Whole-Genome Comparison Reveals Divergent IR Borders and Mutation Hotspots in Chloroplast Genomes of Herbaceous Bamboos (Bambusoideae: Olyreae)

Abstract
1. Introduction
2. Results
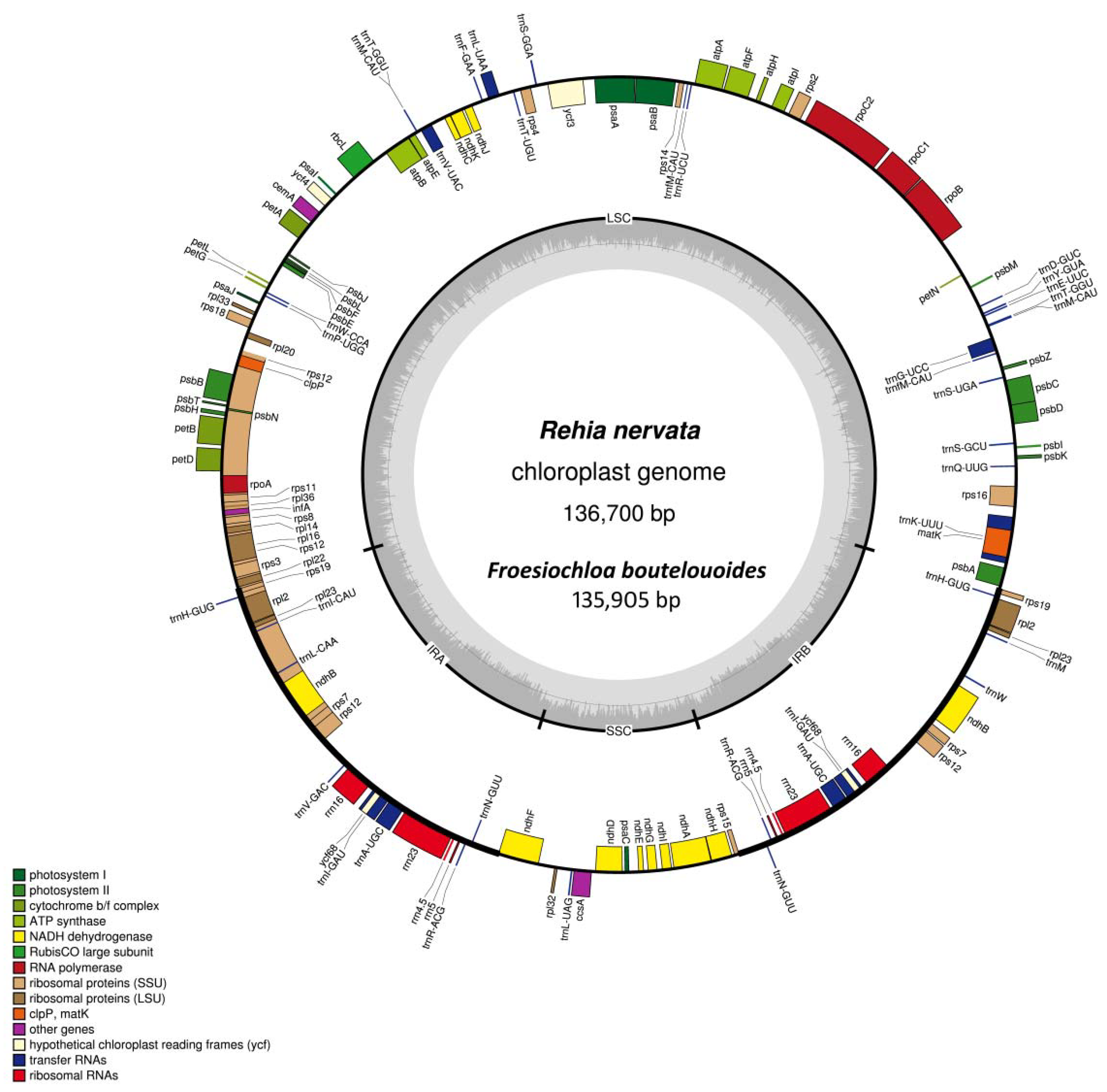
2.1. Genome Assembly and Features
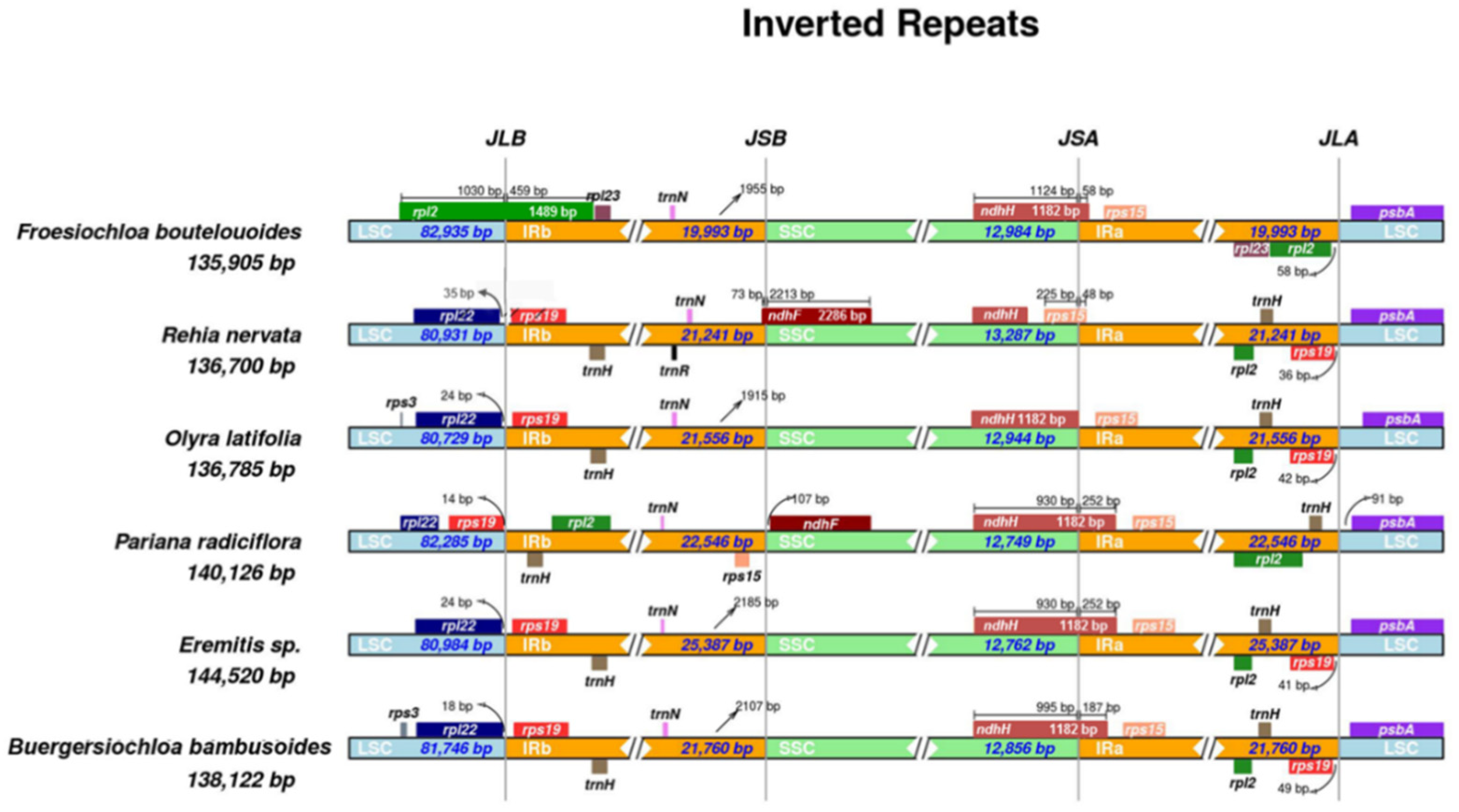
2.2. Whole Chloroplast Genome Comparison among Olyreae
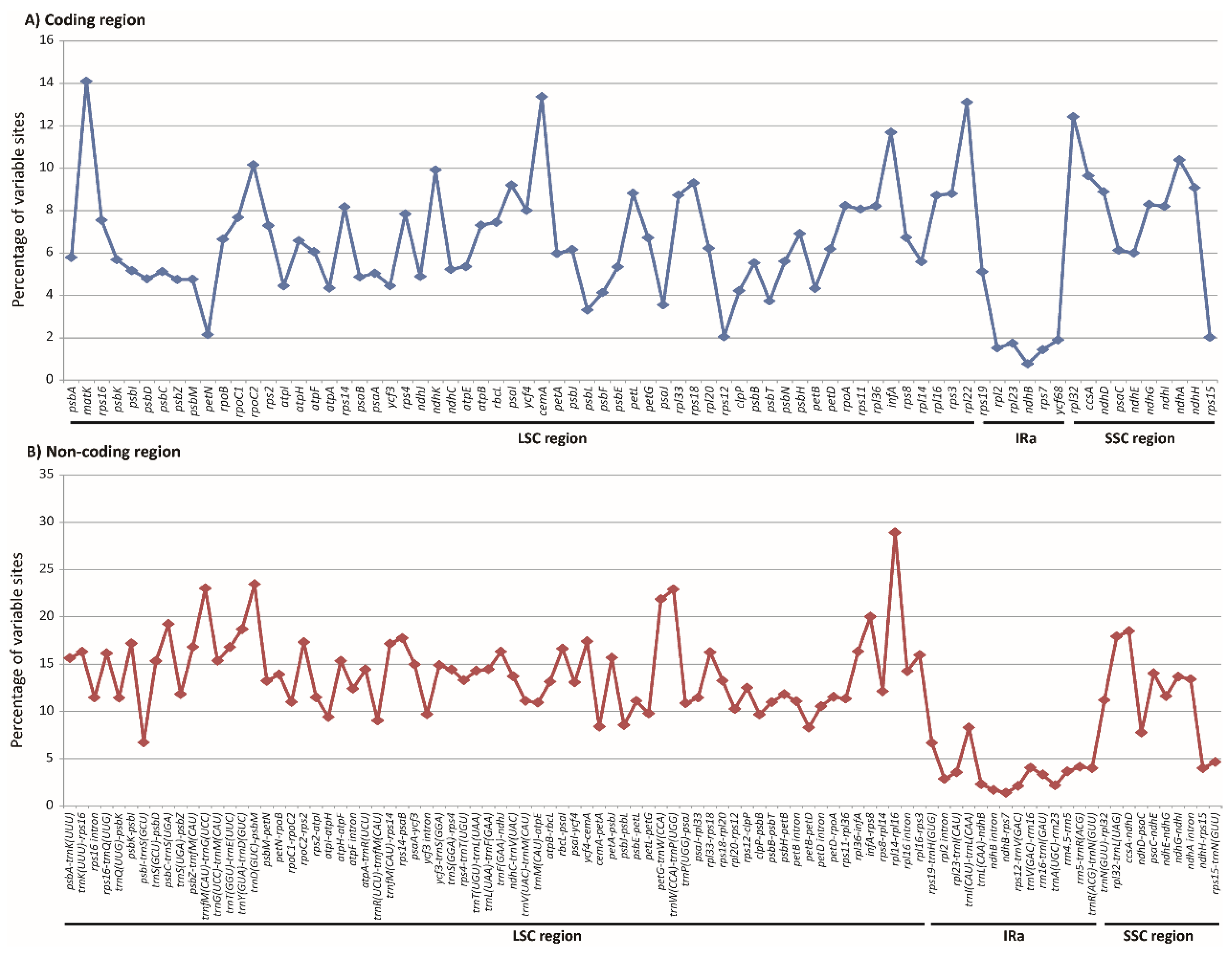
2.3. Molecular Marker Development
2.4. Phylogenomic Inference
3. Discussion
3.1. Dynamic Chloroplast Genome IR Borders in Olyreae
3.2. Heterogeneous Sequence Divergence in Olyreae Chloroplast Genome
3.3. Chloroplast Phylogenomic Estimation of Bamboos
4. Materials and Methods
4.1. Plant Materials and DNA Sequencing
4.2. Genome Assembly, Annotation and Repeat Analysis
4.3. Whole-Genome Comparison and Mutation Hotspot Identification
4.4. Phylogenomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clark, L.G.; Londoño, X.; Ruiz-Sanchez, E. Bamboo taxonomy and habitat. In Bamboo; Liese, W., Köhl, M., Eds.; Springer: Cham, Switzerland, 2015; Chapter 1. [Google Scholar]
- Zhang, X.Z.; Zeng, C.X.; Ma, P.F.; Haevermans, T.; Zhang, Y.X.; Zhang, L.N.; Guo, Z.H.; Li, D.Z. Multi-locus plastid phylogenetic biogeography supports the asian hypothesis of the temperate woody bamboos (Poaceae: Bambusoideae). Mol. Phylogenet. Evolut. 2015, 96, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Bamboo Phylogeny Group (BPG). An updated tribal and subtribal classification of the bamboos (poaceae: Bambusoideae). Bamboo Sci. Cult. 2012, 24, 1–10. [Google Scholar]
- Kelchner, S.A.; Bamboo Phylogeny Group (BPG). Higher level phylogenetic relationships within the bamboos (Poaceae: Bambusoideae) based on five plastid markers. Mol. Phylogenet. Evolut. 2013, 67, 404. [Google Scholar] [CrossRef] [PubMed]
- Judziewicz, E.J.; Clark, L.G.; Londoño, X.; Stern, M.J. American Bamboos; Smithsonian Institution Press: Washington, DC, USA, 1999; pp. 255–319. [Google Scholar]
- Triplett, J.K.; Clark, L.G.; Fisher, A.E.; Wen, J. Independent allopolyploidization events preceded speciation in the temperate and tropical woody bamboos. New Phytol. 2014, 204, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Zhao, L.; Larson-Rabin, Z.; Li, D.Z.; Guo, Z.H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Lan, S.; Cai, B.; Chen, S.; Chen, H.; Zhou, S. The complete chloroplast genome of Guadua angustifolia and comparative analyses of neotropical-paleotropical bamboos. PLoS ONE 2015, 10, e0143792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.; Chen, S.Y. Genome skimming reveals the complete chloroplast genome of Ampelocalamus naibunensis (Poaceae: Bambusoideae: Arundinarieae) with phylogenomic implication. Mitochondrial DNA Part B Resour. 2016, 1, 635–637. [Google Scholar] [CrossRef]
- Oliveira, R.P.; Clark, L.G.; Schnadelbach, A.S.; Monteiro, S.H.; Borba, E.L.; Longhi-Wagner, H.M.; van den Berg, C. A molecular phylogeny of Raddia and its allies within the tribe Olyreae (Poaceae, Bambusoideae) based on noncoding plastid and nuclear spacers. Mol. Phylogenet. Evolut. 2014, 78, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, W.P.; Clark, L.G.; Attigala, L.; Ruiz-Sanchez, E.; Duvall, M.R. Evolution of the bamboos (Bambusoideae; Poaceae): A full plastome phylogenomic analysis. BMC Evolut. Biol. 2015, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Schelkunov, M.I.; Shtratnikova, V.Y.; Nuraliev, M.S.; Selosse, M.A.; Penin, A.A.; Logacheva, M.D. Exploring the limits for reduction of plastid genomes: A case study of the mycoheterotrophic orchids Epipogium aphyllum and Epipogium roseum. Genome Biol. Evolut. 2015, 7, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium× hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evolut. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.M.; Wang, J.; Feng, L.; Liu, S.; Pang, H.; Qi, L.; Li, J.; Sun, Y.; Qiao, W.; Zhang, L. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Chloroplast genome evolution in Actinidiaceae: ClpP loss, heterogenous divergence and phylogenomic practice. PLoS ONE 2016, 11, e0162324. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Terachi, T.; Sasakuma, T. Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. USA 1988, 85, 8573–8577. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the Apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Syst. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; Depamphilis, C.W. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.K.; Cronn, R.C.; Edwards, C.; Fishbein, M.; Liston, A. Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evolut. 2013, 5, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Cosner, M.E.; Raubeson, L.A.; Jansen, R.K. Chloroplast DNA rearrangements in Campanulaceae: Phylogenetic utility of highly rearranged genomes. BMC Evolut. Biol. 2004, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Ma, P.-F.; Li, D.-Z. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.D.N.; Anjos, K.G.D.; Faoro, H.; Fraga, H.P.D.F.; Greco, T.M.; Pedrosa, F.D.O.; Souza, E.M.D.; Rogalski, M.; Souza, R.F.D.; Guerra, M.P. Phylogenetic inference and SSR characterization of tropical woody bamboos tribe Bambuseae (Poaceae: Bambusoideae) based on complete plastid genome sequences. Curr. Genet. 2015, 62, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two new world bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—Extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Guo, Z.-H.; Li, D.-Z. Evidence for horizontal transfer of mitochondrial DNA to the plastid genome in a bamboo genus. Sci. Rep. 2015, 5, 11608. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W.J. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1996, 92, 11331–11338. [Google Scholar] [CrossRef]
- Ahmed, I.; Matthews, P.J.; Biggs, P.J.; Naeem, M.; Mclenachan, P.A.; Lockhart, P.J. Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta (L.) Schott (Araceae) and closely related taxa. Mol. Ecol. Resour. 2013, 13, 929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Ao, L.; Chen, J.; Li, W.; Hu, W.; Wei, Z.; Kim, K.; Lee, S.C.; Yang, T.J. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, H.-L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.P.; Borba, E.L.; Longhi-Wagner, H.M. Morphometrics of herbaceous bamboos of the Raddia brasiliensis complex (Poaceae-Bambusoideae): Implications for the taxonomy of the genus and new species from Brazil. Plant Syst. Evolut. 2008, 270, 159–182. [Google Scholar] [CrossRef]
- Oliveira, R.P.; Borba, E.L.; Longhiwagner, H.M.; Acs, P.; Lambert, S.M. Genetic and morphological variability in the Raddia brasiliensis complex (Poaceae: Bambusoideae). Plant Syst. Evolut. 2008, 274, 25–35. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M. Next-generation organellar genomics: Potentials and pitfalls of high-throughput technologies for molecular evolutionary studies and plant systematics. In Next-Generation Sequencing in Plant Systematics; Hörandl, E., Appelhans, M.S., Eds.; International Association for Plant Taxonomy (IAPT): Bratislava, Slovakia, 2015; Chapter 1. [Google Scholar]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Cusimano, N.; Wicke, S. Massive intracellular gene transfer during plastid genome reduction in nongreen Orobanchaceae. New Phytol. 2016, 210, 680. [Google Scholar] [CrossRef] [PubMed]
- Logacheva, M.D.; Schelkunov, M.I.; Nuraliev, M.S.; Samigullin, T.H.; Penin, A.A. The plastid genome of mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol. Evolut. 2014, 6, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Duvall, M.R.; Fisher, A.E.; Columbus, J.T.; Ingram, A.L.; Wysocki, W.P.; Burke, S.V.; Clark, L.G.; Kelchner, S.A. Phylogenomics and plastome evolution of the chloridoid grasses (Chloridoideae: Poaceae). Int. J. Plant Sci. 2016, 177, 235–246. [Google Scholar] [CrossRef]
- Grass Phylogeny Working Group II (GPWG II). New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012, 193, 304. [Google Scholar]
- Yang, J.-B.; Yang, S.-X.; Li, H.-T.; Yang, J.; Li, D.-Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Fujimoto, M.; Arimura, S.-I.; Murata, J.; Tsutsumi, N.; Kadowaki, K.-I. Loss of the rpl32 gene from the chloroplast genome and subsequent acquisition of a preexisting transit peptide within the nuclear gene in Populus. Gene 2007, 402, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many parallel losses of infa from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Whole-genome comparison reveals heterogeneous divergence and mutation hotspots in chloroplast genome of Eucommia ulmoides Oliver. Int. J. Mol. Sci. 2018, 19, 1037. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.F.; Baker, W.J.; Comer, J.R.; Conran, J.G.; Lahmeyer, S.C.; Leebens-Mack, J.H.; Li, J.; Lim, G.S.; Mayfield-Jones, D.R.; Perez, L. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 2016, 209, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Givnish, T.J.; Zuluaga, A.; Marques, I.; Lam, V.K.; Gomez, M.S.; Iles, W.J.; Ames, M.; Spalink, D.; Moeller, J.R.; Briggs, B.G. Phylogenomics and historical biogeography of the monocot order Liliales: Out of Australia and through Antarctica. Cladistics 2016, 32, 581–605. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Triant, D.A.; Forrester, N.J.; Bergner, L.M.; Wu, M.; Taylor, D.R. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenet. Evolut. 2014, 72, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare iii. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Wu, M.; Palmer, J.D.; Taylor, D.R. Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus Silene. Genome Biol. Evolut. 2012, 4, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Guisinger, M.M.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc. Natl. Acad. Sci. USA 2008, 105, 18424–18429. [Google Scholar] [CrossRef] [PubMed]
- Bouchenak-Khelladi, Y.; Salamin, N.; Savolainen, V.; Forest, F.; Mv, B.; Chase, M.W.; Hodkinson, T.R. Large multi-gene phylogenetic trees of the grasses (Poaceae): Progress towards complete tribal and generic level sampling. Mol. Phylogenet. Evolut. 2008, 47, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Sungkaew, S.; Stapleton, C.M.; Salamin, N.; Hodkinson, T.R. Non-monophyly of the woody bamboos (Bambuseae; Poaceae): A multi-gene region phylogenetic analysis of Bambusoideae s.s. J. Plant Res. 2009, 122, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Triplett, J.K.; Clark, L.G. Phylogeny of the temperate bamboos (Poaceae: Bambusoideae: Bambuseae) with an emphasis on Arundinaria and allies. Syst. Bot. 2010, 35, 102–120. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare ii: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.X.; Zhang, Y.X.; Triplett, J.K.; Yang, J.B.; Li, D.Z. Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol. Phylogenet. Evolut. 2010, 56, 821–839. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.H.; Kan, D.P.; Lee, S.B.; Daniell, H.; Lee, Y.W.; Lin, C.C.; Lin, N.S.; Lin, C.S. Complete nucleotide sequence of Dendrocalamus latiflorus and Bambusa oldhamii chloroplast genomes. Tree Physiol. 2009, 29, 847. [Google Scholar] [CrossRef] [PubMed]
- Attigala, L.; Wysocki, W.P.; Duvall, M.R.; Clark, L.G. Phylogenetic estimation and morphological evolution of Arundinarieae (Bambusoideae: Poaceae) based on plastome phylogenomic analysis. Mol. Phylogenet. Evolut. 2016, 101, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.M.; Burke, S.V.; Wysocki, W.P.; Barrett, M.D.; Clark, L.G.; Craine, J.M.; Peterson, P.M.; Soreng, R.J.; Vorontsova, M.S.; Duvall, M.R. A 250 plastome phylogeny of the grass family (Poaceae): Topological support under different data partitions. PeerJ 2018, 6, e4299. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.S.; Clark, L.G.; Wendel, J.F.; Muse, S.V. Comparisons of the molecular evolutionary process at rbcL and ndhF in the grass family (Poaceae). Mol. Biol. Evol. 1997, 14, 769. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.V.; Clark, L.G.; Triplett, J.K.; Grennan, C.P.; Duvall, M.R. Biogeography and phylogenomics of New World Bambusoideae (Poaceae), revisited. Am. J. Bot. 2014, 101, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Zhang, X.Z.; Zhang, Y.X.; Zeng, C.X.; Ma, P.F.; Zhao, L.; Guo, Z.H.; Li, D.Z. Identification of putative orthologous genes for the phylogenetic reconstruction of temperate woody bamboos (Poaceae: Bambusoideae). Mol. Ecol. Resour. 2014, 14, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Q.; Yang, J.B.; Peng, Z.H.; Gao, J.; Yang, Y.M.; Peng, S.; Li, D.Z. A molecular phylogenetic and fruit evolutionary analysis of the major groups of the paleotropical woody bamboos (Gramineae: Bambusoideae) based on nuclear ITS, GBSSI gene and plastid trnL-F DNA sequences. Mol. Phylogenet. Evolut. 2008, 48, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Liu, J.; Ge, S.; Xiang, Q.Y.; Zimmer, E.A. Phylogenomic approaches to deciphering the tree of life. J. Syst. Evolut. 2015, 53, 369–370. [Google Scholar] [CrossRef]
- Yang, H.M.; Zhang, Y.X.; Yang, J.B.; Li, D.Z. The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol. Phylogenet. Evolut. 2013, 68, 340–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Zeng, C.X.; Li, D.Z. Complex evolution in Arundinarieae (Poaceae: Bambusoideae): Incongruence between plastid and nuclear gbssi gene phylogenies. Mol. Phylogenet. Evolut. 2012, 63, 777–797. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, W.P.; Ruiz-Sanchez, E.; Yin, Y.; Duvall, M.R. The floral transcriptomes of four bamboo species (Bambusoideae; Poaceae): Support for common ancestry among woody bamboos. BMC Genom. 2016, 17, 384. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chen, S.Y.; Zhang, X.Z. Characterization of the complete chloroplast genome of seabuckthorn (Hippophae rhamnoides l.). Conserv. Genet. Resour. 2017, 9, 623–626. [Google Scholar] [CrossRef]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Characterization of the complete chloroplast genome of Elaeagnus mollis, a rare and endangered oil plant. Conserv. Genet. Resour. 2017, 9, 439–442. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. The chloroplast genome sequence of bittersweet (Solanum dulcamara): Plastid genome structure evolution in Solanaceae. PLoS ONE 2018, 13, e0196069. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evolut. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, bty220. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evolut. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Ge, S. The phylogeny of the BEP clade in grasses revisited: Evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evolut. 2012, 62, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, N.; Ma, P.F.; Liu, Q.; Li, D.Z.; Guo, Z.H. Phylogenomic analyses of nuclear genes reveal the evolutionary relationships within the BEP clade and the evidence of positive selection in Poaceae. PLoS ONE 2013, 8, e64642. [Google Scholar] [CrossRef] [PubMed]
- Soreng, R.J.; Peterson, P.M.; Romaschenko, K.; Davidse, G.; Zuloaga, F.O.; Judziewicz, E.J.; Filgueiras, T.S.; Davis, J.I.; Morrone, O. A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Syst. Evolut. 2015, 53, 117–137. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. ParitionFinder2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evolut. 2017, 34, 772–773. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Haeseler, V.A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evolut. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
Sample Availability: All samples are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Froesiochloa boutelouoides | Rehia nervata | |
|---|---|---|
| Total paired-end reads | 20,158,220 | 20,330,116 |
| Aligned paired-end reads | 432,450 | 425,590 |
| Mean coverage | 318.2 | 310.6 |
| Size (bp) | 135,905 | 136,700 |
| LSC a length (bp) | 82,935 | 80,931 |
| SSC b length (bp) | 12,984 | 13,273 |
| IR c length (bp) | 19,993 | 21,248 |
| Number of genes (unique) | 124 (110) | 130 (112) |
| Protein-coding genes (unique) | 79 (75) | 84 (77) |
| tRNAs (unique) | 37 (31) | 38 (31) |
| rRNAs (unique) | 8 (4) | 8 (4) |
| GC content (%) | 38.7% | 38.8% |
| Coding regions (%) | 41.1% | 44.1% |
| Region | Length (bp) | Aligned Length (bp) | No. VCs a | Percentage of VCs | No. PICs b | Percentage of PICs |
|---|---|---|---|---|---|---|
| trnD(GUC)-psbM | 854–1064 | 1177 | 276 | 23.45 | 139 | 11.81 |
| rpl32-trnL(UAG) | 612–753 | 820 | 147 | 17.93 | 53 | 6.46 |
| ycf4-cemA | 408–457 | 477 | 83 | 17.40 | 27 | 5.66 |
| psbK-psbI | 385–400 | 413 | 71 | 17.19 | 30 | 7.26 |
| psbZ-trnfM(CAU) | 462–818 | 946 | 159 | 16.81 | 61 | 6.45 |
| trnT(GGU)-trnE(UUC) | 463–493 | 542 | 91 | 16.79 | 24 | 4.43 |
| rbcL-psaI | 1117–1219 | 1347 | 224 | 16.63 | 82 | 6.09 |
| trnF(GAA)-ndhJ | 442–604 | 631 | 103 | 16.32 | 36 | 5.71 |
| trnK(UUU)-rps16 | 380–563 | 595 | 97 | 16.30 | 26 | 4.37 |
| rps16-trnQ(UUG) | 721–1091 | 1252 | 202 | 16.13 | 60 | 4.79 |
| petA-psbJ | 956–988 | 1065 | 167 | 15.68 | 56 | 5.26 |
| atpH-atpF | 421–456 | 476 | 73 | 15.34 | 15 | 3.15 |
| trnS(GCU)-psbD | 866–982 | 1077 | 165 | 15.32 | 60 | 5.57 |
| psaA-ycf3 | 583–640 | 736 | 110 | 14.95 | 32 | 4.35 |
| ycf3-trnS(GGA) | 535–595 | 621 | 91 | 14.65 | 38 | 6.12 |
| trnT(UGU)-trnL(UAA) | 733–785 | 853 | 122 | 14.30 | 45 | 5.28 |
| rpl16 intron | 1046–1117 | 1150 | 164 | 14.26 | 52 | 4.52 |
| psaC-ndhE | 462–513 | 592 | 83 | 14.02 | 30 | 5.07 |
| ndhC-trnV(UAC) | 777–917 | 1036 | 142 | 13.71 | 50 | 4.83 |
| ndhA intron | 1002–1017 | 1067 | 143 | 13.40 | 44 | 4.12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Chen, S.; Zhang, X. Whole-Genome Comparison Reveals Divergent IR Borders and Mutation Hotspots in Chloroplast Genomes of Herbaceous Bamboos (Bambusoideae: Olyreae). Molecules 2018, 23, 1537. https://doi.org/10.3390/molecules23071537
Wang W, Chen S, Zhang X. Whole-Genome Comparison Reveals Divergent IR Borders and Mutation Hotspots in Chloroplast Genomes of Herbaceous Bamboos (Bambusoideae: Olyreae). Molecules. 2018; 23(7):1537. https://doi.org/10.3390/molecules23071537
Chicago/Turabian StyleWang, Wencai, Siyun Chen, and Xianzhi Zhang. 2018. "Whole-Genome Comparison Reveals Divergent IR Borders and Mutation Hotspots in Chloroplast Genomes of Herbaceous Bamboos (Bambusoideae: Olyreae)" Molecules 23, no. 7: 1537. https://doi.org/10.3390/molecules23071537
APA StyleWang, W., Chen, S., & Zhang, X. (2018). Whole-Genome Comparison Reveals Divergent IR Borders and Mutation Hotspots in Chloroplast Genomes of Herbaceous Bamboos (Bambusoideae: Olyreae). Molecules, 23(7), 1537. https://doi.org/10.3390/molecules23071537