16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
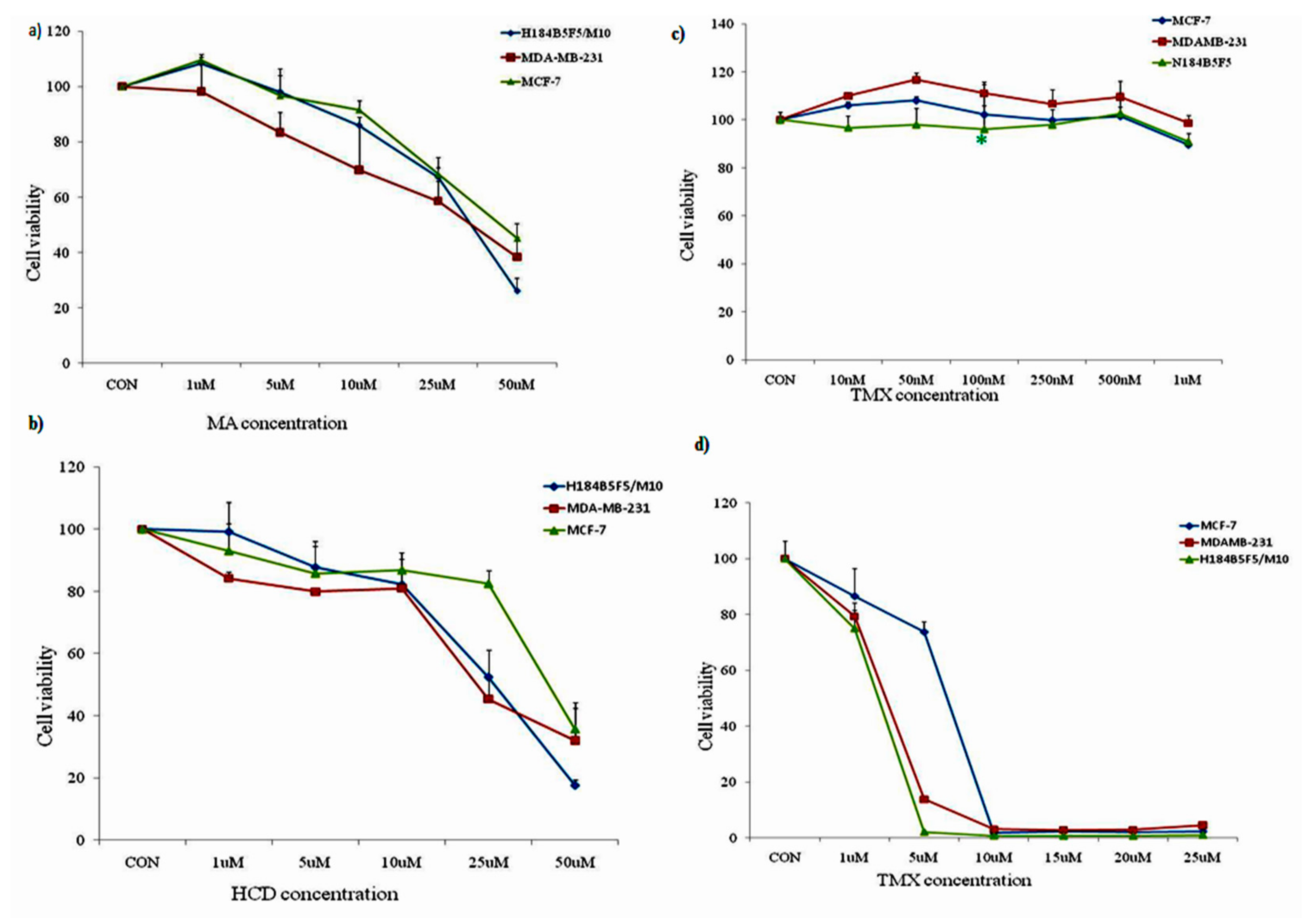
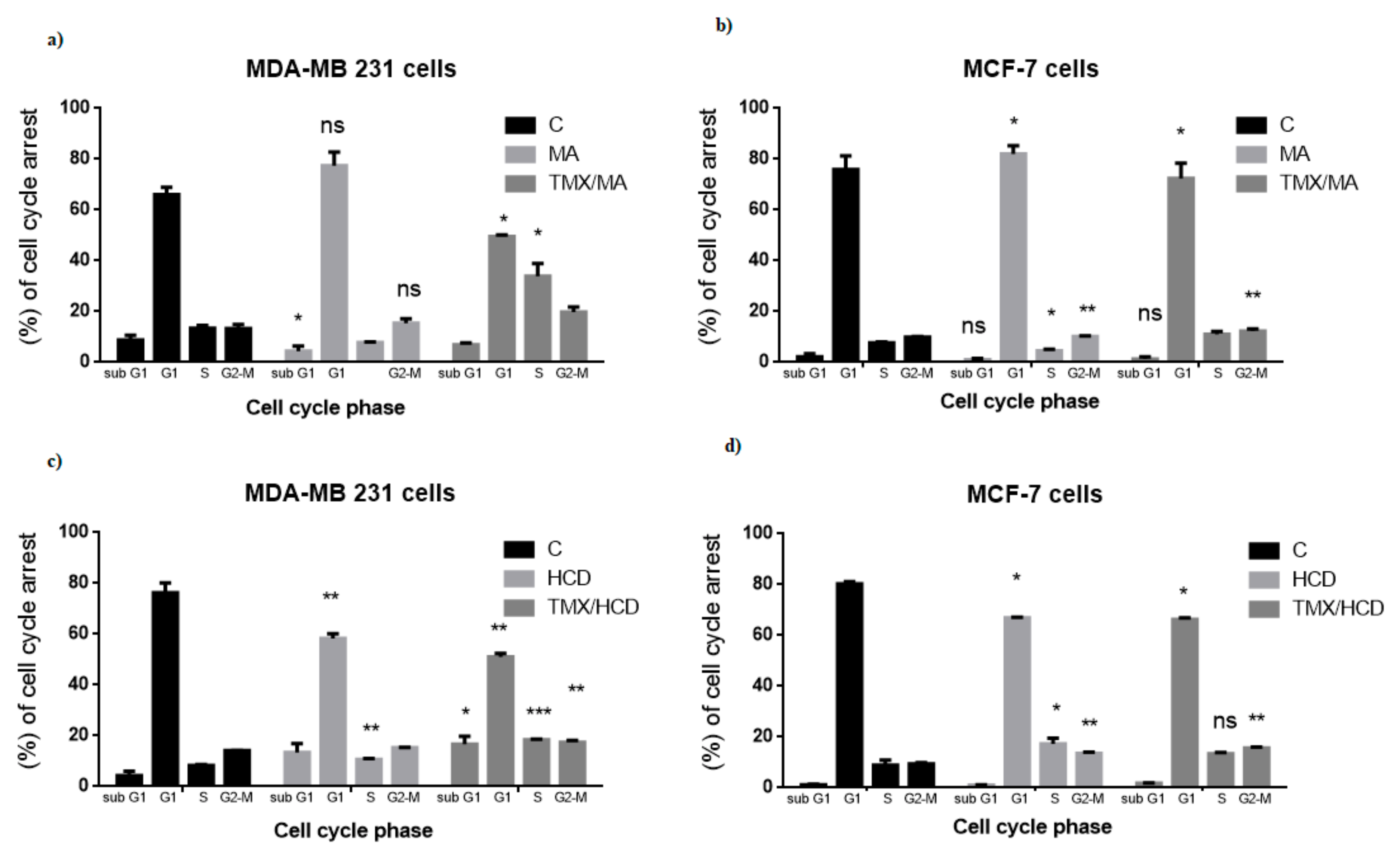
2. Results and Discussion
2.1. HCD, MA, and Tamoxifen Effects on Breast Cancer and Human Mammary Epithelial Cell Viability
2.2. MA or HCD Pretreatment Could Enhance the Growth Inhibition of TMX in MDA-MB 231 Breast Cancer Cells
2.3. Effect of MA or HCD on Tamoxifen Treated Breast Cancer Cell Cycle Distribution
2.4. Effect of MA/HCD and or TMX on Apoptotic Markers
3. Materials and Methods
3.1. Cell Culture
3.2. Cell Viability Assays
3.3. Analysis of Cell Cycle Distribution
3.4. RT-PCR
3.5. Western Blot Analysis
3.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.J.; Dutkowski, C.M.; Barrow, D.; Mottram, H.J.; Hutcheson, I.R.; Nicholson, R.I.; Guichard, S.M.; Gee, J.M. Impact of dual mtorc1/2 mtor kinase inhibitor azd8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014, 16, R12. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.L.; Lewis-Wambi, J.S. Mechanisms of endocrine resistance in breast cancer: An overview of the proposed roles of noncoding RNA. Breast Cancer Res. 2015, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rasool, M.; Chaoudhry, H.; Pushparaj, N.P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, E.; Armengol-Alonso, A.; Munoz, M.; Segui-Palmer, M.A. Current status of hormone therapy in patients with hormone receptor positive (hr+) advanced breast cancer. Breast 2014, 23, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Darakhshan, S.; Ghanbari, A.; Gholami Rad, F.; Bidmeshki Pour, A. Tamoxifen and tranilast show a synergistic effect against breast cancer in vitro. Bratisl. Lek. Listy 2015, 116, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Kumar, B.N.; Sarkar, S.; Das, S.; Azab, B.; Santhekadur, P.K.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B.; et al. Targeted apoptotic effects of thymoquinone and tamoxifen on xiap mediated akt regulation in breast cancer. PLoS ONE 2013, 8, e61342. [Google Scholar] [CrossRef] [PubMed]
- Garza-Morales, R.; Gonzalez-Ramos, R.; Chiba, A.; Montes de Oca-Luna, R.; McNally, L.R.; McMasters, K.M.; Gomez-Gutierrez, J.G. Temozolomide enhances triple-negative breast cancer virotherapy in vitro. Cancers 2018, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chang, F.R.; Shih, Y.C.; Hsieh, T.J.; Chia, Y.C.; Tseng, H.Y.; Chen, H.C.; Chen, S.J.; Hsu, M.C.; Wu, Y.C. Cytotoxic constituents of Polyalthia longifolia var. Pendula. J. Nat. Prod. 2000, 63, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.R.; Hwang, T.L.; Yang, Y.L.; Li, C.E.; Wu, C.C.; Issa, H.H.; Hsieh, W.B.; Wu, Y.C. Anti-inflammatory and cytotoxic diterpenes from formosan Polyalthia longifolia var. Pendula. Planta Med. 2006, 72, 1344–1347. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Lee, C.C.; Chan, W.L.; Chang, W.H.; Wu, Y.C.; Chang, J.G. 16-hydroxycleroda-3,13-dien-15,16-olide deregulates pi3k and aurora b activities that involve in cancer cell apoptosis. Toxicology 2011, 285, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Lee, C.C.; Chang, F.R.; Chang, W.H.; Wu, Y.C.; Chang, J.G. 16-hydroxycleroda-3,13-dien-15,16-olide regulates the expression of histone-modifying enzymes prc2 complex and induces apoptosis in cml k562 cells. Life Sci. 2011, 89, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, V.; Lin, S.X.; Lee, C.H.; Weng, C.F. A focal adhesion kinase inhibitor 16-hydroxy-cleroda-3,13-dien-16,15-olide incorporated into enteric-coated nanoparticles for controlled anti-glioma drug delivery. Colloids Surf. B Biointerfaces 2016, 141, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Thiyagarajan, V.; Lin, S.H.; Chia, Y.C.; Weng, C.F. A novel inhibitor, 16-hydroxy-cleroda-3,13-dien-16,15-olide, blocks the autophosphorylation site of focal adhesion kinase (y397) by molecular docking. Biochim. Biophys. Acta 2013, 1830, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.J.; Liu, T.Z.; Lu, F.J.; Hsieh, P.Y.; Chen, C.H. Actinodaphnine induces apoptosis through increased nitric oxide, reactive oxygen species and down-regulation of nf-kappab signaling in human hepatoma mahlavu cells. Food Chem. Toxicol. 2006, 44, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Singh, S.P.; Shukla, P.K. Antimicrobial evaluation of clerodane diterpenes from Polyalthia longifolia var. Pendula. Nat. Prod. Commun. 2009, 4, 327–330. [Google Scholar] [PubMed]
- Azumi, S.; Tanimura, A.; Tanamoto, K. A novel inhibitor of bacterial endotoxin derived from cinnamon bark. Biochem. Biophys. Res. Commun. 1997, 234, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hengli, Z.; Qiheng, G.; Jiahui, Z.; Li, P. Different cell activities in breast cancer based on different estrogen receptor-alpha(+)/estrogen receptor-alpha(−) ratios. J. Clin. Oncol. 2016, 34, e12004. [Google Scholar]
- Chang, J. Scientific evaluation of traditional Chinese medicine under dshea: A conundrum. Dietary supplement health and education act. J. Altern. Complement. Med. 1999, 5, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Hara, I.; Gohji, K.; Arakawa, S.; Kamidono, S. P53 modulation of fas/apo-1 mediated apoptosis in a human renal cell carcinoma cell line. Int. J. Oncol. 1998, 12, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Xavier, J.M.; Steer, C.J.; Rodrigues, C.M. The role of p53 in apoptosis. Discov. Med. 2010, 9, 145–152. [Google Scholar] [PubMed]
- Darakhshan, S.; Ghanbari, A. Tranilast enhances the anti-tumor effects of tamoxifen on human breast cancer cells in vitro. J. Biomed. Sci. 2013, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Storr, S.J.; Zhang, Y.; Rakha, E.A.; Green, A.R.; Ellis, I.O.; Martin, S.G. Caspase-3 and caspase-8 expression in breast cancer: Caspase-3 is associated with survival. Apoptosis 2017, 22, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, Y.; Chen, Y. Predicting drug combination index and simulating the network-regulation dynamics by mathematical modeling of drug-targeted egfr-erk signaling pathway. Sci. Rep. 2017, 7, 40752. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Harvey, H.A.; Hamilton, R.W. Venous thrombosis as a side effect of tamoxifen treatment. Cancer Treat. Rep. 1984, 68, 887–889. [Google Scholar] [PubMed]
- Sakhri, J.; Ben Salem, C.; Harbi, H.; Fathallah, N.; Ltaief, R. Severe acute pancreatitis due to tamoxifen-induced hypertriglyceridemia with positive rechallenge. JOP 2010, 11, 382–384. [Google Scholar] [PubMed]
- Latourelle, J.C.; Dybdahl, M.; Destefano, A.L.; Myers, R.H.; Lash, T.L. Risk of Parkinson’s disease after tamoxifen treatment. BMC Neurol. 2010, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Biswas, J.; Majumdar, S.; Nayak, S.; Alam, N.; Mukherjee, K.K.; Gupta, S. Tamoxifen use in indian women—Adverse effects revisited. Asian Pac. J. Cancer Prev. 2009, 10, 609–612. [Google Scholar] [PubMed]
- Pasquini, M.; Speca, A.; Biondi, M. Quetiapine for tamoxifen-induced insomnia in women with breast cancer. Psychosomatics 2009, 50, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Holz, M.K. Tamoxifen action in er-negative breast cancer. Signal Transduct. Insights 2016, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafiz, H.A. Epigenetic mechanisms of tamoxifen resistance in luminal breast cancer. Diseases 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velmurugan, B.K.; Wang, P.-C.; Weng, C.-F. 16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer. Molecules 2018, 23, 1966. https://doi.org/10.3390/molecules23081966
Velmurugan BK, Wang P-C, Weng C-F. 16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer. Molecules. 2018; 23(8):1966. https://doi.org/10.3390/molecules23081966
Chicago/Turabian StyleVelmurugan, Bharath Kumar, Po-Chih Wang, and Ching-Feng Weng. 2018. "16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer" Molecules 23, no. 8: 1966. https://doi.org/10.3390/molecules23081966
APA StyleVelmurugan, B. K., Wang, P.-C., & Weng, C.-F. (2018). 16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer. Molecules, 23(8), 1966. https://doi.org/10.3390/molecules23081966