||||
| Ser266/2.9Å | |||||||
| b | ZINC12126699 N-[5-cyclopropyl-2-(4-isopropyl-6-oxo-1H-pyrimidin-2-yl)pyrazol-3-yl]-2-[(2,2-dimethyl-3H-benzofuran | −9.0 | 4.07 | 462.53 | 1/9 | Ser266/3.3Å |  |
| Ser140/3.0Å | |||||||
| Gly232/3.4Å | |||||||
| His340/3.3Å | |||||||
| Leu194/2.9Å | |||||||
| c | ZINC19961402 (5R,6S)-5(3-chlorophenyl)-4,7-dioxo-2-(phenylamino)-3,4,5,6.7,8-hexahydroquinazoline-6-carbonitrile | −9.0 | 1.61 | 391.818 | 3/7 | His143/3.2Å |  |
| Gly/234/3.5Å | |||||||
| Gly232/3.1Å | |||||||
| d | ZINC16283531 (NE)-N-[[(4,6-dimethylpyrimidin-2-yl)amino]-[[3-(trifluoromethyl)phenyl]amino]methylene]-2,2-dimethyl | −8.7 | 4.33 | 392.405 | 1/6 | Gly234/3.2Å |  |
| e | ZINC58191888 (4Z)-4-[[4-[(1-methylimidazol-2-yl)methoxy]phenyl]-methyslene]isoquinoline-1,3-dione | −8.6 | 2.5 | 359.385 | 1/6 | His103/3.2Å |  |
| His340/3.5Å | |||||||
| Cys235/3.1Å | |||||||
| f | ZINC12929396 3-isopropyl-N-methyl-N-[(4-oxo-3H-quinazolin-2-yl)methyl]isoxazolo[4,5-e]pyridine-5-carboxamide | −8.6 | 3.46 | 377.404 | 1/8 | Gly232/3.4Å |  |
| His340/3.1Å | |||||||
| Gly192/3.5Å |
| Compound | Substituent Group | Herbicidal Activity | Fungicide Activity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R | X | Brassica campestris | Echinochloca crus-galli | C. arachidicola | B. cinerea | G. zeae | S. sclerotiorum | R. cerealis | P. sasakii | |||
| Root | Stem | Root | Stem | |||||||||
| 4a | 7-ethoxybenzofuran | / | 0 | 0 | 0 | 0 | 46.24 ± 1.63 | 30.24 ± 2.94 | 19.18 ± 1.63 | 97.18 ± 2.16 | 0 | 39.65 ± 4.32 |
| 4b | pyridin-2-yl | / | 0 | 0 | 4.15 ± 1.63 | 0 | 31.24 ± 1.63 | 50.95 ± 0.82 | 26.34 ± 3.56 | 85.35 ± 2.94 | 9.46 ± 1.41 | 43.96 ± 2.16 |
| 4c | pyridin-3-yl | / | 31.30 ± 2.16 | 6.12 ± 1.63 | 14.24 ± 2.45 | 25.59 ± 2.45 | 8.57 ± 1.41 | 0 | 19.57 ± 1.63 | 96.09 ± 2.94 | 9.94 ± 3.74 | 30.28 ± 1.41 |
| 4d | pyridin-4-yl | / | 43.12 ± 5.35 | 0 | 26.32 ± 2.94 | 10.37 ± 1.41 | 31.36 ± 2.16 | 21.28 ± 0.82 | 26.02 ± 0.82 | 98.12 ± 1.63 | 9.42 ± 7.12 | 0 |
| 4e | 6-CF3-pyridin-2-yl | / | 0 | 0 | 73.85 ± 9.27 | 36.22 ± 1.41 | 27.97 ± 1.63 | 60.43 ± 0.82 | 43.86 ± 2.16 | 81.39 ± 1.63 | 14.97 ± 2.16 | 28.66 ± 3.56 |
| 4f | thiophene-2-yl | / | 0 | 0 | 0 | 0 | 46.52 ± 1.41 | 35.28 ± 1.63 | 37.56 ± 1.41 | 73.92 ± 3.56 | 39.38 ± 7.12 | 63.17 ± 1.63 |
| 4g | furan-2-yl | / | 27.21 ± 3.74 | 0 | 25.24 ± 1.41 | 10.26 ± 2.16 | 42.30 ± 2.16 | 33.37 ± 0.82 | 65.26 ± 5.89 | 100 | 17.49 ± 2.83 | 52.28 ± 2.94 |
| 4h | Ph | / | 0 | 0 | 23.32 ± 4.32 | 7.15 ± 2.94 | 31.61 ± 0.82 | 70.29 ± 0.82 | 30.15 ± 0.82 | 100 | 9.28 ± 4.32 | 0 |
| 4i | 2-CF3-Ph | / | 0 | 0 | 0 | 0 | 15.23 ± 2.83 | 50.49 ± 0.82 | 22.50 ± 8.29 | 96.01 ± 2.83 | 0 | 0 |
| 4j | 3-CF3-Ph | / | 0 | 0 | 22.18 ± 3.74 | 5.18 ± 2.16 | 23.06 ± 0.82 | 30.58 ± 2.94 | 15.35 ± 1.63 | 100 | 4.55 ± 1.41 | 43.09 ± 6.98 |
| 4k | 4-CF3-Ph | / | 17.02 ± 1.63 | 0 | 0 | 0 | 8.41 ± 0.82 | 50.09 ± 1.63 | 26.64 ± 1.41 | 100 | 9.37 ± 8.52 | 33.17 ± 2.45 |
| 4l | 3-F-Ph | / | 19.65 ± 2.94 | 23.11 ± 1.41 | 0 | 0 | 46.52 ± 2.94 | 55.98 ± 1.63 | 41.53 ± 2.16 | 96.94 ± 1.63 | 0 | 33.92 ± 10.2 |
| 4m | 4-F-Ph | / | 71.14 ± 1.63 | 45.20 ± 3.56 | 61.25 ± 2.45 | 35.38 ± 4.32 | 0 | 30.94 ± 2.94 | 4.29 ± 3.56 | 96.37 ± 2.83 | 9.25 ± 1.63 | 22.40 ± 3.27 |
| 4n | 2-F-Ph | / | 69.30 ± 2.94 | 22.45 ± 2.94 | 12.14 ± 2.94 | 0 | 31.61 ± 0.82 | 50.37 ± 0.82 | 59.58 ± 2.45 | 94.38 ± 3.74 | 9.34 ± 2.94 | 52.38 ± 5.89 |
| 4o | 2-Cl-Ph | / | 19.08 ± 3.27 | 8.18 ± 3.74 | 21.15 ± 1.63 | 5.15 ± 2.94 | 36.99 ± 2.16 | 40.29 ± 0.82 | 57.30 ± 5.35 | 93.28 ± 2.45 | 23.36 ± 2.16 | 24.19 ± 2.94 |
| 4p | 3-Cl-Ph | / | 0 | 0 | 0 | 0 | 27.16 ± 1.63 | 45.38 ± 2.16 | 43.91 ± 0.82 | 71.30 ± 0.82 | 14.63 ± 2.94 | 20.28 ± 0.82 |
| 4q | 4-Cl-Ph | / | 32.25 ± 3.60 | 57.14 ± 3.56 | 6.18 ± 2.16 | 0 | 27.12 ± 3.56 | 50.49 ± 4.08 | 48.64 ± 1.41 | 69.07 ± 4.97 | 18.28 ± 2.45 | 8.07 ± 2.16 |
| 4r | 2,6-F2-Ph | / | 59.66 ± 3.27 | 40.02 ± 2.16 | 0 | 0 | 36.35 ± 1.41 | 60.38 ± 0.82 | 24.28 ± 0.82 | 64.32 ± 1.41 | 18.91 ± 11.43 | 20.59 ± 2.94 |
| 4s | 2,4-Cl2-Ph | / | 60.29 ± 1.63 | 25.30 ± 2.16 | 2.92 ± 2.16 | 0 | 18.81 ± 0.82 | 65.94 ± 1.63 | 29.17 ± 1.63 | 79.59 ± 4.32 | 14.28 ± 5.72 | 24.94 ± 1.63 |
| 4t | PhOCH2CH2 | / | 54.48 ± 2.94 | 41.01 ± 2.45 | 33.45 ± 4.32 | 10.18 ± 4.32 | 55.49 ± 2.94 | 50.28 ± 2.16 | 48.61 ± 1.41 | 62.27 ± 5.72 | 36.97 ± 3.56 | 32.97 ± 0.82 |
| 4u | 2,4-Cl2-PhOCH2CH2 | / | 66.70 ± 0.82 | 73.15 ± 2.83 | 83.52 ± 2.16 | 55.26 ± 2.45 | 27.61 ± 0.82 | 65.37 ± 0.82 | 62.28 ± 1.63 | 50.38 ± 2.94 | 36.29 ± 2.94 | 12.08 ± 2.94 |
| 4v | 4-NO2-Ph | / | 25 ± 2.45 | 0 | 0 | 0 | 36.35 ± 1.63 | 55.28 ± 1.63 | 43.09 ± 1.41 | 69.29 ± 2.45 | 14.52 ± 1.41 | 28.33 ± 3.74 |
| 4w | 4-MeO-Ph | / | 0 | 0 | 58.24 ± 2.45 | 33.23 ± 2.16 | 45.19 ± 1.63 | 75.68 ± 2.94 | 30 ± 2.45 | 79.21 ± 1.41 | 23.88 ± 2.16 | 12.28 ± 2.16 |
| 4x | cyclopropyl | / | 0 | 11.04 ± 1.63 | 26.40 ± 4.24 | 10.12 ± 2.16 | 64.56 ± 2.94 | 70.64 ± 1.63 | 33 ± 0.82 | 79.28 ± 3.27 | 23.34 ± 3.74 | 32.17 ± 0.82 |
| 8a | 3-CF3-Ph | H | 53.15 ± 3.56 | 17.10 ± 2.16 | 9.20 ± 3.27 | 20.02 ± 6.48 | 47.89 ± 4.97 | 25.28 ± 4.32 | 37.17 ± 0.82 | 85.59 ± 3.56 | 24.09 ± 2.16 | 39.16 ± 4.08 |
| 8b | 3-Cl-Ph | H | 56.22 ± 1.63 | 41.24 ± 2.16 | 47.48 ± 1.63 | 16.65 ± 3.74 | 40.39 ± 0.82 | 58.46 ± 0.82 | 40.76 ± 1.41 | 98.03 ± 1.63 | 10.29 ± 2.94 | 43.24 ± 3.56 |
| 8c | PhOCH2CH2 | H | 73.27 ± 2.94 | 60.55 ± 1.41 | 38.17 ± 3.56 | 10.15 ± 1.63 | 53.19 ± 2.16 | 67.35 ± 1.41 | 43.16 ± 0.82 | 93.49 ± 2.16 | 19.87 ± 4.32 | 35.49 ± 1.63 |
| 8d | 2,4-Cl2-PhOCH2CH2 | H | 76.25 ± 0.82 | 29.58 ± 2.16 | 21.18 ± 4.08 | 5.47 ± 4.32 | 27.18 ± 1.63 | 55.88 ± 1.63 | 40.25 ± 0.82 | 76.58 ± 2.16 | 5.24 ± 1.63 | 24.28 ± 1.41 |
| 8e | 4-MeO-Ph | H | 27.49 ± 2.16 | 13.22 ± 2.83 | 43.54 ± 5.89 | 31.24 ± 4.55 | 18.49 ± 2.83 | 50.92 ± 0.82 | 57.28 ± 6.53 | 76.19 ± 2.16 | 9.91 ± 1.41 | 16.57 ± 2.16 |
| 8f | 6-CF3-pyridin-2-yl | H | 31.54 ± 3.74 | 0 | 21.40 ± 1.41 | 5.18 ± 1.63 | 36.48 ± 3.74 | 55.38 ± 0.82 | 38.66 ± 3.74 | 71.49 ± 0.82 | 27.28 ± 3.74 | 36.39 ± 3.56 |
| 8g | 3-CF3-Ph | CF3 | 52.22 ± 6.48 | 19.44 ± 4.24 | 15 ± 2.94 | 0 | 27.18 ± 1.63 | 54.19 ± 0.82 | 69.19 ± 1.63 | 93.38 ± 2.16 | 0 | 35.02 ± 0.82 |
| 8h | PhOCH2CH2 | CF3 | 82.48 ± 7.26 | 72.25 ± 4.55 | 41.15 ± 1.63 | 11.08 ± 4.08 | 27.19 ± 1.41 | 25.28 ± 2.16 | 37.17 ± 2.94 | 95.06 ± 3.74 | 19.21 ± 2.83 | 26.89 ± 3.56 |
| 8i | 3-Cl-Ph | CF3 | 48.47 ± 2.94 | 26.15 ± 2.16 | 8.18 ± 2.16 | 0 | 27.49 ± 3.27 | 50.94 ± 2.16 | 62.91 ± 8.83 | 74.15 ± 1.41 | 0 | 24.34 ± 2.83 |
| 8j | 2-Cl-Ph | CF3 | 35.26 ± 4.32 | 40.25 ± 2.16 | 20.18 ± 2.16 | 1.08 ± 0.82 | 36.52 ± 1.63 | 20.35 ± 1.41 | 37.09 ± 2.45 | 74.27 ± 1.41 | 14.19 ± 1.63 | 20.28 ± 9.42 |
| 8k | 4-Cl-Ph | CF3 | 18.30 ± 2.94 | 29.02 ± 2.45 | 3.24 ± 1.41 | 0 | 9.87 ± 1.41 | 50.97 ± 2.16 | 24.28 ± 0.82 | 83.59 ± 4.55 | 23.28 ± 1.41 | 24.19 ± 2.45 |
| 8l | 4-NO2-Ph | CF3 | 2.19 ± 1.41 | 0 | 3.20 ± 2.16 | 9.20 ± 3.56 | 27.91 ± 3.27 | 65.97 ± 0.82 | 38.17 ± 3.56 | 57.14 ± 2.94 | 27.09 ± 4.55 | 20.28 ± 1.41 |
| 8m | 4-MeO-Ph | CF3 | 12.18 ± 2.94 | 11.15 ± 3.27 | 44.18 ± 2.16 | 13.30 ± 2.45 | 45.49 ± 1.63 | 60.68 ± 1.63 | 62.29 ± 0.82 | 52.79 ± 7.35 | 14.68 ± 1.41 | 16.47 ± 2.16 |
| 8n | 6-CF3-pyridin-2-yl | CF3 | 4.24 ± 2.94 | 2.99 ± 2.16 | 0 | 0 | 9.35 ± 2.16 | 50.94 ± 2.16 | 38.89 ± 4.55 | 67.29 ± 8.04 | 9.53 ± 2.94 | 36.35 ± 2.16 |
| ZINC12007063 | / | / | 78.48 ± 2.16 | 63.22 ± 2.83 | 66.18 ± 2.16 | 61.18 ± 3.27 | 17.29 ± 3.56 | 24.35 ± 0.82 | 16.21 ± 1.41 | 63.94 ± 3.56 | 36.27 ± 2.16 | 13.02 ± 1.63 |
| Atrazine | / | / | 96.54 ± 1.63 | 92.45 ± 4.32 | 92.62 ± 4.32 | 90.23 ± 2.83 | / | / | / | / | / | / |
| Azoxystrobin | / | / | / | / | / | / | 56.49 ± 1.41 | 71.29 ± 1.63 | 75.19 ± 2.16 | 100 | 88.10 ± 1.41 | 84.34 ± 2.45 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, J.; Zhao, B.; Zhang, Z.; Xing, J.; Zhang, J.; Dong, J.; Fan, Z. Structure-Based Discovery and Synthesis of Potential Transketolase Inhibitors. Molecules 2018, 23, 2116. https://doi.org/10.3390/molecules23092116
Huo J, Zhao B, Zhang Z, Xing J, Zhang J, Dong J, Fan Z. Structure-Based Discovery and Synthesis of Potential Transketolase Inhibitors. Molecules. 2018; 23(9):2116. https://doi.org/10.3390/molecules23092116
Chicago/Turabian StyleHuo, Jingqian, Bin Zhao, Zhe Zhang, Jihong Xing, Jinlin Zhang, Jingao Dong, and Zhijin Fan. 2018. "Structure-Based Discovery and Synthesis of Potential Transketolase Inhibitors" Molecules 23, no. 9: 2116. https://doi.org/10.3390/molecules23092116
APA StyleHuo, J., Zhao, B., Zhang, Z., Xing, J., Zhang, J., Dong, J., & Fan, Z. (2018). Structure-Based Discovery and Synthesis of Potential Transketolase Inhibitors. Molecules, 23(9), 2116. https://doi.org/10.3390/molecules23092116