Bioactive Indanes: Proof of Concept Study for Enantioselective Synthetic Routes to PH46A, a New Potential Anti-Inflammatory Agent

Abstract
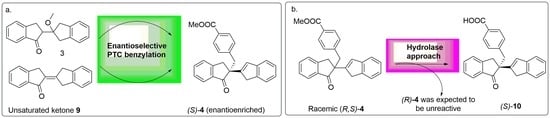
1. Introduction
2. Results and Discussion
2.1. Synthesis of Chiral Standards: (S)-4, (R)-4, (S)-10 and (R)-10
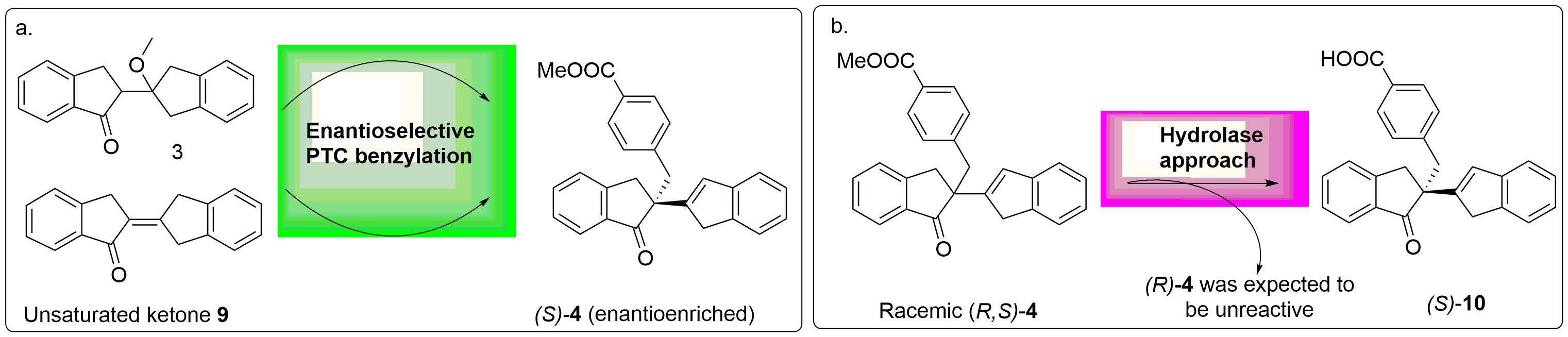
2.2. PTC-Promoted Alkylation of Ketone 3
2.3. PTC-Promoted Alkylation of Unsaturated Ketone 9
2.4. Hydrolase Screening
3. Materials and Methods
3.1. General Information
3.2. General Producre for the Sysnthesis of Chiral Standards: (S)-4 and (R)-4
3.2.1. Methyl (S)-4-((1′-oxo-1′,3′-dihydro-1H,2′H-[2,2′-biinden]-2′-yl)methyl)benzoate (S)-4
3.2.2. Methyl (R)-4-((1′-oxo-1′,3′-dihydro-1H,2′H-[2,2′-biinden]-2′-yl)methyl)benzoate (R)-4
3.3. General Producre of Hydrolysis of Chiral Standards: (S)-10 and (R)-10
3.3.1. (S)-4-((1′-Axo-1′,3′-dihydro-1H,2′H-[2,2′-biinden]-2′-yl)methyl)benzoic acid (S)-10
3.3.2. (R)-4-((1′-Oxo-1′,3′-dihydro-1H,2′H-[2,2′-biinden]-2′-yl)methyl)benzoic acid (R)-10
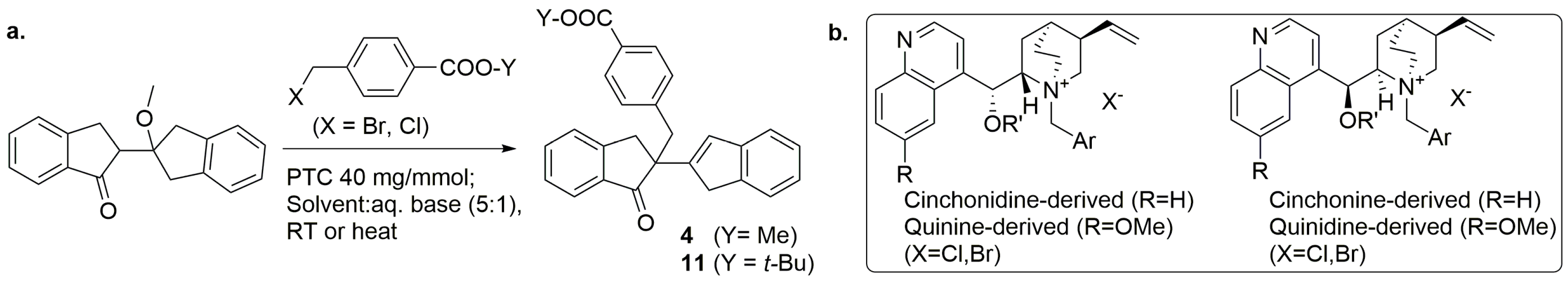
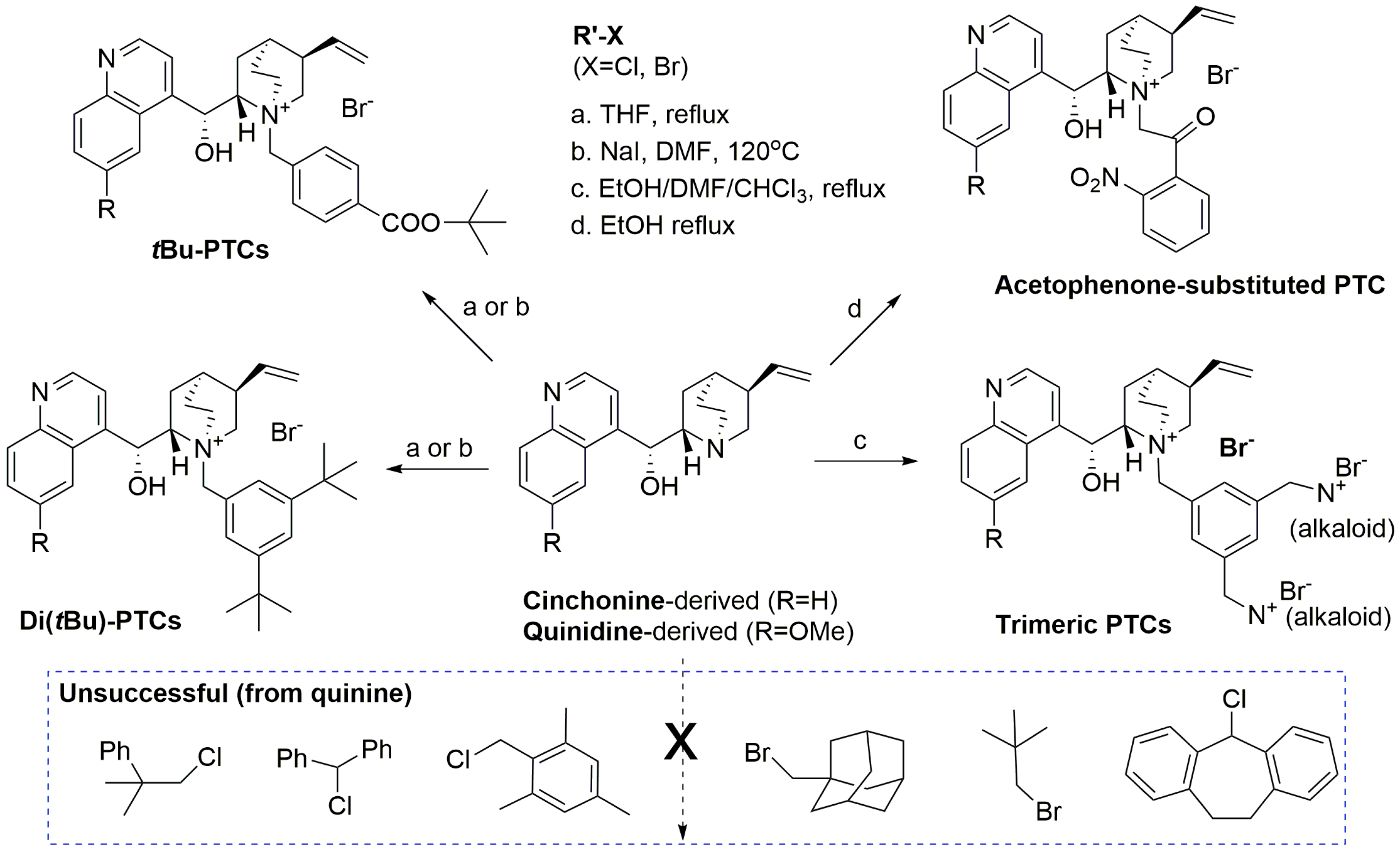
3.4. General Procedure of Synthesis of PTCs (Cinchona Alkaloids Derived)
3.5. Representative Procedure for PTC Alkylations Using MBMB (Entry 5R)
3.6. Hydrolase Screening Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frankish, N.; Farrell, R.; Sheridan, H. Investigation into the mast cell stabilizing activity of nature-identical and synthetic indanones. J. Pharm. Pharmacol. 2004, 56, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, H.; Walsh, J.J.; Cogan, C.; Jordan, M.; McCabe, T.; Passante, E.; Frankish, N.H. Diastereoisomers of 2-benzyl-2,3-dihydro-2-(1H-inden-2-yl)-1H-inden-1-ol: Potential anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2009, 19, 5927–5930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; McCabe, T.; Marzec, B.; Frankish, N.; Sheridan, H. N-cyclopentyl-N-(3-oxo-2,3-dihydro-1H-inden-1-yl)acetamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, o958. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Paluch, K.; Scalabrino, G.; Frankish, N.; Healy, A.-M.; Sheridan, H. Molecular structure studies of (1S,2S)-2-benzyl-2,3-dihydro-2-(1H-inden-2-yl)-1H-inden-1-ol. J. Mol. Struct. 2015, 1083, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, H.; Lemon, S.; Frankish, N.; McArdle, P.; Higgins, T.; James, J.P.; Bhandari, P. Synthesis and antispasmodic activity of nature identical substituted indanes and analogues. Eur. J. Med. Chem. 1990, 25, 603–608. [Google Scholar] [CrossRef]
- Sheridan, H.; Frankish, N.; Farrell, R. Synthesis and antispasmodic activity of analogues of natural pterosins. Eur. J. Med. Chem. 1999, 34, 953–966. [Google Scholar] [CrossRef]
- Frankish, N.; Sheridan, H. 6-(Methylamino)hexane-1,2,3,4,5-pentanol 4-(((1S,2S)-1-hydroxy-2,3-dihydro-1H,1′H-[2,2-biinden]-2-yl)methyl)benzoate (PH46A): A novel small molecule with efficacy in murine models of colitis. J. Med. Chem. 2012, 55, 5497–5505. [Google Scholar] [CrossRef] [PubMed]
- Frampton, C.S.; Zhang, T.; Scalabrino, G.; Frankish, N.; Sheridan, H. (1S)-1-Phenylethanaminium 4-{[(1S,2S)-1-hydroxy-2,3-dihydro-1H,1′H-[2,2′-biinden]-2-yl]methyl}benzoate. Acta Crystallogr. C 2012, 68, o323–o326. [Google Scholar] [CrossRef] [PubMed]
- Therapeutics, T. A Study to Assess the Safety and Tolerability of PH46A in Healthy Volunteers, to Measure Drug levels in These Subjects and to Determine the Effect of Food on the Drug’s Absorption, 11 March 2014 ed.; BioMed Central ISRCTN Registry: London, UK, 2014. [Google Scholar]
- Cumming, G.R.; Zhang, T.; Scalabrino, G.; Frankish, N.; Sheridan, H. Investigation of the stereoselective synthesis of the indane dimer PH46A, a new potential anti-inflammatory agent. Org. Process Res. Dev. 2017, 21, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Dolling, U.H.; Davis, P.; Grabowski, E.J.J. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)−indacrinone via chiral phase-transfer catalysis. J. Am. Chem. Soc. 1984, 106, 446–447. [Google Scholar] [CrossRef]
- Hughes, D.L.; Dolling, U.H.; Ryan, K.M.; Schoenewaldt, E.F.; Grabowski, E.J.J. Efficient catalytic asymmetric alkylations. 3. A kinetic and mechanistic study of the enantioselective phase-transfer methylation of 6,7-dichloro-5-methoxy-2-phenyl-1-indanone. J. Org. Chem. 1987, 52, 4745–4752. [Google Scholar] [CrossRef]
- Milner, S.E.; Brossat, M.; Moody, T.S.; Elcoate, C.J.; Lawrence, S.E.; Maguire, A.R. Efficient kinetic bioresolution of 2-nitrocyclohexanol. Tetrahedron Asymmetry 2010, 21, 1011–1016. [Google Scholar] [CrossRef]
- Brossat, M.; Moody, T.S.; Taylor, S.J.C.; Wiffen, J.W. Simple one-pot process for the bioresolution of tertiary amino ester protic ionic liquids using subtilisin. Tetrahedron Asymmetry 2009, 20, 2112–2116. [Google Scholar] [CrossRef]
- Brossat, M.; Moody, T.S.; de Nanteuil, F.; Taylor, S.J.C.; Vaughan, F. Development of an acid-washable tag for the separation of enantiomers from bioresolutions. Org. Process Res. Dev. 2009, 13, 706–709. [Google Scholar] [CrossRef]
- Schmid, R.D.; Verger, R. Lipases: Interfacial enzymes with attractive applications. Angew. Chem. Int. Ed. 1998, 37, 1609–1633. [Google Scholar] [CrossRef]
- Wolfenden, R.; Snider, M.J. The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Wells, A. 9.11 industrial applications of biocatalysis: An overview. In Comprehensive Chirality; Elsevier: Amsterdam, The Netherlands, 2012; pp. 253–287. [Google Scholar]
- Woodley, J.M. New opportunities for biocatalysis: Making pharmaceutical processes greener. Trends Biotechnol. 2008, 26, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Mangan, D.; Miskelly, I.; Moody, T.S. A facile stereoselective biocatalytic route to the precursor of woody acetate. Org. Process Res. Dev. 2011, 15, 1036–1039. [Google Scholar] [CrossRef]
- Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A new generation of chiral phase-transfer catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef] [PubMed]
- Maruoka, K.; Ooi, T. Enantioselective amino acid synthesis by chiral phase-transfer catalysis. Chem. Rev. 2003, 103, 3013–3028. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.J. The enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral schiff base esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Lygo, B.; Beynon, C.; Lumley, C.; McLeod, M.C.; Wade, C.E. Co-catalyst enhancement of enantioselective PTC michael additions involving glycine imines. Tetrahedron Lett. 2009, 50, 3363–3365. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent development and application of chiral phase-transfer catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, Z.-T.; Zheng, Q.-Y. Organic base-promoted enantioselective electrophilic cyanation of β-keto esters by using chiral phase-transfer catalysts. Org. Biomol. Chem. 2015, 13, 8812–8816. [Google Scholar] [CrossRef] [PubMed]
- Siva, A.; Murugan, E. New trimeric cinchona alkaloid-based quaternary ammonium salts as efficient chiral phase transfer catalysts for enantioselective synthesis of α-amino acids. J. Mol. Catal. A Chem. 2006, 248, 1–9. [Google Scholar] [CrossRef]
- Sóti, P.L.; Telkes, L.; Rapi, Z.; Tóth, A.; Vigh, T.; Nagy, Z.K.; Bakó, P.; Marosi, G. Synthesis of an aza chiral crown ether grafted to nanofibrous silica support and application in asymmetric michael addition. J. Inorg. Organomet. Polym. Mater. 2014, 24, 713–721. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yasuda, N. Contemporary asymmetric phase transfer catalysis: Large-scale industrial applications. Org. Process Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- Scorzelli, F.; Di Mola, A.; Palombi, L.; Massa, A. Isoindolinones as michael donors under phase transfer catalysis: Enantioselective synthesis of phthalimidines containing a tetrasubstituted carbon stereocenter. Molecules 2015, 20, 8484–8498. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-G.; Jeong, B.-S.; Yoo, M.-S.; Park, M.-K.; Huh, H.; Jew, S.-S. Trimeric cinchona alkaloid phase-transfer catalyst: A,α′,α″-tris[O(9)-allylcinchonidinium]mesitylene tribromide. Tetrahedron Lett. 2001, 42, 4645–4648. [Google Scholar] [CrossRef]
- Jian, L.; Liping, Z.; Lei, L.; Yongmei, W. A new class of acetophenone-based cinchona alkaloids as phase-transfer catalysts: Application to the enantioselective synthesis of α-amino acids. Chem. Lett. 2007, 36, 1354–1355. [Google Scholar]
- O’Donnell, M.J.; Wu, S.; Huffman, J.C. A new active catalyst species for enantioselective alkylation by phase-transfer catalysis. Tetrahedron 1994, 50, 4507–4518. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Vasques, T.; Ramirez, T.; Plata, R.E.; Wu, J. Pseudoenzymatic catalyst–substrate interactions in ion-pair mediated chiral phase transfer catalysis. Tetrahedron Lett. 2006, 47, 5581–5583. [Google Scholar] [CrossRef]
- Lygo, B.; Andrews, B.I. Asymmetric phase-transfer catalysis utilizing chiral quaternary ammonium salts: Asymmetric alkylation of glycine imines. Acc. Chem. Res. 2004, 37, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Frankish, N.; Sheridan, H.; Farrell, R.; Byrne, W. Indane Dimmer Compounds and Their Pharmaceutical Use. U.S. Patent US 6300376 B1, 9 October 2001. [Google Scholar]
- Cipiciani, A.; Bellezza, F.; Fringuelli, F.; Stillitano, M. Enantioselectivity of alcohol-treated candida rugosa lipase in the kinetic resolution of racemic methyl 2-aryloxypropionates in water and aqueous organic media. Tetrahedron Asymmetry 1999, 10, 4599–4605. [Google Scholar] [CrossRef]
- Berglund, P.; Vallikivi, I.; Fransson, L.; Dannacher, H.; Holmquist, M.; Martinelle, M.; Björkling, F.; Parve, O.; Hult, K. Switched enantiopreference of humicola lipase for 2-phenoxyalkanoic acid ester homologs can be rationalized by different substrate binding modes. Tetrahedron Asymmetry 1999, 10, 4191–4202. [Google Scholar] [CrossRef]
- Moreno, J.; Sinisterra, J. A systematic analysis of the variables that control a highly stereoselective resolution of racemic non-steroidal antiinflammatory drugs using immobilized lipase from candida cylindracea. J. Mol. Catal. A Chem. 1995, 98, 171–184. [Google Scholar] [CrossRef]
- Morrone, R.; Nicolosi, G.; Patti, A.; Piattelli, M. Resolution of racemic flurbiprofen by lipase-mediated esterification in organic solvent. Tetrahedron Asymmetry 1995, 6, 1773–1778. [Google Scholar] [CrossRef]
- Arroyo, M.; Sinisterra, J.V. High enantioselective esterification of 2-arylpropionic acids catalyzed by immobilized lipase from candida antarctica: A mechanistic approach. J. Org. Chem. 1994, 59, 4410–4417. [Google Scholar] [CrossRef]
- Faber, K. Biotransformations in Organic Chemistry, 6th ed.; Springer: Heidelberg, Germany, 2011; pp. 38–43. [Google Scholar]
- Sih, C.J.; Wu, S.-H. Resolution of enantiomers via biocatalysis. In Topics in Stereochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1989; pp. 63–125. [Google Scholar]
- Keith, J.M.; Larrow, J.F.; Jacobsen, E.N. Practical considerations in kinetic resolution reactions. Adv. Synth. Catal. 2001, 343, 5–26. [Google Scholar] [CrossRef]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative analyses of biochemical kinetic resolutions of enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
- Martin, V.S.; Woodard, S.S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K.B. Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity? J. Am. Chem. Soc. 1981, 103, 6237–6240. [Google Scholar] [CrossRef]
- Bredig, G.; Fajans, K. Zur stereochemie der katalyse. Berichte Deutschen Chemischen Gesellschaft 1908, 41, 752–763. [Google Scholar] [CrossRef]
- Straathof, A.J.J.; Rakels, J.L.L.; Heijnen, J.J. Kinetics of the enzymatic resolution of racemic compounds in bi-bi reactions. Biocatalysis 1992, 7, 13–27. [Google Scholar] [CrossRef]
- Bert, J.; van Tol, A.; Jongejan, J.A.; Geerlof, A.; Duine, J.A. Enantioselective enzymatic catalysis.: 2. Applicability of methods for enantiomeric ratio determinations. Recueil Travaux Chimiques Pays-Bas 1991, 110, 255–262. [Google Scholar] [CrossRef]
- Hussain, I.; Bäckvall, J.-E. Chemoenzymatic dynamic kinetic resolution and related dynamic asymmetric transformations. In Enzyme Catalysis in Organic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 1777–1806. [Google Scholar]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | PTC & Conditions | Time | e.e. (Major Enantiomer a) |
|---|---|---|---|
| 1A | N-Bn-quininium chloride, TOL:25% aq. NaOH (5:1) | <2 h | 11% (S) |
| 1B | N-Bn-cinchonidinium chloride, TOL:25% aq. NaOH (5:1) | <2 h | 12% (S) |
| 1C | N-(2-MeO-Bn)-quinidinium bromide, TOL:25% aq. NaOH (5:1) | <2 h | 12% (R) |
| 1D | N-(2-NO2-Bn)-quinidinium bromide, TOL:25% aq. NaOH (5:1) | 6 < t < 20 h | 24% (R) |
| 1E | N,N′-Me2-ephedrinium bromide, TOL:25% aq. NaOH (5:1) | 2 < t < 6 h | 4% (R) b |
| 1F | N-Bn-N′-Me-ephedrinium bromide, TOL:25% aq. NaOH (5:1) | 2 < t < 6 h | 12% (S) |
| 2A | N-(2-NO2-Bn)-quinidinium bromide, TOL/25% aq. NaOH (5:1) | <16 h RT | 22% (R) |
| 2B | N-(2-NO2-Bn)-quinidinium bromide, TOL/12% aq. NaOH (5:1) | <16 h RT | 25% (R) |
| 2C | N-(2-NO2-Bn)-quinidinium bromide, TOL/6% aq. NaOH (5:1) | <16 h RT | 25% (R) |
| 2D | N-(2-NO2-Bn)-quinidinium bromide, TOL/25% aq. K2CO3 (5:1) | 16 h RT + <6 h 70 °C | 34% (R) c |
| 2E | N-(2-NO2-Bn)-quinidinium bromide, TOL/6% aq. KOH (5:1) | <16 h RT | 23% (R) |
| 2F | N-(2-NO2-Bn)-quinidinium bromide, THF/MtBE (1:1)/25% aq. NaOH | <16 h RT | No expected product |
| 2G | N-(2-NO2-Bn)-quinidinium bromide, DCM/25% aq. NaOH (5:1) | <16 h RT | 9% (S) |
| 2H | N-(2-NO2-Bn)-quinidinium bromide, THF/solid NaOH (1:50, v:w) | <16 h RT | No expected product |
| 2I | N-(2-NO2-Bn)-quinidinium bromide, MtBE/25% aq. NaOH (5:1) | <16 h RT | 14% (R) |
| 2J | N-(2-NO2-Bn)-quinidinium bromide, TOL/CyH (1:1), 25% aq. NaOH | <16 h RT | 19% (R) |
| 2K | N-(2-NO2-Bn)-quinidinium bromide, TOL/25% aq. Cs2CO3 (5:1) | >3 h 60 °C | 30% (R) d |
| Entry | PTC Core | -CH2Ar | -OR′ | X− | Time | e.e. (Major Enantiomer a) |
|---|---|---|---|---|---|---|
| 3G | Cinchonine | -CH2(2-NO2-Ph) | -OH | Br− | >16 h b | 32% (R) |
| 3H | Cinchonine | -CH2(2-MeO-Ph) | -OH | Br− | <16 h | 28% (R) |
| 3I | Cinchonine | -CH2(2-CN-Ph) | -OH | Br− | <16 h | 15% (R) |
| 3J | Cinchonine | -CH2(1-Np) | -OH | Cl− | <16 h | 27% (R) |
| 3K | Cinchonine | -CH2(8-Me-1-Np) | -OH | Cl− | <16 h | 42% (R) |
| 3L | Quinidine | -CH2(2-CN-Ph) | -OH | Br− | <16 h | 4% (R) |
| 3M | Quinidine | -CH2(1-Np) | -OH | Cl− | <16 h | 33% (R) |
| 3N | Quinidine | -CH2(2,4-Cl2-Ph) | -OH | Br− | <16 h | 7% (R) |
| 3O | Quinidine | -CH2(2-Pyr) | -OH | Cl− | <16 h | 18% (S) |
| 3P | Cinchonidine | -CH2Ph | -O-allyl | Br− | <16 h | 13% (S) |
| 3Q | Cinchonidine | -CH2(9-anthryl) | -O-allyl | Br− | >16 h b | 7% (S) |
| 3R | Cinchonidine | -CH2(9-anthryl) | -OH | Cl− | <16 h | 41% (S) |
| 4S | Quinine | -CH2(3,5-di(tBu)-Ph) | -OH | Br− | <16 h | 3% (S) |
| 4T | Cinchonidine | -CH2(3,5-di(tBu)-Ph) | -OH | Br− | <16 h | 11% (S) |
| 4U | Quinine | -CH2C(O)(2-NO2-Ph) | -OH | Br− | ~16 h | 33% (S) |
| 4V | Cinchonidine | -CH2C(O)(2-NO2-Ph) | -OH | Br− | >>16 h c | 23% (S) |
| 4W | Quinine | -CH2(3,5-alkaloid+-Ph) d | -OH | Br− | ~16 h | 9% (S) |
| 4X | Cinchonidine | -CH2(3,5-alkaloid+-Ph) d | -OH | Br− | <16 h | 12% (S) |
| 4Y | Quinine | -CH2(4-tBuO2C-Ph) e | -OH | Br− | >16 h | 26% (S) |
| 4Z | Cinchonidine | -CH2(4-tBuO2C-Ph) e | -OH | Br− | <16 h | 35% (S) |
| 5K | Cinchonine | -CH2(8-Me-1-Np) | -OH | Cl− | >16 h | 46% (R) |
| 5R | Cinchonidine | -CH2(9-anthryl) | -OH | Br− | >16 h | 38% (S) |
| Entry | PTC Core | -CH2Ar | -OR′ | e.e. (Major Enantiomer a) | X− | Reference e.e. of Reaction of Ketone 3 |
|---|---|---|---|---|---|---|
| 6D | Quinidine | -CH2(2-NO2-Ph) | -OH | 28% (R) | Br− | 24% (R), 1D |
| 6K | Cinchonine | -CH2(8-Me-1-Np) | -OH | 50% (R) | Cl− | 42% (R), 3K |
| 6R | Cinchonidine | -CH2(9-anthryl) | -OH | 50% (S) | Cl− | 41% (S), 3R |
| 6Z | Cinchonidine | -CH2(4-tBuO2C-Ph) b | -OH | 43% (S) | Br− | 35% (S), 4Z |
| Entry | Hydrolase | e.e. (Substrate) | e.e. (Product) | Major Enantiomer | E | Conversion |
|---|---|---|---|---|---|---|
| 7 | AH-06 | N/A | N/A | Racemic | N/A | Trace |
| 8 | AH-09 | 3% | 20% | S | 1.5 | 16% |
| 9 | AH-24 | N/A | N/A | S | N/A | Trace |
| 10 | AH-46 | 46% | 46% | R | 3 | 17% |
| Entry | Hydrolase | Solvent | e.e. (Substrate) | e.e. (Product) | Major Product | E | Conversion |
|---|---|---|---|---|---|---|---|
| 11 | AH-09 | MtBE | 1% | 2% | S | <1 | 3% |
| 12 | AH-09 | Diethyl ether | 2% | 28% | S | 1.8 | 6% |
| 13 | AH-09 | Pentane | 1% | 8% | S | 1.2 | 9% |
| 14 | AH-09 | Hexane | 1% | 35% | S | 2.1 | 5% |
| 15 | AH-09 | DMSO | 3% | 20% | S | 1.5 | 16% |
| 16 | AH-09 | DMF | - | - | - | - | Trace |
| 17 | AH-09 | Dioxane | - | - | - | - | Trace |
| 18 | AH-46 | MtBE | 9% | 77% | R | 8.4 | 10% |
| 19 | AH-46 | Toluene | 6% | 73% | R | 6.8 | 8% |
| 20 | AH-46 | Pentane | 1% | 7% | R | 1.2 | 9% |
| 21 | AH-46 | Hexane | 1% | 13% | R | 1.3 | 9% |
| 22 | AH-46 | DMSO | 9% | 46% | R | 3 | 17% |
| 23 | AH-46 | Diethyl ether | - | - | - | - | Trace |
| 24 | AH-46 | Dioxane | - | - | - | - | Trace |
| 25 | AH-24 | DMSO | - | - | S | - | Trace |
| 26 | AH-24 | Ethanol | - | - | - | - | Trace |
| 27 | AH-24 | None | - | - | - | - | Trace |
| Code | Hydrolase | Code | Hydrolase |
|---|---|---|---|
| AH-01 | Lipase A from Alcaligenes sp. | AH-25 | Lipase from Rhizopus niveus |
| AH-02 | Lipase B from Alcaligenes sp. | AH-26 | Protease from Bacillus stearothermophilus |
| AH-03 | Lipase C from Alcaligenes sp. | AH-27 | Lipase from Aspergillus niger |
| AH-04 | Lipase from Pseudomonas stutzeri | AH-28 | Lipase from Penicillium roquefort |
| AH-05 | Lipase from Pseudomonas cepacia | AH-29 | Protease from Aspergillus niger |
| AH-06 | Lipase A from Candida rugosa | AH-30 | Lipase from Aspergillus oryzae |
| AH-07 | Lipase D from Alcaligenes sp. | AH-31 | Protease from Aspergillus melleus |
| AH-08 | Lipase E from Alcaligenes sp. | AH-32 | Lipase from Penicillium camembertii |
| AH-09 | Lipase B from Candida rugosa | AH-33 | Protease C from Bacillus subtilis |
| AH-10 | Lipase F from Alcaligenes sp. | AH-34 | Protease B from Aspergillus oryzae |
| AH-11 | Lipase from fungal source | AH-35 | Lipase from Pseudomonas fluorescens |
| AH-12 | Protease A from Bacillus subtilis | AH-36 | Lipase A from Burkholderia cepacia |
| AH-13 | Phytase | AH-37 | Lipase B from Burkholderia cepacia |
| AH-14 | Alkaline protease A | AH-38 | Lipase A from Rhizomucor miehei |
| AH-15 | Alkaline lipase A | AH-39 | Lipase from Candida antarctica |
| AH-16 | Lipase from Bromeliaceae sp. | AH-40 | Lipase from Thermomyces lanuginosus |
| AH-17 | Lipase from Carica papaya | AH-41 | Protease A from Bacillus sp. |
| AH-18 | Neutral protease A | AH-42 | Lipase B from Candida Antarctica (liq) |
| AH-19 | Alkaline protease B | AH-43 | Lipase A from Candida antarctica |
| AH-20 | Acidic protease A | AH-44 | Protease B from Bacillus sp. |
| AH-21 | Protease A from Aspergillus oryzae | AH-45 | Lipase from Thermomyces lanuginosus |
| AH-22 | Protease B from Bacillus subtilis | AH-46 | Lipase C from Rhizomucor miehei |
| AH-23 | Acylase from Aspergillus sp. | AH-47 | Lipase Porcine Pancrease Type II |
| AH-24 | Lipase B from Candida rugosa | AH-48 | Ficin |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Scalabrino, G.; Frankish, N.; Sheridan, H. Bioactive Indanes: Proof of Concept Study for Enantioselective Synthetic Routes to PH46A, a New Potential Anti-Inflammatory Agent. Molecules 2018, 23, 1503. https://doi.org/10.3390/molecules23071503
Zhang T, Scalabrino G, Frankish N, Sheridan H. Bioactive Indanes: Proof of Concept Study for Enantioselective Synthetic Routes to PH46A, a New Potential Anti-Inflammatory Agent. Molecules. 2018; 23(7):1503. https://doi.org/10.3390/molecules23071503
Chicago/Turabian StyleZhang, Tao, Gaia Scalabrino, Neil Frankish, and Helen Sheridan. 2018. "Bioactive Indanes: Proof of Concept Study for Enantioselective Synthetic Routes to PH46A, a New Potential Anti-Inflammatory Agent" Molecules 23, no. 7: 1503. https://doi.org/10.3390/molecules23071503
APA StyleZhang, T., Scalabrino, G., Frankish, N., & Sheridan, H. (2018). Bioactive Indanes: Proof of Concept Study for Enantioselective Synthetic Routes to PH46A, a New Potential Anti-Inflammatory Agent. Molecules, 23(7), 1503. https://doi.org/10.3390/molecules23071503