Tetramethylpyrazine-Inducible Promoter Region from Rhodococcus jostii TMP1


 ,
,
Abstract
1. Introduction
2. Results
2.1. Analysis of the Promoter Activity in Rhodococcus josti TMP1 Cells
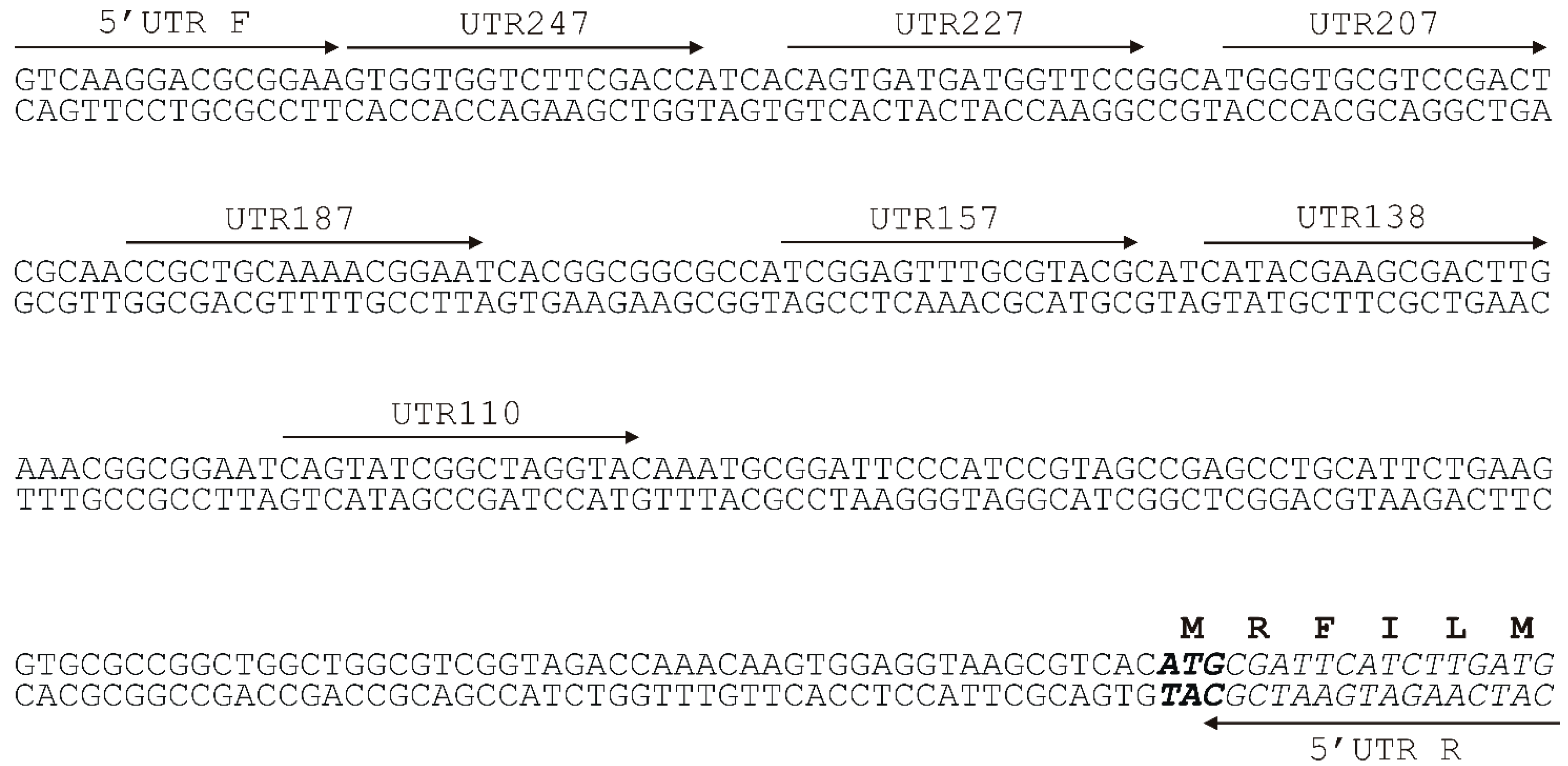
2.2. Analysis of the Minimal Promoter Sequence and Transcription Start Sites
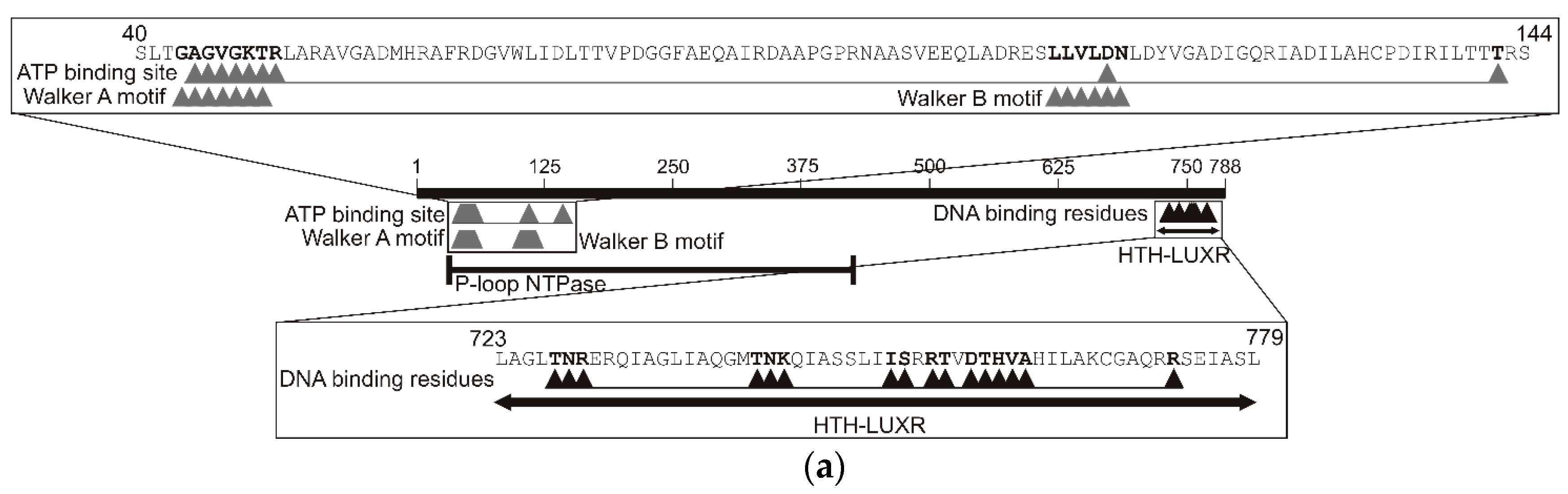
2.3. Analysis of the TpdR Regulator
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Growth Media and Culture Conditions
4.3. Genomic DNA Isolation
4.4. PCR
4.5. Preparation of the Electro-Competent Cells and Conditions of the Electroporation
4.6. Enhanced GFP (EGFP) Fluorescence Measurement
4.7. RNR Isolation and the Identification of the Transcription Start Site
4.8. Mutagenesis of the PTTMP Promoter Sequence
Author Contributions
Funding
Conflicts of Interest
References
- Alvarez, H.M. Central metabolism of species of genus Rhodococcus. In The Biology of Rhodococcus; Alvarez, H.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 91–109. ISBN 978-3-642-12937-7. [Google Scholar]
- Larkin, M.J.; Kulakov, L.A.; Allen, C.C.R. Biodegradation by members of the genus Rhodococcus: Biochemistry, physiology, and genetic adaptation. Adv. Appl. Microbiol. 2006, 59, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.; Prieto, M.A. Bacterial promoters triggering biodegradation of aromatic pollutants. Curr. Opin. Biotechnol. 2000, 11, 467–475. [Google Scholar] [CrossRef]
- Amouric, A.; Quéméneur, M.; Grossi, V.; Liebgott, P.P.; Auria, R.; Casalot, L. Identification of different alkane hydroxylase systems in Rhodococcus ruber strain SP2B, a hexane-degrading actinomycete. J. Appl. Microbiol. 2010, 108, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Cappelletti, M.; Fedi, S.; Frascari, D.; Ohtake, H.; Turner, R.J.; Zannoni, D. Analyses of both the alkB gene transcriptional start site and alkB promoter-inducing properties of Rhodococcus sp. strain BCP1 grown on n-alkanes. Appl. Environ. Microbiol. 2011, 77, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Veselý, M.; Knoppová, M.; Nešvera, J.; Pátek, M. Analysis of catRABC operon for catechol degradation from phenol-degrading Rhodococcus erythropolis. Appl. Microbiol. Biotechnol. 2007, 76, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Szőköl, J.; Rucká, L.; Šimčíková, M.; Halada, P.; Nešvera, J.; Pátek, M. Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl. Microbiol. Biotechnol. 2014, 98, 8267–8279. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Hara, N.; Sakai, M.; Yamada, A.; Miyauchi, K.; Masai, E.; Fukuda, M. Biphenyl-inducible promoters in a polychlorinated biphenyl-degrading bacterium, Rhodococcus sp. RHA1. Biosci. Biotechnol. Biochem. 2004, 68, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Waki, T.; Nakamura, K.; Mukouzaka, Y.; Kudo, T. The GAF-like-domain-containing transcriptional regulator DfdR is a sensor protein for dibenzofuran and several hydrophobic aromatic compounds. J. Bacteriol. 2009, 191, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Nakashima, N.; Tamura, N.; Tamura, T. Isolation and characterization of the Rhodococcus opacus thiostrepton-inducible genes tipAL and tipAS: Application for recombinant protein expression in Rhodococcus. FEMS Microbiol. Lett. 2004, 237, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Namashima, N.; Tamura, T. Isolation and characterization of a rolling-circle-type plasmid from Rhodococcus erythropolis and application of the plasmid to multiple-recombinant-protein expression. Appl. Environ. Microbiol. 2004, 70, 5557–5568. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, Y.; Mitani, Y.; Yun, H-Y.; Nakashima, N.; Tamura, N.; Tamura, T. Identification of a methanol-inducible promoter from Rhodococcus erythropolis PR4 and its use as an expression vector. J. Biosci. Bioeng. 2012, 113, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Tauch, A.; Kirchner, O.; Löffler, B.; Götker, S.; Pühler, A.; Kalinowski, J. Efficient electrotransformation of Corynebacterium diphtheriae with a mini-replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr. Microbiol. 2002, 45, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Triccas, J.A.; Parish, T.; Britton, W.J.; Gicquel, B. An inducible expression system permitting the efficient purification of a recombinant antigen from Mycobacterium smegmatis. FEMS Microbiol. Lett. 1998, 167, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Hetzler, S.; Bröker, D.; Steinbüchel, A. Saccharification of cellulose by recombinant Rhodococcus opacus PD630 strains. Appl. Environ. Microbiol. 2013, 79, 5159–5166. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, J.; Schmidt-Dannert, C. Construction of a BioBrick™ compatible vector system for Rhodococcus. Plasmid 2017, 90, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Michener, J.K.; Thodey, K.; Liang, J.C.; Smolke, C.D. Applications of genetically-encoded biosensors for the construction and control of biosynthetic pathways. Metab. Eng. 2012, 14, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-L.; Rha, E.; Lee, S.J.; Kim, H.; Kwon, K.; Jeong, Y.-S.; Rhee, Y.H.; Song, J.J.; Kim, H.-S.; Lee, S.-G. Toward a generalized and high-throughput enzyme screening system based on artificial genetic circuits. ACS Synth. Biol. 2014, 3, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.K.; Guzman, C.D.; Taylor, N.D.; Raman, S.; Anderson, K.; Church, G.M. Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 2015, 43, 7648–7660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jensen, M.K.; Keasling, J.D. Development of biosensors and their application in metabolic engineering. Curr. Opin. Chem. Biol. 2015, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, L.; Bott, M.; Marienhagen, J. Novel screening methods—Biosensors. Curr. Opin. Biotechnol. 2015, 35, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Mahr, R.; Frunzke, J. Transcription factor-based biosensors in biotechnology: Current state and future prospects. Appl. Microbiol. Biotechnol. 2016, 100, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.K.; Church, G.M. Genetically encoded sensors enable real-time observation of metabolite production. Proc. Natl. Acad. Sci. USA 2016, 113, 2388–2393. [Google Scholar] [CrossRef] [PubMed]
- Kutanovas, S.; Stankeviciute, J.; Urbelis, G.; Tauraite, D.; Rutkiene, R.; Meskys, R. Identification and characterization of a tetramethylpyrazine catabolic pathway in Rhodococcus jostii TMP1. Appl. Environ. Microbiol. 2013, 79, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Kutanovas, S.; Rutkiene, R.; Urbelis, G.; Tauraite, D.; Stankeviciute, J.; Meskys, R. Bioconversion of methylpyrazines and pyridines using novel pyrazines-degrading microorganisms. Chemija 2013, 24, 67–73. [Google Scholar]
- De Schrijver, A.; De Mot, R. A subfamily of MalT related ATP-dependent regulators in the LuxR family. Microbiology 1999, 145, 1287–1288. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Squires, C.H.; Monticello, D.J.; Childs, J.D. Genetic analysis of the dsz promoter and associated regulatory regions of Rhodococcus erythropolis IGTS8. J. Bacteriol. 1996, 178, 6409–6418. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Gallardo, L.; Santero, E.; Camafeita, E.; Calvo, E.; Schlömann, M.; Floriano, B. Molecular and biochemical characterization of the tetralin degradation pathway in Rhodococcus sp. strain TFB. J. Microb. Biotech. 2009, 2, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Hansmeier, N.; Albersmeier, A.; Tauch, A.; Damberg, T.; Ros, R.; Anselmetti, D.; Pühler, A.; Kalinowski, J. The surface (S)-layer gene cspB of Corynebacterium glutamicum is transcriptionally activated by a LuxR-type regulator and located on a 6 kb genomic island absent from the type strain ATCC 13032. Microbiology 2006, 152, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Snider, J.; Thibault, G.; Houry, W.A. The AAA+ superfamily of functionally diverse proteins. Genome Biol. 2008, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Enemark, E.J. Fundamental characteristics of AAA+ protein family structure and function. Archaea 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Labbé, D.; Garnon, J.; Lau, P.C. Characterization of the genes encoding a receptor-like histidine kinase and a cognate response regulator from a biphenyl/polychlorobiphenyl-degrading bacterium, Rhodococcus sp. strain M5. J. Bacteriol. 1997, 179, 2772–2776. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Chae, J.C.; Zylstra, G.J.; Sohn, H.Y.; Kwon, G.S.; Kim, E. Identification of two-component regulatory genes involved in o-xylene degradation by Rhodococcus sp. strain DK17. J. Microbiol. 2005, 43, 49–53. [Google Scholar] [PubMed]
- Patankar, A.V.; González, J.E. Orphan LuxR regulators of quorum sensing. FEMS Microbiol. Rev. 2009, 33, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Ingigliardi, D.; Richet, E.; Raibaud, O. Two MalT binding sites in direct repeat. A structural motif involved in the activation of all the promoters of the maltose regulons in Escherichia coli and Klebsiella pneumoniae. J. Mol. Biol. 1991, 218, 323–334. [Google Scholar] [CrossRef]
- Darwin, A.J.; Li, J.; Stewart, V. Analysis of nitrate regulatory protein NarL-binding sites in the fdnG and narG operon control regions of Escherichia coli K-12. Mol. Microbiol. 1996, 20, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Ducros, V.M.; Lewis, R.J.; Verma, C.S.; Dodson, E.J.; Leonard, G.; Turkenburg, J.P.; Murshudov, G.N.; Wilkinson, A.J.; Brannigan, J.A. Crystal structure of GerE, the ultimate transcriptional regulator of spore formation in Bacillus subtilis. J. Mol. Biol. 2001, 306, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Quan, S.; Dabbs, E.R. Nocardioform arsenic resistance plasmid characterization and improved Rhodococcus cloning vectors. Plasmid 1993, 29, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Sandu, C.; Chiribau, C.-B.; Sachelaru, P.; Brandsch, R. Plasmids for nicotine-dependent and -independent gene expression in Arthrobacter nicotinovorans and other Arthrobacter species. Appl. Environ. Microbiol. 2005, 71, 8920–8924. [Google Scholar] [CrossRef] [PubMed]
- Woo, T.H.; Cheng, A.F.; Ling, J.M. An application of a simple method for the preparation of bacterial DNA. BioTechniques 1992, 13, 696–698. [Google Scholar] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; ISBN 978-0879695774. [Google Scholar]
- Sharma, R.C.; Schimke, R.T. Preparation of electrocompetent E. coli using salt-free growth medium. BioTechniques 1996, 20, 42–44. [Google Scholar] [PubMed]
- Gartemann, K.-H.; Eichenlaub, R. Isolation and characterization of IS 1409, an insertion element of 4-chlorobenzoate-degrading Arthrobacter sp. strain TM1, and development of a system for transposon mutagenesis. J. Bacteriol. 2001, 183, 3729–3736. [Google Scholar] [CrossRef] [PubMed]
- Truncaite, L.; Piešiniene, L.; Kolesinskiene, G.; Zajanckauskaite, A.; Driukas, A.; Klausa, V.; Nivinskas, R. Twelve new MotA-dependent middle promoters of bacteriophage T4: Consensus sequence revised. J. Mol. Biol. 2003, 327, 335–346. [Google Scholar] [CrossRef]
Sample Availability: Samples of the plasmid DNAs are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Description | Reference |
|---|---|---|
| Escherichia coli DH5α | ϕ80dlacZΔM15 Δ(lacZY-argF) U169 deoR recA1 endA1 hsdR17(rK-mK+) sup E44 thi-1 gyrA96 relA1 | Thermo Fisher Scientific, Vilnius, Lithuania |
| Rhodococcus jostii TMP1 | Utilise tetramethylpyrazine (TTMP) as a sole source of carbon and energy | [25] |
| Rhodococcus erythropolis SQ1 | [39] | |
| Plasmids | ||
| pART3gfp | KmR; hybrid vector for nicotine-inducible enhanced GFP (EGFP) protein expression in Arthrobacter sp.; 6.1 kb | [40] |
| pART3-5′UTR-gfp | 277 bp upstream tpdA gene (envolved in TTMP degradation) fragment was amplified and cloned to pART3gfp via BamHI | [24] |
| pART3-5′UTR-gfp-R | tpdR gene (encoding transcription regulator) with upstream region (3 kb fragment) was amplified and cloned to pART3-5′UTR-gfp via XbaI | This work |
| Primers | Sequence 5′ → 3′ | Purpose | Reference |
|---|---|---|---|
| 5′UTR_F | TACGTGGATCCGTCAAGGAC | direct primer of the upstream tpdA region | [25] |
| UTR247 | GATGGATCCGTGGTGGTCTTCGACC | determination of a minimal promoter sequence | This work |
| UTR227 | GATGGATCCAGTGATGATGGTTCCGG | determination of a minimal promoter sequence | This work |
| UTR207 | GATGGATCCTGGGTGCGTCCGACTC | determination of a minimal promoter sequence | This work |
| UTR187 | GATGGATCCGCTGCAAAACGGAATC | determination of a minimal promoter sequence | This work |
| UTR157 | GATGGATCCTCGGAGTTTGCGTACG | determination of a minimal promoter sequence | This work |
| UTR138 | GATGGATCCATACGAAGCGACTTGAAAC | determination of a minimal promoter sequence | This work |
| UTR110 | GATGGATCCAGTATCGGCTAGGTACA | determination of a minimal promoter sequence | This work |
| 5′UTR_R | CACATGGATCCATCAAGATGAATCGC | reverse primer of the upstream tpdA region | [25] |
| Reg-F_Xba | GGATCTAGACCGAAGAACGAACG | tpdR amplification | This work |
| Reg-R_Xba | GTCTAGATCACAAACCAGTTCGC | tpdR amplification | This work |
| P_GFP_R | GGTGAACAGCTCCTCG | determination of transcription start site | This work |
| P-del-F | ACGAAGCGACTTGAATCAGTATCGGCTAG | PTTMP mutagenesis | This work |
| P-del-atv | CTAGCCGATACTGATTCAAGTCGCTTCGT | PTTMP mutagenesis | This work |
| P-TA-F | GGATTCCCAACCGTAGCCGAG | PTTMP mutagenesis | This work |
| P-TA-atv | CTCGGCTACGGTTGGGAATCC | PTTMP mutagenesis | This work |
| P-CA1-F | GGATTCCCATACGTAGCCGAGC | PTTMP mutagenesis | This work |
| P-CA1-atv | GCTCGGCTACGTATGGGAATCC | PTTMP mutagenesis | This work |
| P-CA2-F | GGATTCCCATCAGTAGCCGAGC | PTTMP mutagenesis | This work |
| P-CA2-atv | GCTCGGCTACTGATGGGAATCC | PTTMP mutagenesis | This work |
| P-GA-F | GGATTCCCATCCATAGCCGAGC | PTTMP mutagenesis | This work |
| P-GA-atv | GCTCGGCTATGGATGGGAATCC | PTTMP mutagenesis | This work |
| P-AS5-F | GGATTCCCGAAAATAGCCGAGC | PTTMP mutagenesis | This work |
| P-AS5-atv | GCTCGGCTATTTTCGGGAATCC | PTTMP mutagenesis | This work |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanislauskienė, R.; Kutanovas, S.; Kalinienė, L.; Bratchikov, M.; Meškys, R. Tetramethylpyrazine-Inducible Promoter Region from Rhodococcus jostii TMP1. Molecules 2018, 23, 1530. https://doi.org/10.3390/molecules23071530
Stanislauskienė R, Kutanovas S, Kalinienė L, Bratchikov M, Meškys R. Tetramethylpyrazine-Inducible Promoter Region from Rhodococcus jostii TMP1. Molecules. 2018; 23(7):1530. https://doi.org/10.3390/molecules23071530
Chicago/Turabian StyleStanislauskienė, Rūta, Simonas Kutanovas, Laura Kalinienė, Maksim Bratchikov, and Rolandas Meškys. 2018. "Tetramethylpyrazine-Inducible Promoter Region from Rhodococcus jostii TMP1" Molecules 23, no. 7: 1530. https://doi.org/10.3390/molecules23071530
APA StyleStanislauskienė, R., Kutanovas, S., Kalinienė, L., Bratchikov, M., & Meškys, R. (2018). Tetramethylpyrazine-Inducible Promoter Region from Rhodococcus jostii TMP1. Molecules, 23(7), 1530. https://doi.org/10.3390/molecules23071530