
Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand


Abstract

1. Introduction
2. Results and Discussion
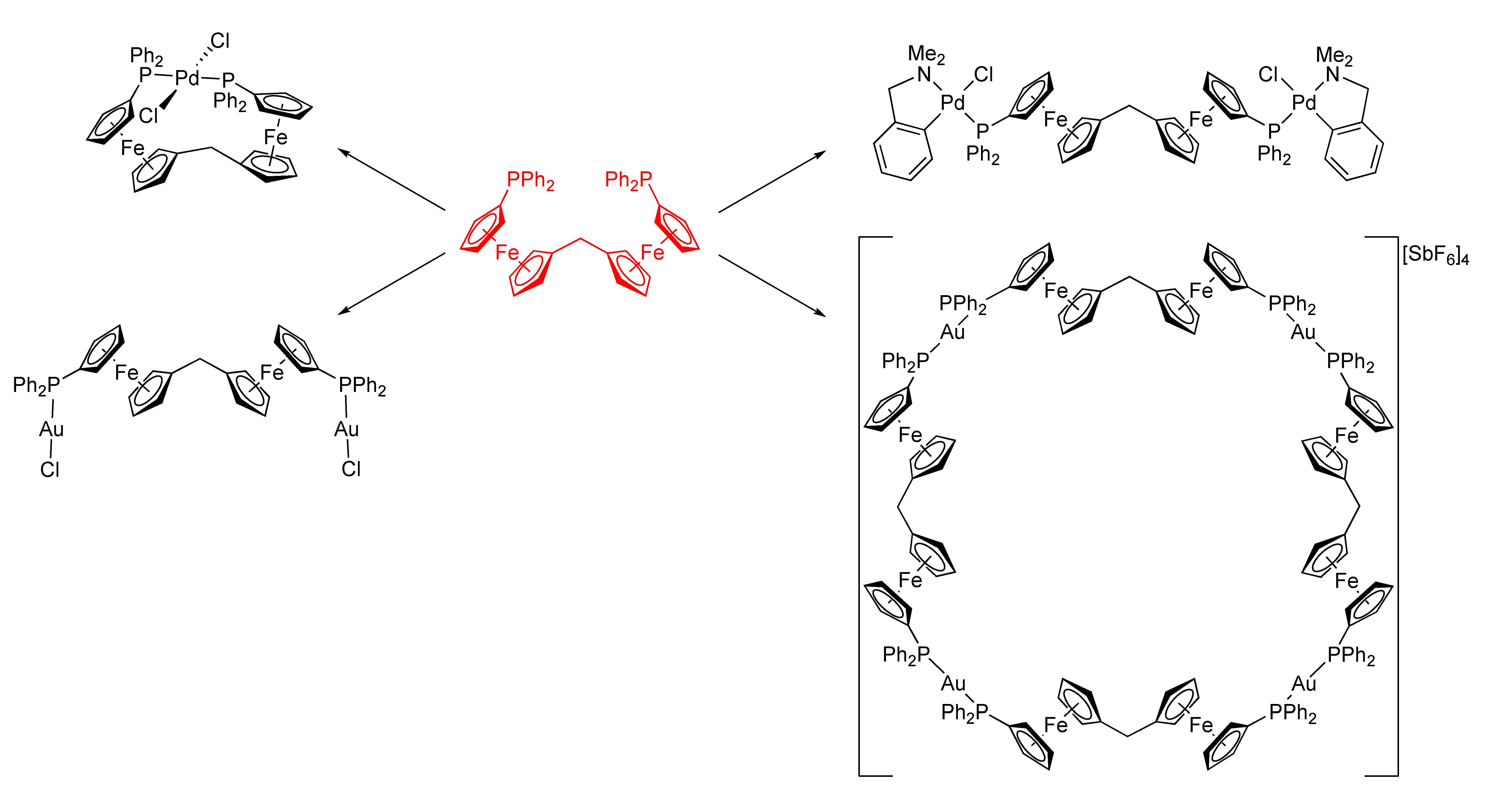
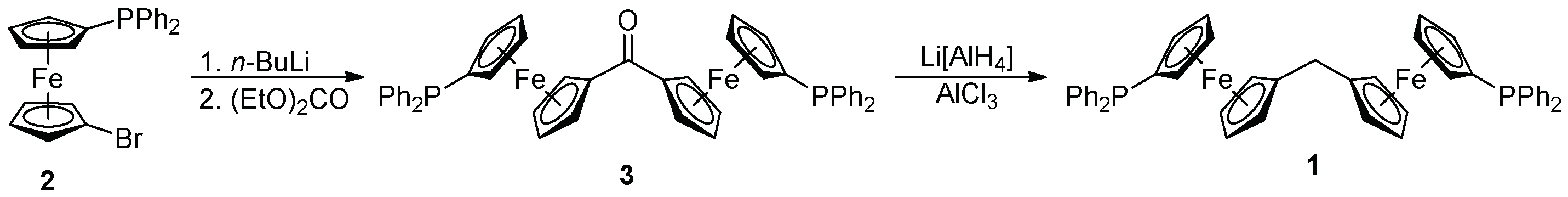
2.1. Synthesis of Ligand 1
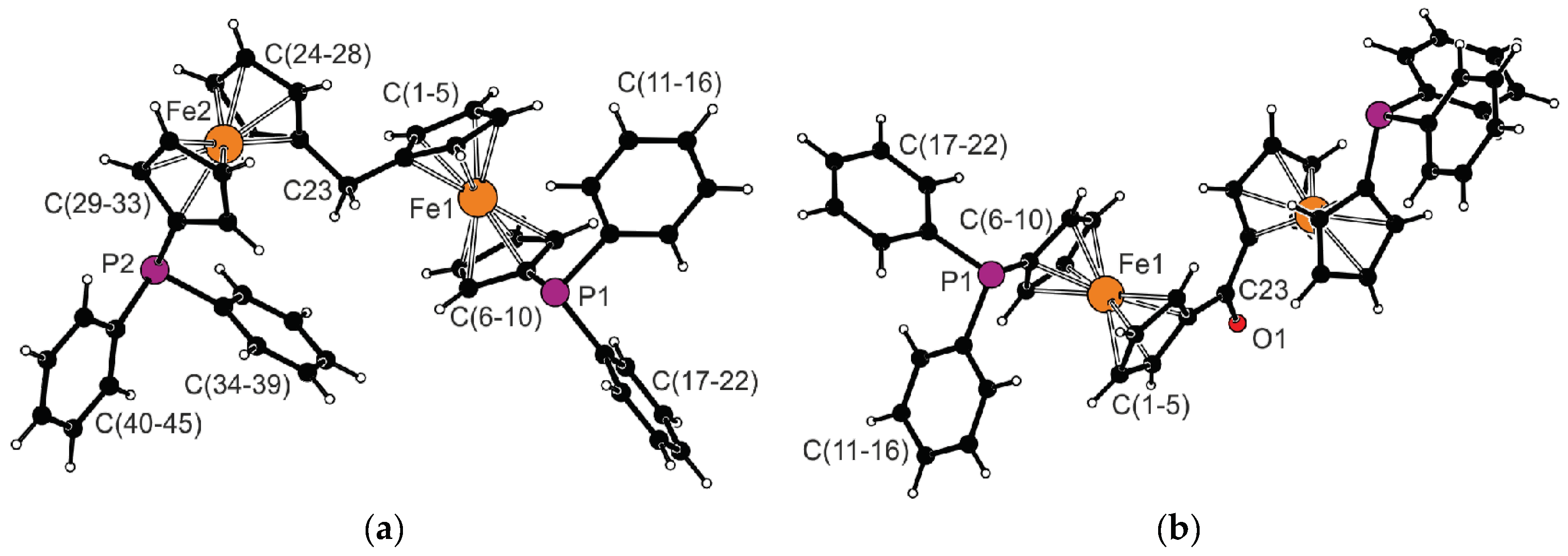
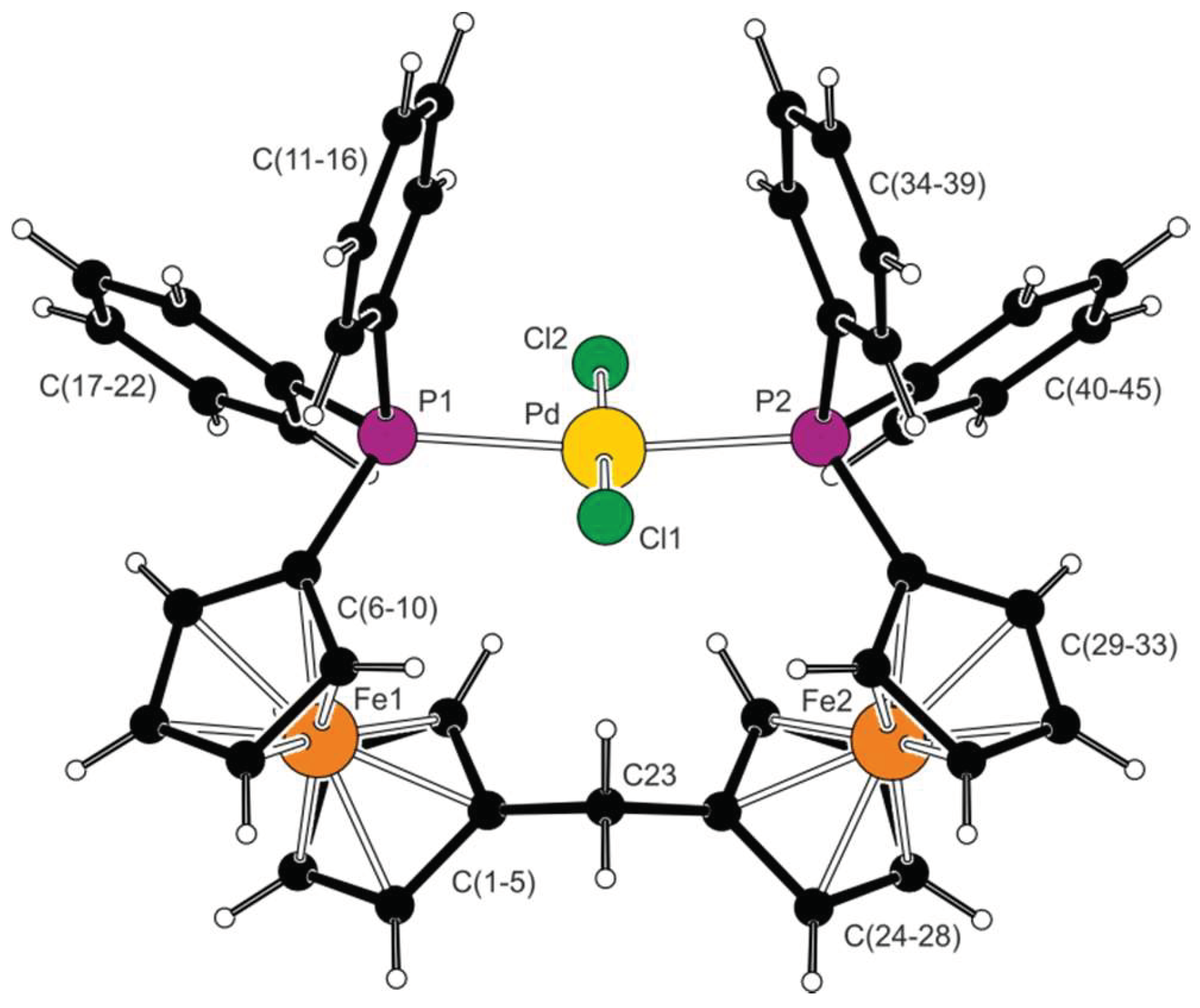
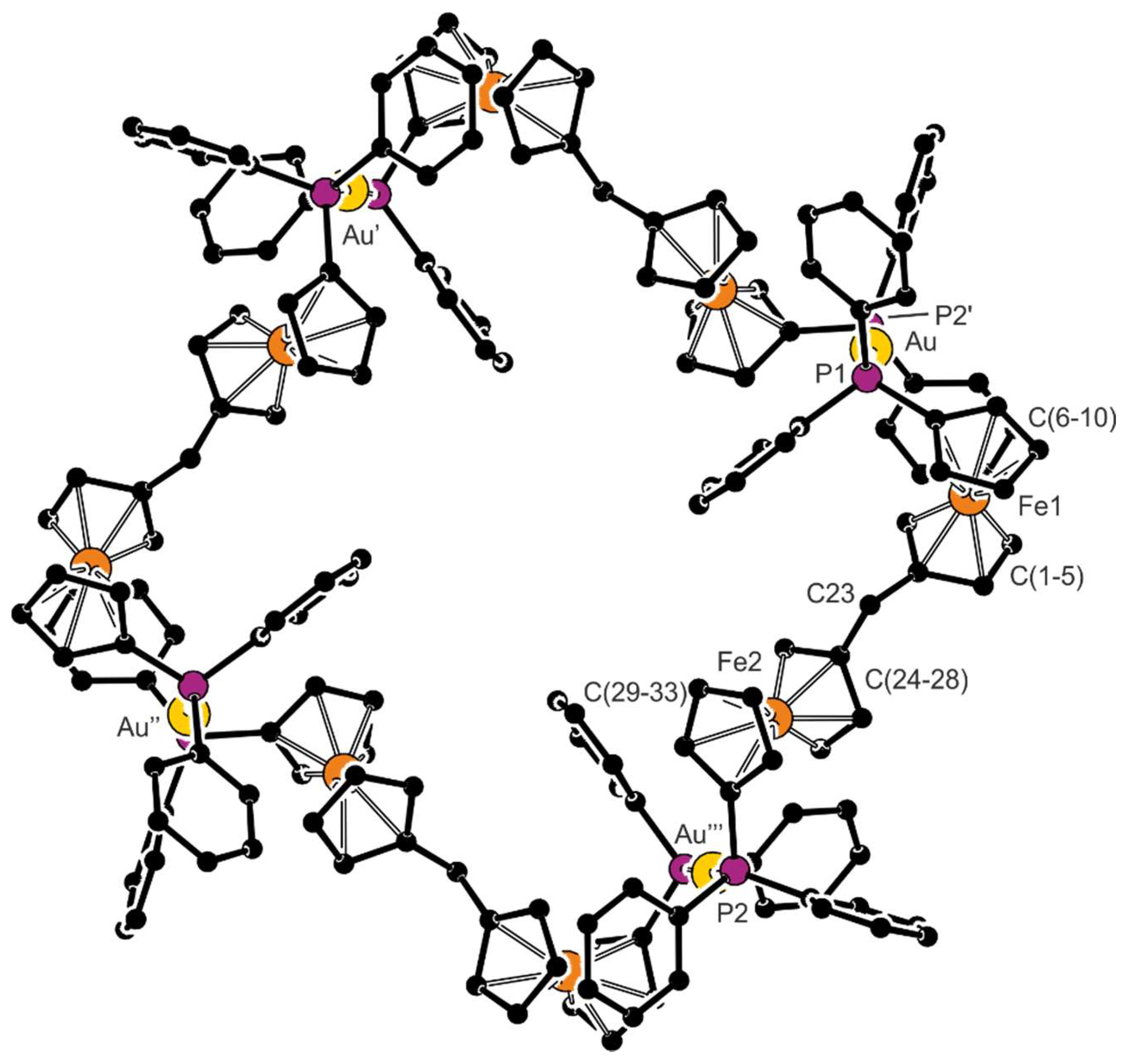
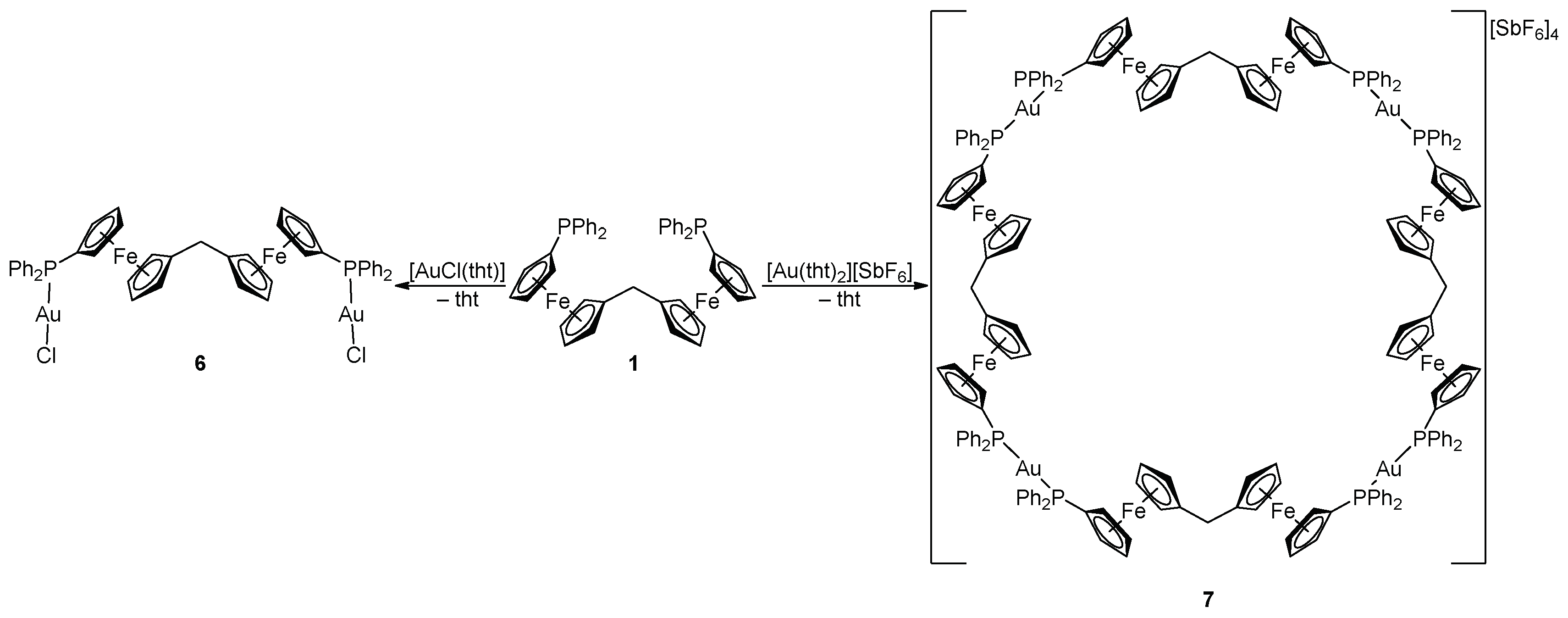
2.2. Preparation and Characterization of Pd(II) and Au(I) Complexes
3. Experimental
3.1. Materials and Methods
3.2. Syntheses
3.2.1. Synthesis of 3
3.2.2. Synthesis of Diphosphine 1
3.2.3. Synthesis of Complex 4
3.2.4. Synthesis of Complex 5
3.2.5. Synthesis of Complex 6
3.2.6. Synthesis of Complex 7
3.3. X-ray Crystallography
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Chemistry; VCH: Weinheim, Germany, 1995; ISBN 978-3-527-61559-9. [Google Scholar]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; Wiley: Chichester, UK, 2008; ISBN 978-0-470-03585-6. [Google Scholar]
- Atkinson, R.C.J.; Gibson, V.C.; Long, N.J. The syntheses and catalytic applications of unsymmetrical ferrocene ligands. Chem. Soc. Rev. 2004, 33, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Gómez Arrayás, R.; Adrio, J.; Carretero, J.C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, M.; Yamauchi, A.; Takegawa, T.; Ito, Y. Synthesis of 2,2′′-bis(diphenylphosphino)-1,1′′-biferrocene, a planar chiral bisphosphine, and its palladium(II) complex. J. Chem. Soc. Chem. Commun. 1991, 874–875. [Google Scholar] [CrossRef]
- Espino, G.; Xiao, L.; Puchberger, M.; Mereiter, K.; Spindler, F.; Manzano, B.R.; Jalón, F.A.; Weissensteiner, W. Synthesis, coordination behaviour, structural features and use in asymmetric hydrogenations of bifep-type biferrocenes. Dalton Trans. 2009, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, M.; Hamashima, H.; Ito, Y. Catalytic asymmetric synthesis with trans-chelating chiral diphosphine ligand TRAP: rhodium-catalyzed asymmetric Michael addition of. alpha-cyano carboxylates. J. Am. Chem. Soc. 1992, 114, 8295–8296. [Google Scholar] [CrossRef]
- Sawamura, M.; Hamashima, H.; Sugawara, M.; Kuwano, R.; Ito, Y. Synthesis and structures of trans-chelating chiral diphosphine ligands bearing aromatic p-substituents, (S,S)-(R,R)- and (R,R)-(S,S)-2,2′′-bis[1-(diarylphosphino)ethyl]-1,1′′-biferrocene (ArylTRAPs) and their transition metal complexes. Organometallics 1995, 14, 4549–4558. [Google Scholar] [CrossRef]
- Trost, B.M.; van Vranken, D.L.; Bingel, C. A modular approach for ligand design for asymmetric allylic alkylations via enantioselective palladium-catalyzed alkylations. J. Am. Chem. Soc. 1992, 114, 9327–9343. [Google Scholar] [CrossRef]
- You, S.-L.; Hou, X.-L.; Dai, L.-X.; Cao, B.-X.; Sun, J. Novel bis-N-[2-(diphenylphosphino)ferrocenyl- carbonyl]diaminocyclohexane ligands: Application in asymmetric allylic alkylation of imino esters with simple allyl carbonate. Chem. Commun. 2000, 1933–1934. [Google Scholar] [CrossRef]
- Longmire, J.M.; Wang, B.; Zhang, X. Highly efficient kinetic resolution of 2-cyclohexenyl acetate in Pd-catalyzed allylic alkylation. Tetrahedron Lett. 2000, 41, 5435–5439. [Google Scholar] [CrossRef]
- Horikoshi, R.; Mochida, T.; Torigoe, R.; Yamamoto, Y. Preparation and electrochemical properties of polynuclear organometallic complexes derived from ferrocene-containing bidentate ligands. Eur. J. Inorg. Chem. 2002, 2002, 3197–3203. [Google Scholar] [CrossRef]
- Lohan, M.; Milde, B.; Heider, S.; Speck, J.M.; Krausse, S.; Schaarschmidt, D.; Rueffer, T.; Lang, H. Synthesis, electrochemistry, spectroelectrochemistry, and solid-state structures of palladium biferrocenylphosphines and their use in C, C cross-coupling reactions. Organometallics 2012, 31, 2310–2326. [Google Scholar] [CrossRef]
- Butler, I.R.; Cullen, W.R. The synthesis of α-N,N-dimethyl-1′-diphenylphosphinoferrocenylethylamine and related ligands. Can. J. Chem. 1983, 61, 147–153. [Google Scholar] [CrossRef]
- Štěpnička, P. 1’-Functionalised ferrocene phosphines: Synthesis, coordination chemistry and catalytic applications. In Ferrocenes: Ligands, Materials and Biomolecules; Štěpnička, P., Ed.; Wiley: Chichester, UK, 2008; Chapter 5; pp. 177–204. ISBN 978-0-470-03585-6. [Google Scholar]
- Barreiro, E.M.; Broggini, D.F.D.; Adrio, L.A.; White, A.J.P.; Schwenk, R.; Togni, A.; Hii, K.K. Gold(I) complexes of conformationally constricted chiral ferrocenyl phosphines. Organometallics 2012, 31, 3745–3754. [Google Scholar] [CrossRef]
- Podlaha, J.; Štěpnička, P.; Gyepes, R.; Mareček, V.; Lhotský, A.; Polášek, M.; Kubišta, J.; Nejezchleba, M. Hydrophobic ferrocene derivatives as potential standards in electrochemistry. Collect. Czech. Chem. Commun. 1997, 62, 185–198. [Google Scholar] [CrossRef]
- Butler, I.R.; Davies, R.L. A rapid convenient synthesis of ferrocene-based triphos analogue ligands. Synthesis 1996, 1350–1354. [Google Scholar] [CrossRef]
- Spectral Database for Organic Compounds SDBS. Benzophenone, SDBS No. 1294. Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/cre_index.cgi (accessed on 23 July 2018).
- Ferguson, G.; Glidewell, C.; Opromolla, G.; Zakaria, C.M.; Zanello, P. The redox behaviour of some bis-ferrocenyl compounds: crystal and molecular structures of diferrocenylmethane and diferrocenylmethanol. J. Organomet. Chem. 1996, 517, 183–190. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Bratych, N.; Hassall, K.; White, J. Redetermination of the structure of diferrocenyl ketone at low temperature. Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, m33–m35. [Google Scholar] [CrossRef]
- Barnard, C.J.; Russell, M.J.H. Palladium. In Comprehesive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Press: Oxford, UK, 1997; Volume 5, Chapter 51; pp. 1099–1170. ISBN 0-08-035948-5. [Google Scholar]
- Puddephatt, R.J. Gold. In Comprehesive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon Press: Oxford, UK, 1997; Volume 5, Chapter 55; pp. 862–923. ISBN 0-08-035948-5. [Google Scholar]
- Zábranský, M.; Císařová, I.; Štěpnička, P. Synthesis, coordination, and catalytic use of 1′-(diphenylphosphino) ferrocene-1-sulfonate anion. Organometallics 2018, 37, 1615–1626. [Google Scholar] [CrossRef]
- Hersh, W.H. False AA’X spin-spin coupling systems in 13C-NMR: Examples involving phosphorus and a 20-year-old mystery in off-resonance decoupling. J. Chem. Educ. 1997, 74, 1485–1488. [Google Scholar] [CrossRef]
- Štěpnička, P.; Schulz, J.; Klemann, T.; Siemeling, U.; Císařová, I. Synthesis, structural characterization, and catalytic evaluation of palladium complexes with homologous ferrocene-based pyridylphosphine ligands. Organometallics 2010, 29, 3187–3200. [Google Scholar] [CrossRef]
- Zábranský, M.; Machara, A.; Císařová, I.; Štěpnička, P. Palladium(II) complexes of homologated ferrocene phosphanylether and thioether ligands. Eur. J. Inorg. Chem. 2017, 2017, 4850–4860. [Google Scholar] [CrossRef]
- Notably, the apparent “molecular” symmetry is not diminished by the solvent molecule, which is located rather symmetrically above the phosphinylated cyclopentadienyl rings.
- Gan, K.-S.; Hor, T.S.A. 1,1′-Bis(diphenylphosphino)ferrocene. Coordination chemistry, organic syntheses, and catalysis. In Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Chemistry; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; Chapter 1; pp. 3–104. ISBN 978-3-527-61559-9. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ 4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Roessler, K.; Rueffer, T.; Walfort, B.; Packheiser, R.; Holze, R.; Zharnikov, M.; Lang, H. Synthesis, characterization and electrochemical behavior of unsymmetric transition metal-terminated biphenyl ethynyl thiols. J. Organomet. Chem. 2007, 692, 1530–1545. [Google Scholar] [CrossRef]
- Aguado, J.E.; Canales, S.; Gimeno, M.C.; Jones, P.G.; Laguna, A.; Villacampa, M.D. Group 11 complexes with unsymmetrical P, S and P, Se disubstituted ferrocene ligands. Dalton Trans. 2005, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Škoch, K.; Císařová, I.; Štěpnička, P. Synthesis and catalytic use of gold(I) complexes containing a hemilabile phosphanylferrocene nitrile donor. Chem. Eur. J. 2015, 21, 15998–16004. [Google Scholar] [CrossRef] [PubMed]
- Dunstan, S.P.C.; Healy, P.C.; Sobolev, A.N.; Tiekink, E.R.T.; White, A.H.; Williams, M.L. Isomorphism in the structural chemistry of two-coordinate adducts of diphenyl(2-formylphenyl)phosphine and triphenylphosphine with gold(I) halides. J. Mol. Struct. 2014, 1072, 253–259. [Google Scholar] [CrossRef]
- Inagaki, F.; Matsumoto, C.; Okada, Y.; Maruyama, N.; Mukai, C. Air-stable cationic gold(I) catalyst featuring a z-type ligand: Promoting enyne cyclizations. Angew. Chem. Int. Ed. 2014, 54, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Appleton, T.G.; Clark, H.C.; Manzer, L.E. The trans-influence: Its measurement and significance. Coord. Chem. Rev. 1973, 10, 335–422. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M. (Tetrahydrothiophene) gold(I) or gold(III) complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Platon squeeze: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors within one year from publication of this article. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1 (Fe1) | Parameter | 1 (Fe2) | Parameter | 3 (Fe1) b |
|---|---|---|---|---|---|
| Fe–C | 2.030(2)–2.057(2) | Fe–C | 2.032 (2)–2.051 (2) | Fe–C | 2.028 (2)–2.055 (2) |
| tilt | 4.1 (1) | tilt | 1.6 (1) | tilt | 2.4 (1) |
| τ | −88.7 (2) | τ | −67.4 (2) | τ | −162.7 (1) |
| P1-C6 | 1.805 (2) | P2-C29 | 1.805 (2) | P1-C6 | 1.817 (2) |
| P1-C11 | 1.838 (2) | P2-C34 | 1.824 (2) | P1-C11 | 1.835 (2) |
| P1-C17 | 1.839 (2) | P2-C40 | 1.838 (2) | P1-C17 | 1.838 (2) |
| C1-C23 | 1.500 (3) | C24-C23 | 1.511 (3) | C1-C23 | 1.472 (2) |
| C1-C23-C24 | 110.8 (2) | – | – | C1-C23-C1′ | 122.4 (2) |
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Pd-P1 | 2.3390 (5) | P1-Pd-Cl1 | 86.14 (2) |
| Pd-P2 | 2.3401 (5) | P1-Pd-Cl2 | 92.95 (2) |
| Pd-Cl1 | 2.3167 (5) | P2-Pd-Cl1 | 86.09 (2) |
| Pd-Cl2 | 2.2906 (5) | P2-Pd-Cl2 | 93.90 (2) |
| Fe1-C | 2.039 (2)–2.061 (2) | Fe2-C | 2.034 (2)–2.058 (2) |
| tilt (Fe1) | 0.71 (9) | tilt (Fe2) | 1.68 (9) |
| τ(Fe1) | 71.2 (1) | τ(Fe2) | −71.2 (1) |
| P1-C6 | 1.805 (2) | P2-C29 | 1.806 (2) |
| P1-C11 | 1.822 (2) | P2-C34 | 1.821 (2) |
| P1-C17 | 1.827 (2) | P2-C40 | 1.824 (2) |
| C1-C23 | 1.509 (2) | C24-C23 | 1.507 (2) |
| Fe1, …, Fe2 | 6.1653 (6) | C1-C23-C24 | 113.0 (1) |
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Au–P | 2.2277 (7) | P–Au–Cl | 176.15 (3) |
| Au–Cl | 2.2881 (7) | Au···Au′ | 12.2322 (7) |
| Fe–C | 2.029 (3)–2.057 (3) | P-C6 | 1.781 (3) |
| tilt (Fe) | 3.5 (2) | P-C11 | 1.819 (3) |
| τ(Fe) | −77.2 (2) | P-C17 | 1.815 (2) |
| Fe···Au | 4.4147 (5) | C1-C23 | 1.502 (3) |
| Fe⋅⋅⋅Fe′ | 6.3509 (8) | C1-C23-C1′ | 109.3 (3) |
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Au-P1 | 2.311 (2) | P1-Au-P2′ | 174.87 (5) |
| Au-P2′ | 2.309 (2) | Au···Au’ b | 11.6898 (9) |
| P1⋅⋅⋅P2 | 10.890 (2) | Au···Au″ c | 16.531 (1) |
| Fe1-C | 2.018 (5)–2.063 (6) | Fe2-C | 2.022 (6)–2.056 (5) |
| tilt (Fe1) | 2.2 (3) | tilt (Fe2) | 3.7 (4) |
| τ(Fe1) | −77.3 (4) | τ(Fe2) | −144.4 (4) |
| P1-C6 | 1.786 (5) | P2-C29 | 1.792 (6) |
| P1-C11 | 1.822 (5) | P2-C34 | 1.813 (5) |
| P1-C17 | 1.817 (5) | P2-C40 | 1.805 (5) |
| C1-C23 | 1.491 (7) | C23-C24 | 1.503 (8) |
| Compound | 1 | 3 | 4·CHCl3 | 6 | 7 |
|---|---|---|---|---|---|
| Formula | C45H38Fe2P2 | C45H36Fe2OP2 | C46H39Cl5Fe2P2Pd | C45H38Au2Cl2Fe2P2 | C180H152Au4F24Fe8P8Sb4 |
| M | 752.39 | 766.38 | 1049.06 | 1217.22 | 4740.43 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic | tetragonal |
| Space group | P21/c (No. 14) | C2/c (No. 15) | P–1 (No. 2) | C2/c (No. 15) | I4/m (No. 87) |
| T/K | 150 (2) | 150 (2) | 150 (2) | 150 (2) | 120(2) |
| a/Å | 15.6501 (4) | 18.7605 (5) | 8.9665 (4) | 16.8308 (5) | 27.6537 (10) |
| b/Å | 13.2675 (3) | 6.5050 (1) | 13.2550 (6) | 8.9762 (3) | 27.6537 (10) |
| c/Å | 17.2689 (4) | 29.1239 (7) | 18.5420 (8) | 26.5919 (9) | 28.6005 (13) |
| α/deg | 90 | 90 | 82.774 (2) | 90 | 90 |
| β/deg | 91.126 (1) | 97.5130 (10) | 81.222 (1) | 94.7570 (10) | 90 |
| γ/deg | 90 | 90 | 77.846 (1) | 90 | 90 |
| V/Å3 | 3585.0 (2) | 3523.7 (1) | 2119.1 (2) | 4003.6 (2) | 21,872 (2) |
| Z | 4 | 4 | 2 | 4 | 4 |
| F(000) | 1560 | 1584 | 1056 | 2328 | 9184 |
| μ (Mo Kα)/mm−1 | 0.931 | 0.950 | 1.520 | 8.262 | 3.787 |
| Diffrns collected | 28,736 | 25,843 | 80,051 | 31,375 | 101,295 |
| Independent diffrns | 8239 | 4054 | 9744 | 4596 | 10,972 |
| Observed diffrns a | 6050 | 3492 | 8989 | 4430 | 8548 |
| Rintb/% | 3.23 | 3.30 | 2.53 | 2.15 | 4.47 |
| No. of param. | 442 | 227 | 505 | 241 | 524 |
| Rc obsd diffrns/% | 3.62 | 3.27 | 2.12 | 1.67 | 3.61 |
| R, wRc all data/% | 5.82, 9.43 | 3.99, 8.72 | 2.43, 5.15 | 1.79, 3.59 | 5.51, 10.1 |
| Δρ/e Å−3 | 0.48, −0.35 | 0.95, −0.25 | 0.65, −0.79 | 1.16, −0.56 | 2.29, −3.45 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schulz, J.; Leitner, Z.; Císařová, I.; Štěpnička, P. Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand. Molecules 2018, 23, 2054. https://doi.org/10.3390/molecules23082054
Schulz J, Leitner Z, Císařová I, Štěpnička P. Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand. Molecules. 2018; 23(8):2054. https://doi.org/10.3390/molecules23082054
Chicago/Turabian StyleSchulz, Jiří, Zdeněk Leitner, Ivana Císařová, and Petr Štěpnička. 2018. "Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand" Molecules 23, no. 8: 2054. https://doi.org/10.3390/molecules23082054
APA StyleSchulz, J., Leitner, Z., Císařová, I., & Štěpnička, P. (2018). Synthesis and Coordination Behavior of a Flexible Bis(phosphinoferrocene) Ligand. Molecules, 23(8), 2054. https://doi.org/10.3390/molecules23082054