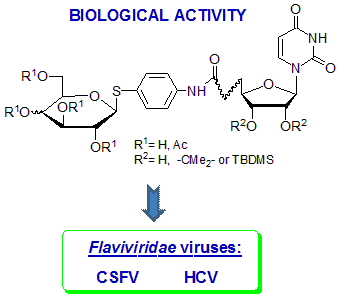
Novel Uridine Glycoconjugates, Derivatives of 4-Aminophenyl 1-Thioglycosides, as Potential Antiviral Compounds

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
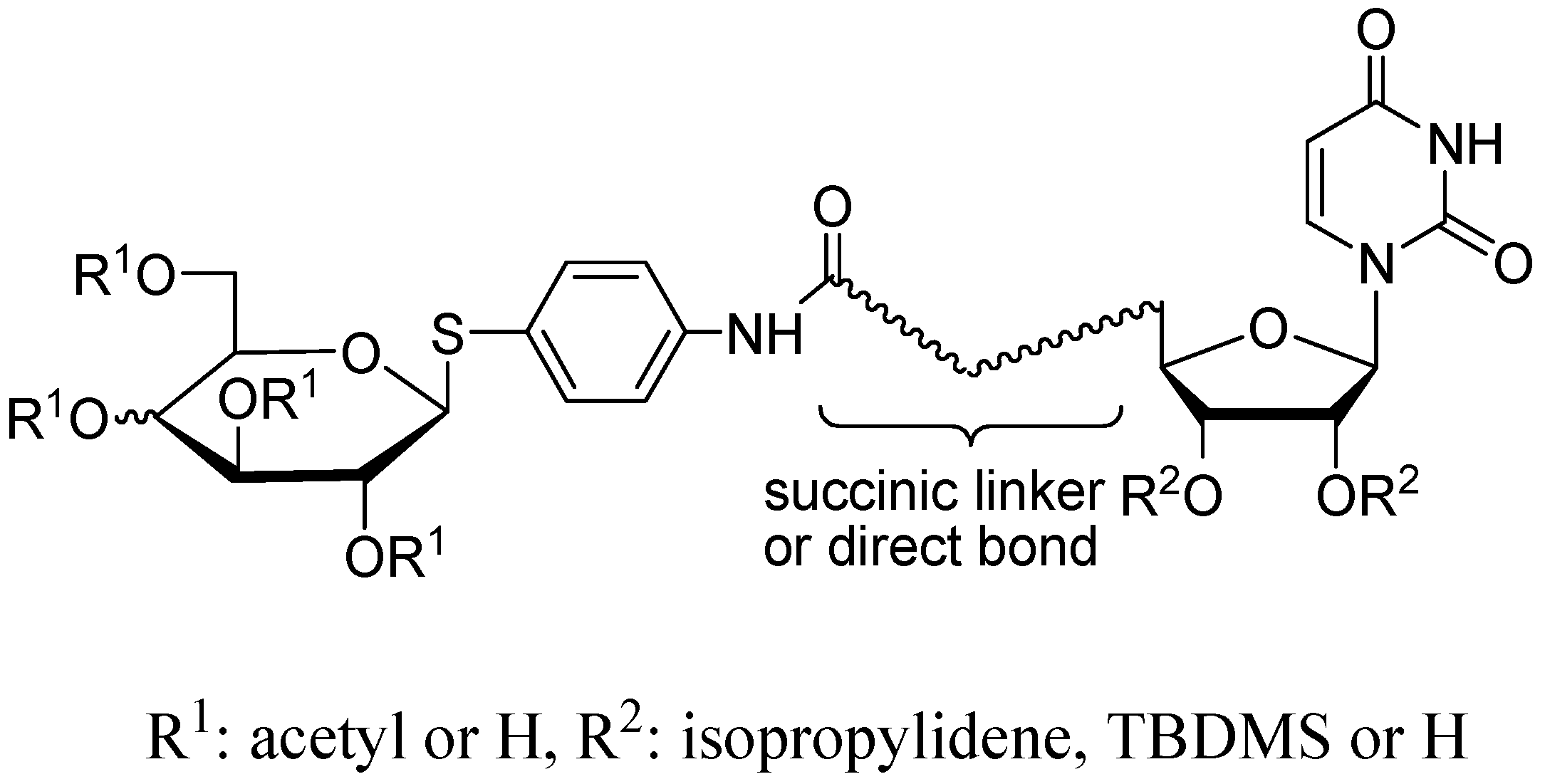
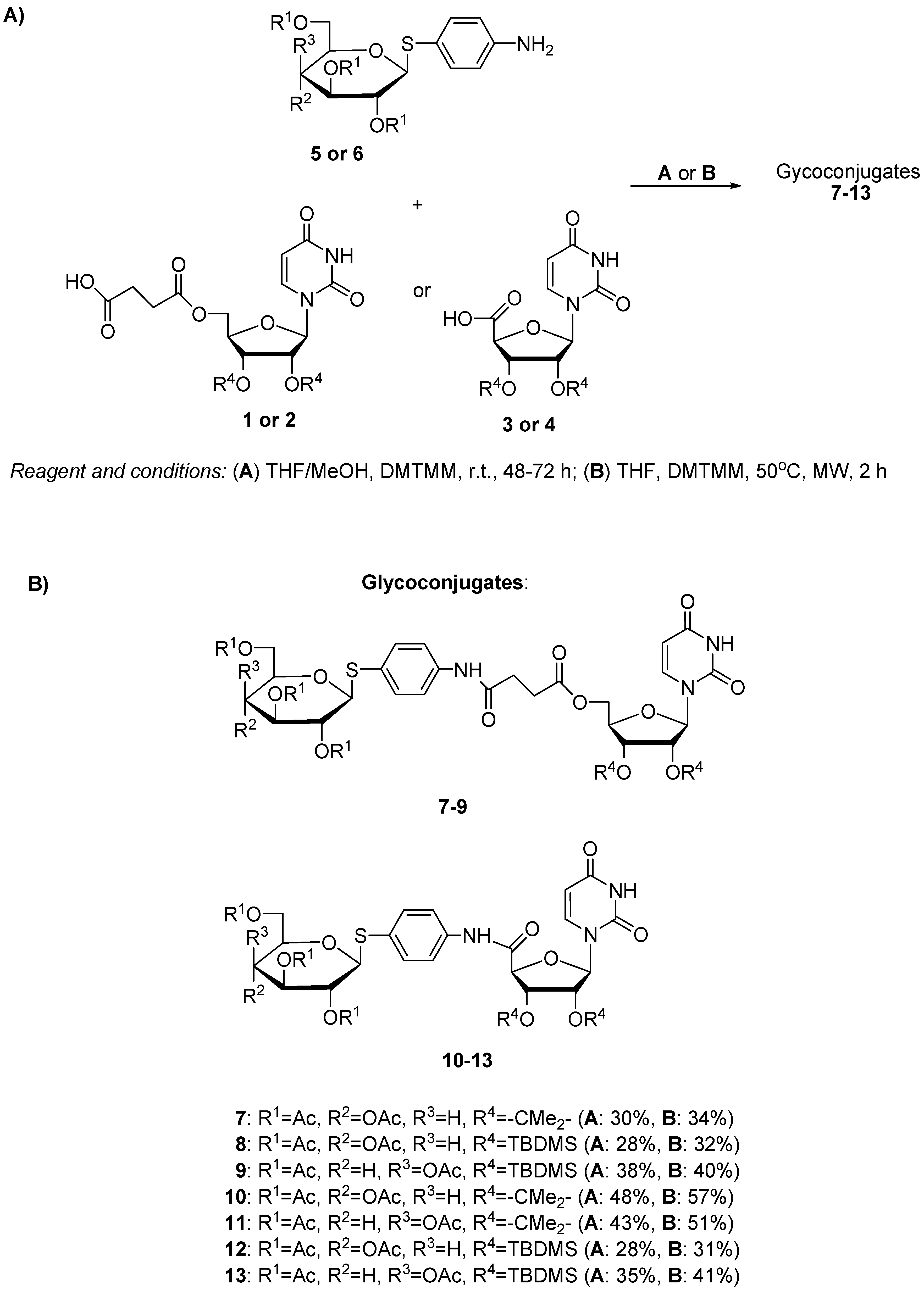
2.1. Chemistry
2.2. Biological Evaluation
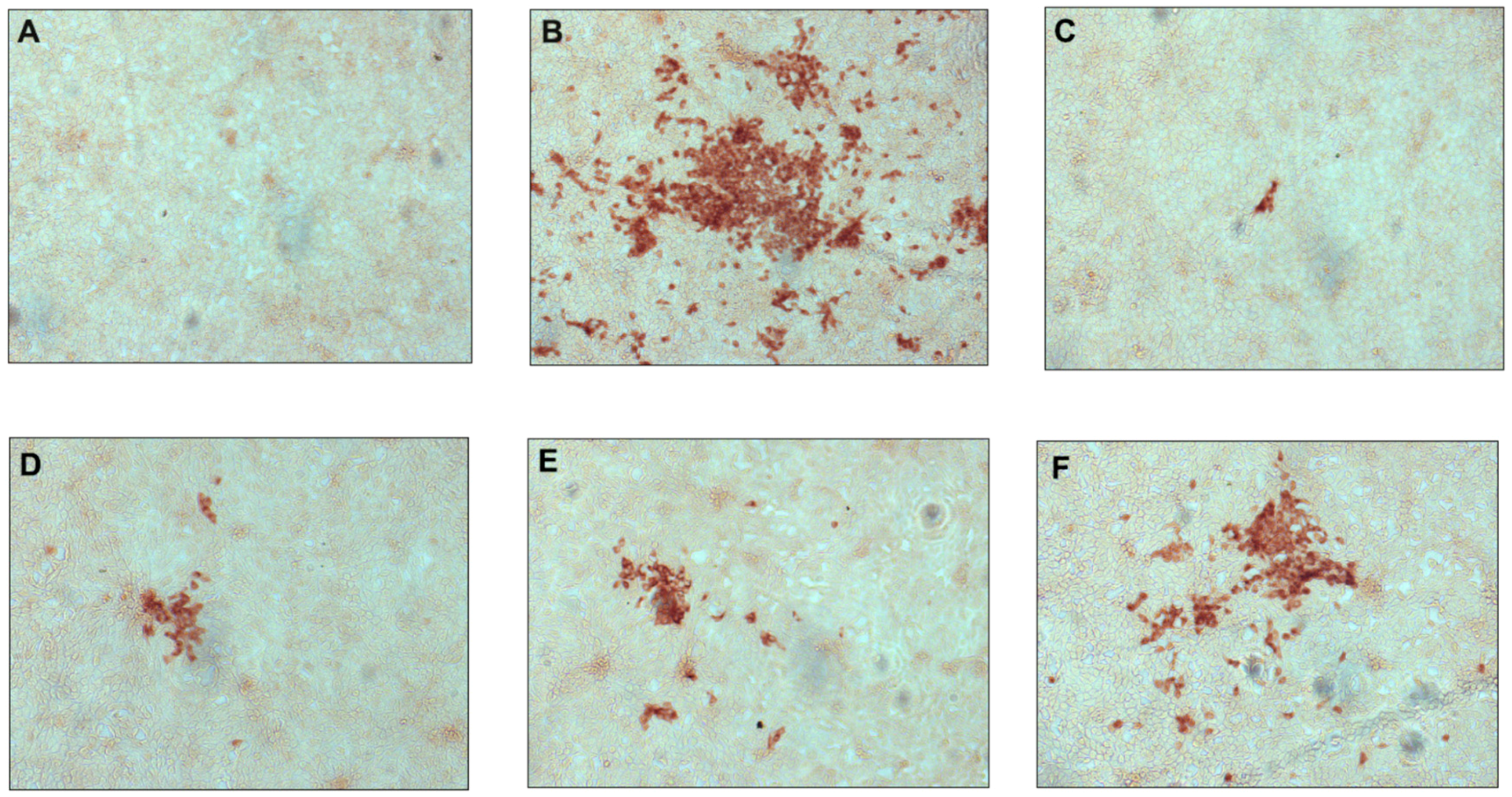
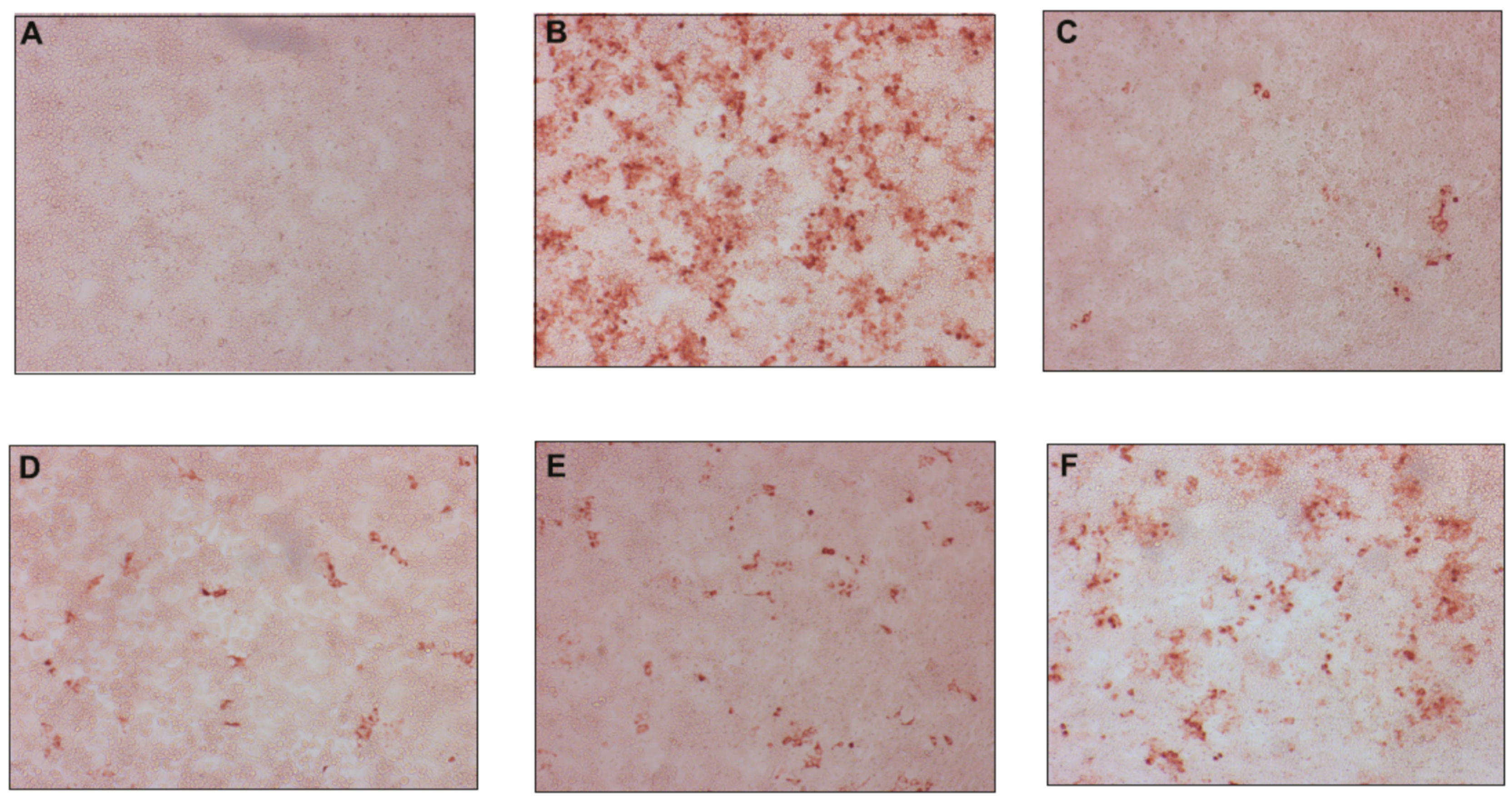
2.2.1. Antiviral Activity against Classical Swine Fever Virus
2.2.2. Antiviral Activity against Hepatitis C Virus
2.2.3. Time-of-Drug Addition Studies
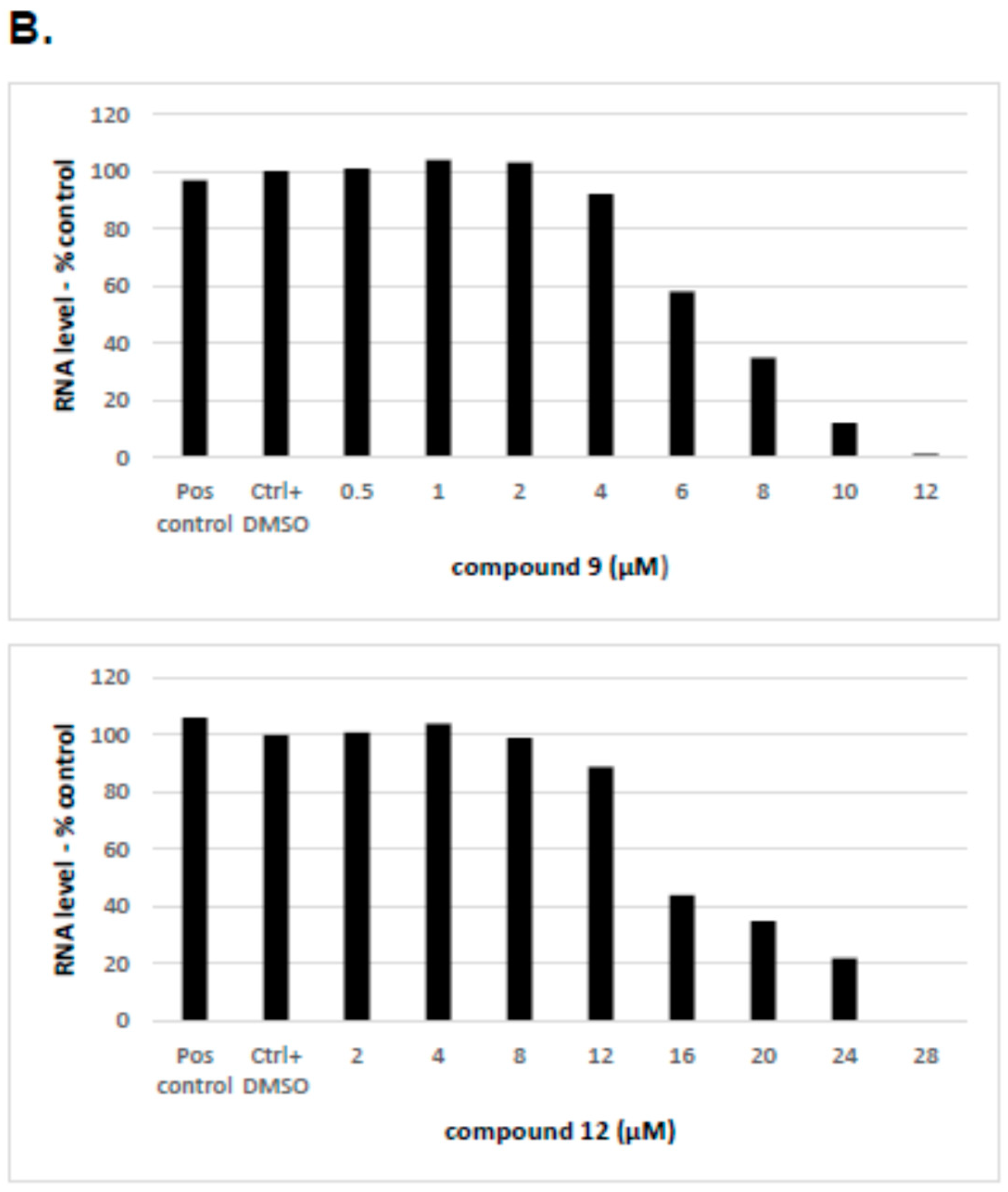
2.2.4. The Inhibitory Effect of Compounds 9 and 12 on HCV Replication
2.2.5. Inhibitory Activity against β-1,4GalT
3. Materials and Methods
3.1. General Experimental Procedures
3.1.1. Synthesis of Succinic Acid Mono-2′,3′-di-O-tert-butyldimethylsilyl-uridin-5′-yl Ester (2)
3.1.2. Synthesis of Glycoconjugates 7–13
Glycoconjugate 7
Glycoconjugate 8
Glycoconjugate 9
Glycoconjugate 10
Glycoconjugate 11
Glycoconjugate 12
Glycoconjugate 13
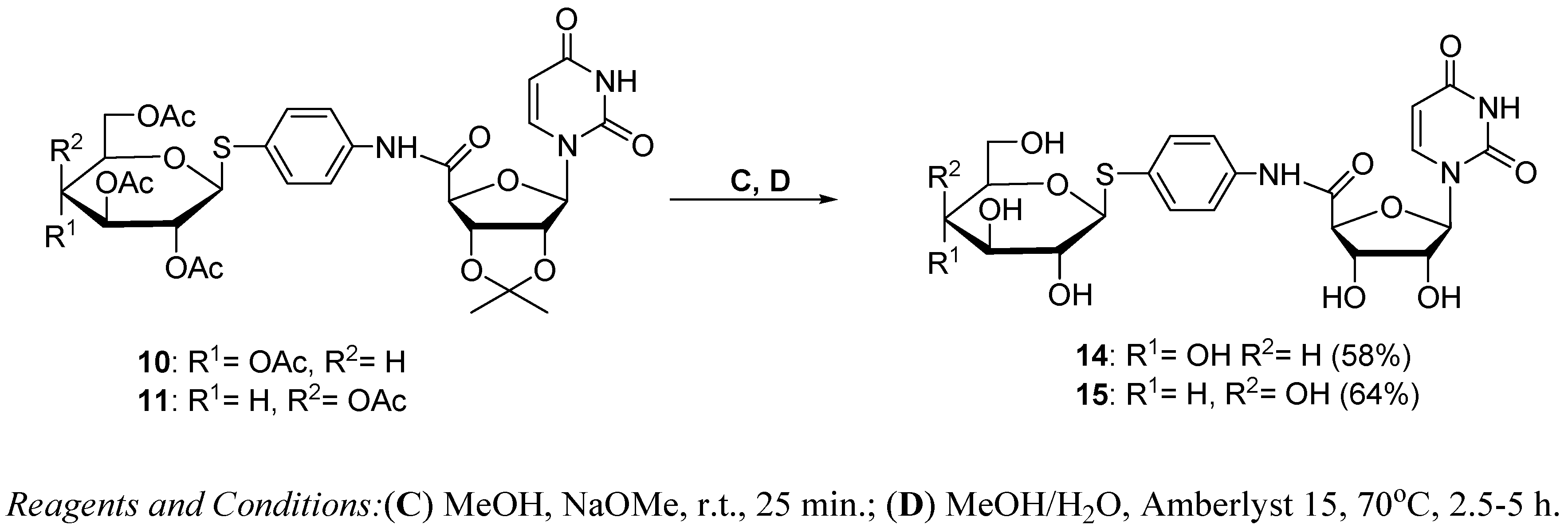
3.1.3. Synthesis of Glycoconjugates 14,15
Glycoconjugate 14
Glycoconjugate 15
3.2. Antiviral Activity
3.2.1. Antiviral Compounds
3.2.2. Cells and Viruses
3.2.3. Cell Viability Assays
3.2.4. CSFV Pseudo-Plaque Reduction Assay
3.2.5. The HCVcc Pseudo-Plaque Reduction Assay
3.2.6. SEAP Reporter Assay
3.2.7. Antiviral Screening Using Replicon Huh7-J17 Cell Line
3.2.8. RNA Inhibition (RT-PCR)
3.2.9. Bovine Milk β-1,4GalT I Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2011, 17, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, E. Pegylated Interferon and Ribavirin Treatment for Hepatitis C Virus Infection. Ther. Adv. Chronic Dis. 2011, 2, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.-M. Hepatitis C Virus: Standard-of-Care Treatment. In Advances in Pharmacology; De Clercq, E., Ed.; Antiviral Agents; Academic Press: Cambridge, MA, USA, 2013; Volume 67, Chapter Five; pp. 169–215. [Google Scholar]
- Migliaccio, G.; Tomassini, J.E.; Carroll, S.S.; Tomei, L.; Altamura, S.; Bhat, B.; Bartholomew, L.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; et al. Characterization of Resistance to Non-obligate Chain-terminating Ribonucleoside Analogs That Inhibit Hepatitis C Virus Replication in Vitro. J. Biol. Chem. 2003, 278, 49164–49170. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.K.; Singh, H.; Grewal, N.; Natt, N.K. Sofosbuvir: A novel treatment option for chronic hepatitis C infection. J. Pharmacol. Pharmacother. 2014, 5, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Bourlière, M.; Oules, V.; Ansaldi, C.; Adhoute, X.; Castellani, P. Sofosbuvir as backbone of interferon free treatments. Dig. Liver Dis. 2014, 46, S212–S220. [Google Scholar] [CrossRef] [PubMed]
- Nkuize, M.; Sersté, T.; Buset, M.; Mulkay, J.-P. Combination ledipasvir-sofosbuvir for the treatment of chronic hepatitis C virus infection: A review and clinical perspective. Ther. Clin. Risk Manag. 2016, 12, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Pol, S.; Corouge, M.; Vallet-Pichard, A. Daclatasvir–sofosbuvir combination therapy with or without ribavirin for hepatitis C virus infection: From the clinical trials to real life. Hepat. Med. Evid. Res. 2016, 8, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Cholongitas, E.; Pipili, C.; Papatheodoridis, G. Interferon-free regimens for the treatment of hepatitis C virus in liver transplant candidates or recipients. World J. Gastroenterol. 2015, 21, 9526–9533. [Google Scholar] [CrossRef] [PubMed]
- Poordad, F.; Khungar, V. Emerging therapeutic options in hepatitis C virus infection. Am. J. Manag. Care 2011, 17 (Suppl. 4), S123–S130. [Google Scholar] [PubMed]
- Asselah, T.; Marcellin, P.; Schinazi, R.F. Treatment of hepatitis C virus infection with direct-acting antiviral agents: 100% cure? Liver Int. 2018, 38, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Barth, H. Hepatitis C virus: Is it time to say goodbye yet? Perspectives and challenges for the next decade. World J. Hepatol. 2015, 7, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-N.; Chen, Y.-H. Marker vaccine strategies and candidate CSFV marker vaccines. Vaccine 2007, 25, 205–230. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Fukusho, A.; Lefèvre, P.C.; Lipowski, A.; Pejsak, Z.; Roehe, P.; Westergaard, J. Classical swine fever: The global situation. Vet. Microbiol. 2000, 73, 103–119. [Google Scholar] [CrossRef]
- Blome, S.; Staubach, C.; Henke, J.; Carlson, J.; Beer, M. Classical Swine Fever—An Updated Review. Viruses 2017, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Stegeman, A.; Elbers, A.; de Smit, H.; Moser, H.; Smak, J.; Pluimers, F. The 1997–1998 epidemic of classical swine fever in the Netherlands. Vet. Microbiol. 2000, 73, 183–196. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Heinz-Jurgen, T.; Lindenbach, B.D.; Rice, C.M. Flaviviridae: The Viruses and Their Replication. In Fields Virology; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2007; 52p. [Google Scholar]
- Breton, C.; Fournel-Gigleux, S.; Palcic, M.M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 2012, 22, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, N.; Honke, K.; Fukuda, M.; Narimatsu, H.; Yamaguchi, Y.; Angata, T. (Eds.) Handbook of Glycosyltransferases and Related Genes, 2nd ed.; Springer: Tokyo, Japan, 2014; ISBN 978-4-431-54239-1. [Google Scholar]
- Hajduch, J.; Nam, G.; Kim, E.J.; Fröhlich, R.; Hanover, J.A.; Kirk, K.L. A convenient synthesis of the C-1-phosphonate analogue of UDP-GlcNAc and its evaluation as an inhibitor of O-linked GlcNAc transferase (OGT). Carbohydr. Res. 2008, 343, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Vaghefi, M.M.; Bernacki, R.J.; Hennen, W.J.; Robins, R.K. Synthesis of certain nucleoside methylenediphosphonate sugars as potential inhibitors of glycosyltransferases. J. Med. Chem. 1987, 30, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Vaghefi, M.M.; Bernacki, R.J.; Dalley, N.K.; Wilson, B.E.; Robins, R.K. Synthesis of glycopyranosylphosphonate analogs of certain natural nucleoside diphosphate sugars as potential inhibitors of glycosyltransferases. J. Med. Chem. 1987, 30, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Bruyère, I.; Malleron, A.; Augé, C.; Praly, J.-P. Non-isosteric C-glycosyl analogues of natural nucleotide diphosphate sugars as glycosyltransferase inhibitors. Bioorg. Med. Chem. 2006, 14, 7293–7301. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Steensma, D.H.; Takaoka, Y.; Yun, J.W.; Kajimoto, T.; Wong, C.H. A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg. Med. Chem. 1997, 5, 661–672. [Google Scholar] [CrossRef]
- Wang, S.; Cuesta-Seijo, J.A.; Lafont, D.; Palcic, M.M.; Vidal, S. Design of glycosyltransferase inhibitors: Pyridine as a pyrophosphate surrogate. Chem. Weinh. Bergstr. Ger. 2013, 19, 15346–15357. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawolek, G.; Bieg, T.; Szeja, W.; Flasz, J. 5-Amino-2-pyridyl 1-thioglycosides in synthesis of analogs of glycosyltransferases substrates. Bioorg. Chem. 2009, 37, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawolek, G.; Chaubey, B.; Szewczyk, B.; Krol, E. Novel thioglycosyl analogs of glycosyltransferase substrates as antiviral compounds against classical swine fever virus and hepatitis C virus. Eur. J. Med. Chem. 2017, 137, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Sehgal, V.; Pradhan, K.L.; Chandna, A.; Gupta, R. Estimation of nickel and chromium in saliva of patients with fixed orthodontic appliances. World J. Orthod. 2008, 9, 196–202. [Google Scholar] [PubMed]
- Wang, S.; Shen, D.L.; Lafont, D.; Vercoutter-Edouart, A.-S.; Mortuaire, M.; Shi, Y.; Maniti, O.; Girard-Egrot, A.; Lefebvre, T.; Pinto, B.M.; et al. Design of glycosyltransferase inhibitors targeting human O-GlcNAc transferase (OGT). MedChemComm 2014, 5, 1172–1178. [Google Scholar] [CrossRef]
- Epp, J.B.; Widlanski, T.S. Facile Preparation of Nucleoside-5′-carboxylic Acids. J. Org. Chem. 1999, 64, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Hwu, J.R.; Jain, M.L.; Tsai, F.-Y.; Tsay, S.-C.; Balakumar, A.; Hakimelahi, G.H. Ceric Ammonium Nitrate on Silica Gel for Efficient and Selective Removal of Trityl and Silyl Groups. J. Org. Chem. 2000, 65, 5077–5088. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawołek, G.; Malarz, K.; Mrozek-Wilczkiewicz, A.; Musioł, M.; Serda, M.; Czaplinska, B.; Musiol, R. Small molecule glycoconjugates with anticancer activity. Eur. J. Med. Chem. 2016, 112, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Krol, E.; Wandzik, I.; Szeja, W.; Grynkiewicz, G.; Szewczyk, B. In vitro antiviral activity of some uridine derivatives of 2-deoxy sugars against classical swine fever virus. Antiviral Res. 2010, 86, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Pastuch-Gawołek, G.; Nidzworski, D.; Rychłowski, M.; Szeja, W.; Grynkiewicz, G.; Szewczyk, B. Synthesis and antiviral activity of a novel glycosyl sulfoxide against classical swine fever virus. Bioorg. Med. Chem. 2014, 22, 2662–2670. [Google Scholar] [CrossRef] [PubMed]
- Laude, H. Hog cholera virus: Art and facts. Ann. Rech. Vet. Ann. Vet. Res. 1987, 18, 127–138. [Google Scholar]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete Replication of Hepatitis C Virus in Cell Culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Micolochick Steuer, H.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and Subtype Profiling of PSI-7977 as a Nucleotide Inhibitor of Hepatitis C Virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef] [PubMed]
- Magri, A.; Ozerov, A.A.; Tunitskaya, V.L.; Valuev-Elliston, V.T.; Wahid, A.; Pirisi, M.; Simmonds, P.; Ivanov, A.V.; Novikov, M.S.; Patel, A.H. Exploration of acetanilide derivatives of 1-(ω-phenoxyalkyl)uracils as novel inhibitors of Hepatitis C Virus replication. Sci. Rep. 2016, 6, 29487. [Google Scholar] [CrossRef] [PubMed]
- Iro, M.; Witteveldt, J.; Angus, A.G.N.; Woerz, I.; Kaul, A.; Bartenschlager, R.; Patel, A.H. A reporter cell line for rapid and sensitive evaluation of hepatitis C virus infectivity and replication. Antivir. Res. 2009, 83, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Angus, A.G.N.; Loquet, A.; Stack, S.J.; Dalrymple, D.; Gatherer, D.; Penin, F.; Patel, A.H. Conserved Glycine 33 Residue in Flexible Domain I of Hepatitis C Virus Core Protein Is Critical for Virus Infectivity. J. Virol. 2012, 86, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Pastuch-Gawolek, G.; Plesniak, M.; Komor, R.; Byczek-Wyrostek, A.; Erfurt, K.; Szeja, W. Synthesis and preliminary biological assay of uridine glycoconjugate derivatives containing amide and/or 1,2,3-triazole linkers. Bioorg. Chem. 2017, 72, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, M.; Kawachi, C.; Hioki, K.; Terao, K.; Tani, S. Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMT-MM. Tetrahedron 2001, 57, 1551–1558. [Google Scholar] [CrossRef]
- Björklund, H.V.; Stadejek, T.; Vilček, Š.; Belák, S. Molecular Characterization of the 3′ Noncoding Region of Classical Swine Fever Virus Vaccine Strains. Virus Genes 1998, 16, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Hierholzer, J.C.; Killington, R.A. Virus isolation and quantitation. In Virology Methods Manual; Mahy, B.W., Kangro, H.O., Eds.; Academic Press: London, UK, 1996; Chapter 2; pp. 25–46. ISBN 978-0-12-465330-6. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 7–15 are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate 1 Amine | Substrate 2 Uridine Deriv. | Product | Procedure | Reaction Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 5 | 1 | 7 | A | 48 | 30 |
| 2 | 5 | 1 | 7 | B | 2 | 34 |
| 3 | 5 | 2 | 8 | A | 48 | 28 |
| 4 | 5 | 2 | 8 | B | 2 | 32 |
| 5 | 6 | 2 | 9 | A | 48 | 38 |
| 6 | 6 | 2 | 9 | B | 2 | 40 |
| 7 | 5 | 3 | 10 | A | 72 | 48 |
| 8 | 5 | 3 | 10 | B | 2 | 57 |
| 9 | 6 | 3 | 11 | A | 72 | 43 |
| 10 | 6 | 3 | 11 | B | 2 | 51 |
| 11 | 5 | 4 | 12 | A | 72 | 28 |
| 12 | 5 | 4 | 12 | B | 2 | 31 |
| 13 | 6 | 4 | 13 | A | 72 | 35 |
| 14 | 6 | 4 | 13 | B | 2 | 41 |
| Compound | CSFV (SK6 Cells) | HCV (Huh-7.5 Cells) | ||||
|---|---|---|---|---|---|---|
| CC50 (μM) a | IC50 (μM) b | SI c | CC50 (μM) a | IC50 (μM) b | SI c | |
| 7 | 103 ± 5.9 | 97 ± 2 | 1.1 | 399 ± 11.4 | 281 ± 4.7 | 1.4 |
| 8 | 42 ± 1.9 | 4.5 ± 0.3 | 9.3 | 16 ± 1.1 | 4 ± 0.2 | 4.0 |
| 9 | 124 ± 6.8 | 4.2 ± 0.5 | 29.5 | 257 ± 5.7 | 4.9 ± 0.3 | 52.4 |
| 10 | 326 ± 11.2 | 149 ± 6.1 | 2.2 | 258 ± 6.2 | 154 ± 2.8 | 1.7 |
| 11 | 340 ± 9.1 | 217 ± 3.9 | 1.6 | 272 ± 4.5 | 167 ± 3.1 | 1.6 |
| 12 | 56 ± 4.3 | 4 ± 0.1 | 14.0 | 270 ± 2.9 | 13.5 ± 0.7 | 20.0 |
| 13 | 49 ± 3.5 | 25 ± 0.9 | 2.0 | 14 ± 0.9 | 7.4 ± 0.2 | 1.9 |
| 14 | 265 ± 8.6 | 241 ± 9.2 | 1.1 | >475 ± 12.5 | 444 ± 12.4 | >1.1 |
| 15 | 278 ± 4.3 | 257 ± 6.9 | 1.1 | 460 ± 9.3 | 454 ± 9.8 | 1.0 |
| I | 86 ± 2.4 | 3 ± 0.1 | 28.7 | 135 ± 2.4 | 7 ± 0.7 | 19.3 |
| II | 151 ± 3.1 | 6 ± 0.4 | 25.2 | 173 ± 3.2 | 7 ± 0.4 | 24.7 |
| SOFOSBUVIR | ND | ND | ND | 31 ± 1.2 | 0.26 ± 0.02 | 119.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krol, E.; Pastuch-Gawolek, G.; Chaubey, B.; Brzuska, G.; Erfurt, K.; Szewczyk, B. Novel Uridine Glycoconjugates, Derivatives of 4-Aminophenyl 1-Thioglycosides, as Potential Antiviral Compounds. Molecules 2018, 23, 1435. https://doi.org/10.3390/molecules23061435
Krol E, Pastuch-Gawolek G, Chaubey B, Brzuska G, Erfurt K, Szewczyk B. Novel Uridine Glycoconjugates, Derivatives of 4-Aminophenyl 1-Thioglycosides, as Potential Antiviral Compounds. Molecules. 2018; 23(6):1435. https://doi.org/10.3390/molecules23061435
Chicago/Turabian StyleKrol, Ewelina, Gabriela Pastuch-Gawolek, Binay Chaubey, Gabriela Brzuska, Karol Erfurt, and Boguslaw Szewczyk. 2018. "Novel Uridine Glycoconjugates, Derivatives of 4-Aminophenyl 1-Thioglycosides, as Potential Antiviral Compounds" Molecules 23, no. 6: 1435. https://doi.org/10.3390/molecules23061435
APA StyleKrol, E., Pastuch-Gawolek, G., Chaubey, B., Brzuska, G., Erfurt, K., & Szewczyk, B. (2018). Novel Uridine Glycoconjugates, Derivatives of 4-Aminophenyl 1-Thioglycosides, as Potential Antiviral Compounds. Molecules, 23(6), 1435. https://doi.org/10.3390/molecules23061435