Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
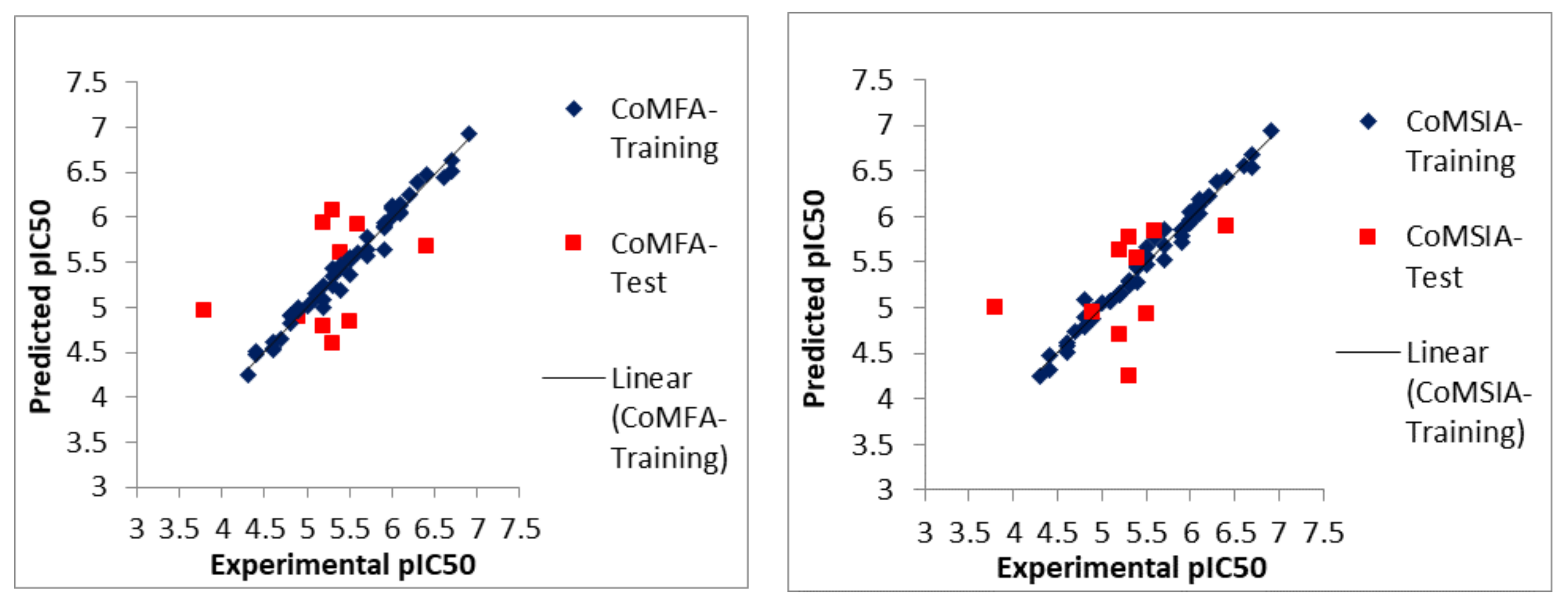
2.1. Statistical Analyses of CoMFA and CoMSIA Models
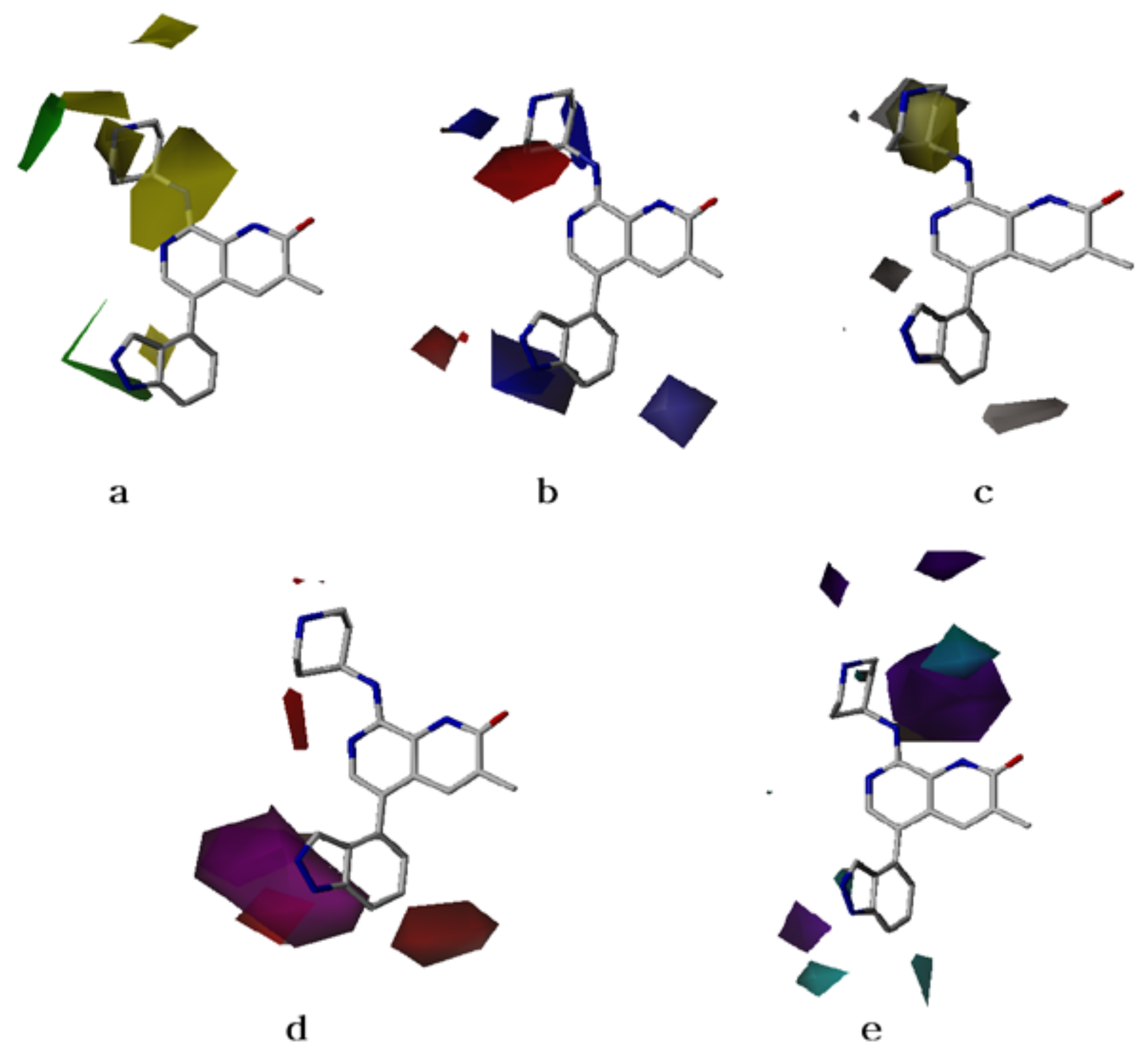
2.2. CoMFA Contour Maps
2.3. CoMSIA Contour Maps
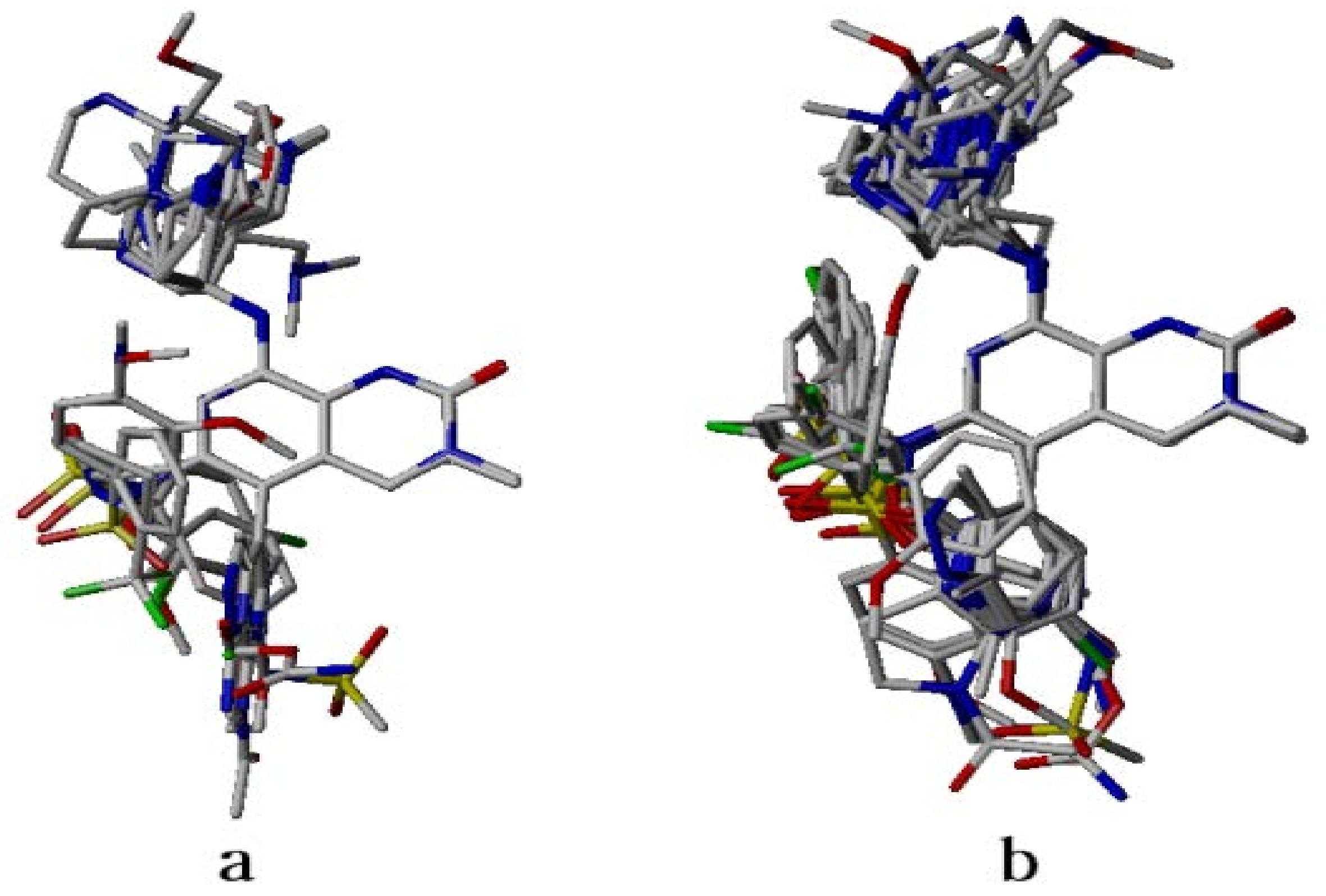
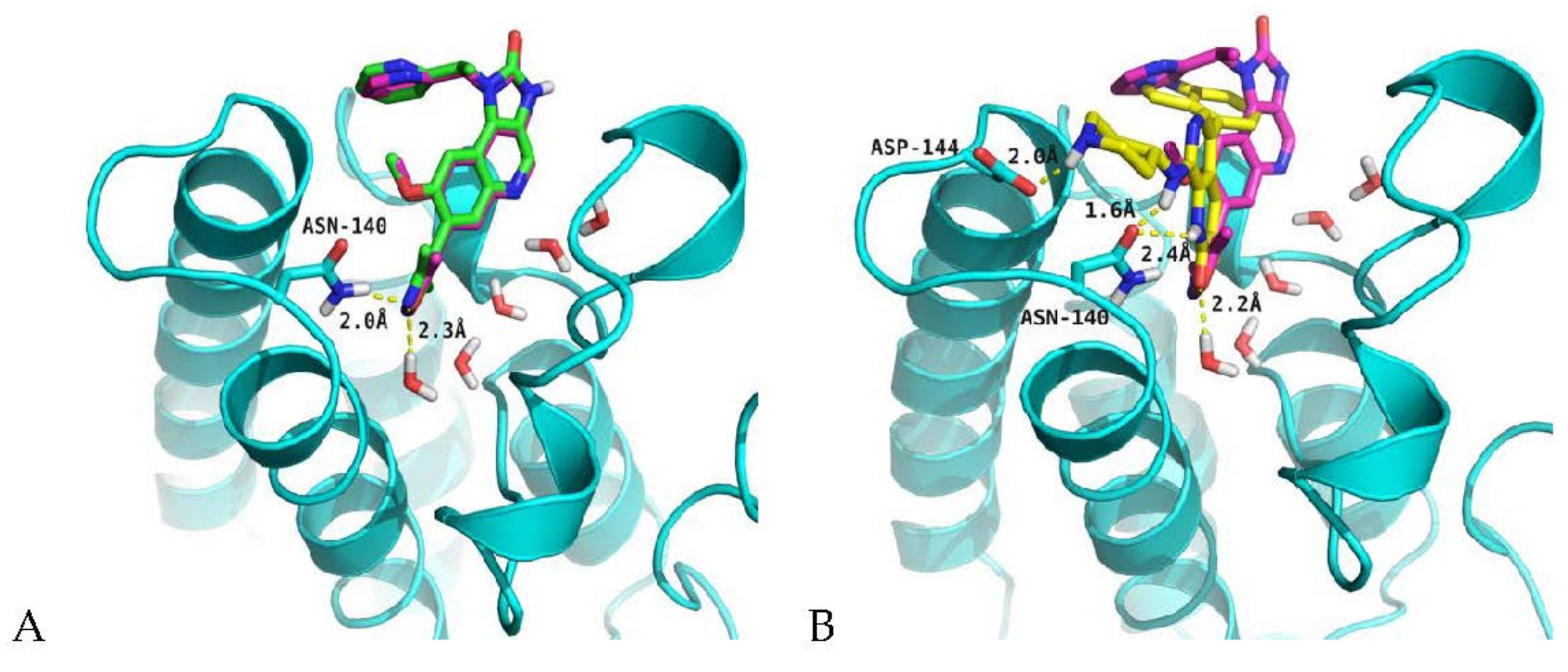
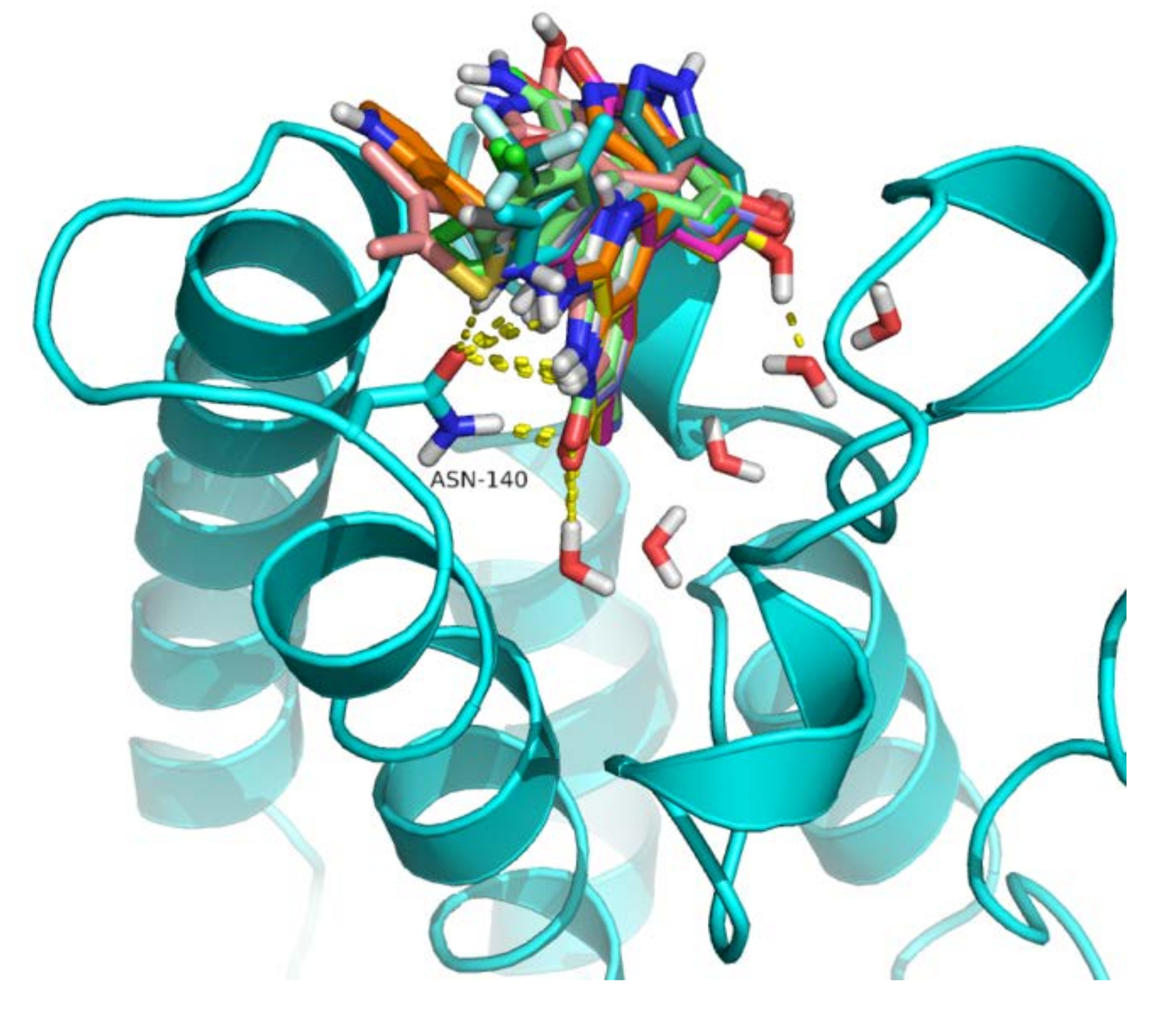
2.4. Analysis of Structure-Based Generated Conformations
2.5. Designing of New Compounds and Their Physicochemical Properties’ Calculations
2.6. Biological Activities Prediction of Newly Designed Compounds
3. Material and Methods
3.1. Biological Data Collection
3.2. Dataset Compounds Modeling and Alignment
3.3. CoMFA and CoMSIA Fields Calculations
3.4. Partial Least Squares (PLS) Regression Analyses and Validations of the Models
3.5. Preparation of Ligands and Protein Structure
3.6. Molecular Docking Protocol
4. Conclusions
Supplementary Materials
Author Contribution
Funding
Acknowledgments
Conflicts of Interest
References
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today. Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, A.; Yugandhar, D.; Srivastava, A.K. BET inhibitors in cancer therapeutics: A patent review. Expert Opin. Ther. Pat. 2016, 26, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Garnier, J.-M.; Sharp, P.P.; Burns, C.J. BET bromodomain inhibitors: A patent review. Expert Opin. Ther. Pat. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Roe, J.-S.; Vakoc, C.R. The Essential Transcriptional Function of BRD4 in Acute Myeloid Leukemia. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.C.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2016, 15, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Sawle, A.D.; Musi, E.; Schwartz, G.K. BRD4-targeted therapy induces Myc-independent cytotoxicity in Gnaq/11-mutatant uveal melanoma cells. Oncotarget 2015, 6, 33397–33409. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xing, J.; Wang, C.; Zhang, H.; Yue, L.; Wan, X.; Chen, W.; Ding, H.; Xie, Y.; Tao, H.; et al. Discovery of novel BET inhibitors by drug repurposing of nitroxoline and its analogues. Org. Biomol. Chem. 2017, 15, 9352–9361. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zheng, X.; Yang, Y.; Wang, X.; Shen, Z. An Overview on Small Molecule Inhibitors of BRD4. Mini Rev. Med. Chem. 2016, 16, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J.; et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhou, J.; Ye, F.; Xiong, H.; Peng, L.; Zheng, Z.; Xu, F.; Cui, M.; Wei, C.; Wang, X.; et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int. J. Mol. Sci. 2015, 16, 1928–1948. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Chen, D.; Qian, D.; Zhang, S.; Zhang, Y.; Guo, S.; Ma, Z.; Wang, S. AZD5153, a novel BRD4 inhibitor, suppresses human thyroid carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2018, 499, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tian, B.; Chen, H.; Wang, P.; Brasier, A.R.; Zhou, J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem. 2018, 151, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Pasha, F.A.; Muddassar, M.; Joo Cho, S. Molecular docking and 3D QSAR studies of Chk2 inhibitors. Chem. Biol. Drug Des. 2009, 73, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Pasha, F.A.; Muddassar, M.; Srivastava, A.K.; Cho, S.J. In silico QSAR studies of anilinoquinolines as EGFR inhibitors. J. Mol. Model. 2010, 16, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Muddassar, M.; Pasha, F.A.; Neaz, M.M.; Saleem, Y.; Cho, S.J. Elucidation of binding mode and three dimensional quantitative structure-activity relationship studies of a novel series of protein kinase B/Akt inhibitors. J. Mol. Model. 2009, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, W.; Shi, Y.; Le, G. Structural analysis of selective agonists of thyroid hormone receptor β using 3D-QSAR and molecular docking. J. Taiwan Inst. Chem. Eng. 2015, 49, 1–18. [Google Scholar] [CrossRef]
- Mittal, R.R.; Harris, L.; McKinnon, R.A.; Sorich, M.J. Partial charge calculation method affects CoMFA QSAR prediction accuracy. J. Chem. Inf. Model. 2009, 49, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Lai, T.; Guo, T.; Chen, F.; Zhang, L.; Ding, W.; Zhang, Y. Synthesis and Acaricidal Activities of Scopoletin Phenolic Ether Derivatives: QSAR, Molecular Docking Study and in Silico ADME Predictions. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.W.; Korenchuk, S.; Bai, Y.; Barbash, O.; Le, B.; Craggs, P.D.; McCabe, M.T.; et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 2013, 8, e72967. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Philpott, M.; Müller, S.; Schulze, J.; Badock, V.; Eberspächer, U.; Moosmayer, D.; Bader, B.; Schmees, N.; Fernández-Montalván, A.; et al. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y. Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg. Med. Chem. Lett. 2016, 26, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Gosmini, R.; Nguyent, V.L.; Toum, J.; Simon, C.; Brusq, J.M.G.; Krysa, G.; Mirguet, O.; Riou-Eymard, A.M.; Boursier, E.V.; Trottet, L.; et al. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 2014, 57, 8111–8131. [Google Scholar] [CrossRef] [PubMed]
- Fish, P.V.; Filippakopoulos, P.; Bish, G.; Brennan, P.E.; Bunnage, M.E.; Cook, A.S.; Federov, O.; Gerstenberger, B.S.; Jones, H.; Knapp, S.; et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55, 9831–9837. [Google Scholar] [CrossRef] [PubMed]
- Demont, E.H.; Chung, C.; Furze, R.C.; Grandi, P.; Michon, A.; Wellaway, C.; Barrett, N.; Bridges, A.M.; Craggs, P.D.; Diallo, H.; et al. Fragment-based discovery of low-micromolar ATAD2 bromodomain inhibitors. J. Med. Chem. 2015, 58, 5649–5673. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.T.; Roy, K. On selection of training and test sets for the development of predictive QSAR models. QSAR Comb. Sci. 2006, 25, 235–251. [Google Scholar] [CrossRef]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Sainy, J.; Sharma, R. QSAR analysis of thiolactone derivatives using HQSAR, CoMFA and CoMSIA. SAR QSAR Environ. Res. 2015, 26, 873–892. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Ashraf, S.; Al-Majid, A.M.; Barakat, A. 3D-QSAR studies on barbituric acid derivatives as urease inhibitors and the effect of charges on the quality of a model. Int. J. Mol. Sci. 2016, 17, 657. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, J.; Peng, C.; Chang, Y.; Guo, L.; Hou, J.; Huang, M.; Wu, B.; Zheng, J.; Lin, L. New molecular insights into the tyrosyl-tRNA synthase inhibitors: CoMFA, CoMSIA analyses and molecular docking studies. Sci. Rep. 2017, 7, 11525. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Barbault, F.; Delamar, M.; Zhang, R. Receptor-and ligand-based 3D-QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R. Atomic Physicochemical Parameters for 3 Dimensional Structure Directed Quantitative Structure—Activity Relationships .4. Additional Parameters for Hydrophobic and Dispersive Interactions and Their Application for an Automated Superposition of Certain Naturally-Occurring Nucleoside Antibiotics. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar]
- Doddareddy, M.R.; Cho, Y.S.; Koh, H.Y.; Pae, A.N. CoMFA and CoMSIA 3D QSAR analysis on N1-arylsulfonylindole compounds as 5-HT6 antagonists. Bioorg. Med. Chem. 2004, 12, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MMFF94 Charges | Structure-Based Model | Ligand-Based Model | ||
|---|---|---|---|---|
| 50 Compounds | ||||
| Parameters | CoMFA | CoMSIA | CoMFA | CoMSIA |
| ONC | 5 | 6 | 2 | 6 |
| q2(loo) | 0.569 | 0.500 | 0.399 | 0.403 |
| r2(ncv) | 0.979 | 0.982 | 0.873 | 0.873 |
| SEE | 0.102 | 0.094 | 0.251 | 0.251 |
| F | 336.723 | 396.442 | 49.120 | 49.133 |
| Pred-r2 | 0.816 | 0.834 | 0.762 | 0.769 |
| Steric (S) | 0.470 | 0.130 | 0.474 | 0.120 |
| Electrostatic (E) | 0.530 | 0.345 | 0.526 | 0.328 |
| Hydrophobic (H) | - | 0.254 | - | 0.213 |
| Donor (D) | - | 0.127 | - | 0.166 |
| Acceptor (A) | - | 0.144 | - | 0.173 |
| r2 bs | 0.988 | 0.988 | 0.915 | 0.927 |
| SDbs | 0.004 | 0.005 | 0.066 | 0.058 |
| Comp | MW | HBD | HBA | QPlogPo/w | QPlogHERG | QPCaco2 | QPlogBB | QPlogKhsa |
|---|---|---|---|---|---|---|---|---|
| 1 | 405.41 | 6 | 7 | 1.232 | −5.899 | 15.239 | −2.972 | −0.046 |
| 2 | 416.43 | 5 | 6 | 2.732 | −6.434 | 76.171 | −2.227 | 0.358 |
| 3 | 404.38 | 6 | 6 | 1.606 | −6.432 | 23.449 | −2.752 | −0.022 |
| 4 | 403.39 | 7 | 5 | 1.373 | −6.352 | 13.145 | −3.049 | −0.067 |
| 5 | 432.49 | 4 | 8 | 2.371 | −6.23 | 62.067 | −2.187 | 0.246 |
| 6 | 432.49 | 5 | 5 | 3.191 | −6.294 | 80.298 | −2.084 | 0.478 |
| 7 | 451.31 | 4 | 4 | 4.433 | −6.894 | 195.227 | −1.354 | 0.717 |
| 8 | 420.47 | 3 | 5 | 3.653 | −6.669 | 107.71 | −1.933 | 0.722 |
| 9 | 431.88 | 6 | 5 | 2.937 | −6.857 | 44.197 | −2.357 | 0.332 |
| 10 | 451.41 | 4 | 5 | 3.546 | −6.692 | 75.418 | −1.887 | 0.54 |

| Substituents | Glide-Score | Predicted pIC50 | |||
|---|---|---|---|---|---|
| No. | R | R′ | CoMFA Model | CoMSIA Model | |
| 1 |  |  | −7.348 | 7.675 | 6.757 |
| 2 |  |  | −7.140 | 7.293 | 6.523 |
| 3 |  |  | −7.775 | 7.116 | 6.219 |
| 4 |  |  | −7.010 | 7.113 | 6.074 |
| 5 |  |  | −5.953 | 6.922 | 6.255 |
| 6 |  |  | −6.691 | 6.740 | 6.614 |
| 7 |  |  | −6.959 | 6.664 | 6.612 |
| 8 |  |  | −6.421 | 6.367 | 7.058 |
| 9 |  |  | −6.429 | 6.347 | 7.035 |
| 10 |  |  | −7.352 | 6.302 | 6.123 |

| No. | R1 | X | pIC50 | No. | R1 | X | pIC50 |
|---|---|---|---|---|---|---|---|
| 1 |  | N | 5.3 | 8 |  | N | 6.4 |
| 2 |  | N | 5.7 | 9 |  | N | 6.3 |
| 3 |  | N | 5.3 | 10 |  | N | 6.1 |
| 4 |  | N | 6.7 | 11 |  | N | 6.0 |
| 5 |  | N | 6.6 | 12 |  | N | 6.0 |
| 6 |  | N | 6.6 | 13 |  | N | 5.6 |
| 7* |  | N | 6.4 | ||||
| No. | R2 | X | pIC50 | No. | R2 | X | pIC50 |
| 14 |  | 4.6 | 27 |  | N | 4.6 | |
| 15 |  | 4.4 | 28 * |  | N | 3.8 | |
| 16* |  | 5.3 | 29 |  | N | 4.8 | |
| 17 |  | 4.3 | 30 * |  | N | 4.9 | |
| 18 |  | CH | 5.0 | 31 |  | N | 4.9 |
| 19 |  | N | 4.7 | 32 |  | N | 5.2 |
| 20 |  | CH | 5.2 | 33 * |  | N | 5.5 |
| 21 |  | N | 5.4 | 34 |  | N | 5.3 |
| 22* |  | CH | 5.2 | 35 |  | N | 5.1 |
| 23 |  | N | 4.8 | 36 |  | N | 5.4 |
| 24 |  | N | 4.4 | 37 |  | N | 5.5 |
| 25 |  | N | 4.9 | 38 |  | N | 5.4 |
| 26 |  | N | 4.6 | ||||
| No. | R3 | X | pIC50 | No. | R3 | X | pIC50 |
| 39 |  | N | 6.1 | 50 * |  | N | 5.6 |
| 40 |  | N | 6.0 | 51 |  | N | 5.5 |
| 41 |  | N | 6.2 | 52 |  | N | 5.9 |
| 42 |  | N | 6.9 | 53 |  | N | 5.9 |
| 43 |  | N | 5.7 | 54 |  | N | 5.2 |
| 44 |  | N | 6.1 | 55 * |  | N | 5.4 |
| 45 |  | N | 5.5 | 56 |  | N | 5.1 |
| 46 * |  | N | 5.3 | 57 |  | N | 5.7 |
| 47 |  | N | 5.9 | 58 |  | N | 6.1 |
| 48 * |  | N | 5.2 | 59 |  | N | 5.9 |
| 49 |  | N | 6.0 | 60 |  | N | 4.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahir, A.; Alharthy, R.D.; Naseem, S.; Mahmood, N.; Ahmed, M.; Shahzad, K.; Akhtar, M.N.; Hameed, A.; Sadiq, I.; Nawaz, H.; et al. Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches. Molecules 2018, 23, 1527. https://doi.org/10.3390/molecules23071527
Tahir A, Alharthy RD, Naseem S, Mahmood N, Ahmed M, Shahzad K, Akhtar MN, Hameed A, Sadiq I, Nawaz H, et al. Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches. Molecules. 2018; 23(7):1527. https://doi.org/10.3390/molecules23071527
Chicago/Turabian StyleTahir, Adeena, Rima D. Alharthy, Saadia Naseem, Natasha Mahmood, Mahmood Ahmed, Khuram Shahzad, Malik Nadeem Akhtar, Abdul Hameed, Irfan Sadiq, Haq Nawaz, and et al. 2018. "Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches" Molecules 23, no. 7: 1527. https://doi.org/10.3390/molecules23071527
APA StyleTahir, A., Alharthy, R. D., Naseem, S., Mahmood, N., Ahmed, M., Shahzad, K., Akhtar, M. N., Hameed, A., Sadiq, I., Nawaz, H., & Muddassar, M. (2018). Investigations of Structural Requirements for BRD4 Inhibitors through Ligand- and Structure-Based 3D QSAR Approaches. Molecules, 23(7), 1527. https://doi.org/10.3390/molecules23071527