Secondary Metabolites from the Root Rot Biocontrol Fungus Phlebiopsis gigantea

Abstract
:
1. Introduction
2. Results and Discussion
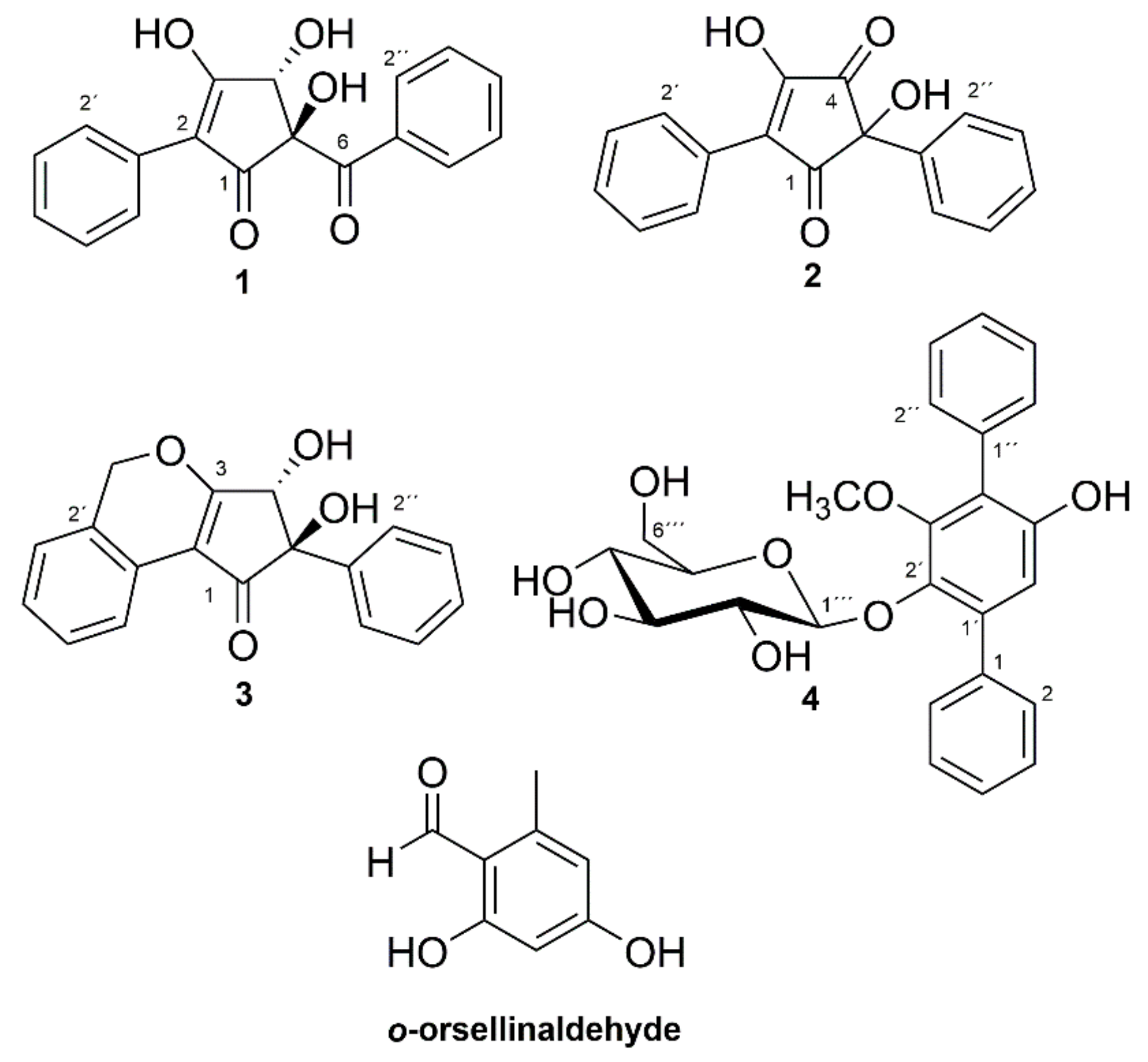
2.1. Isolation of Compounds
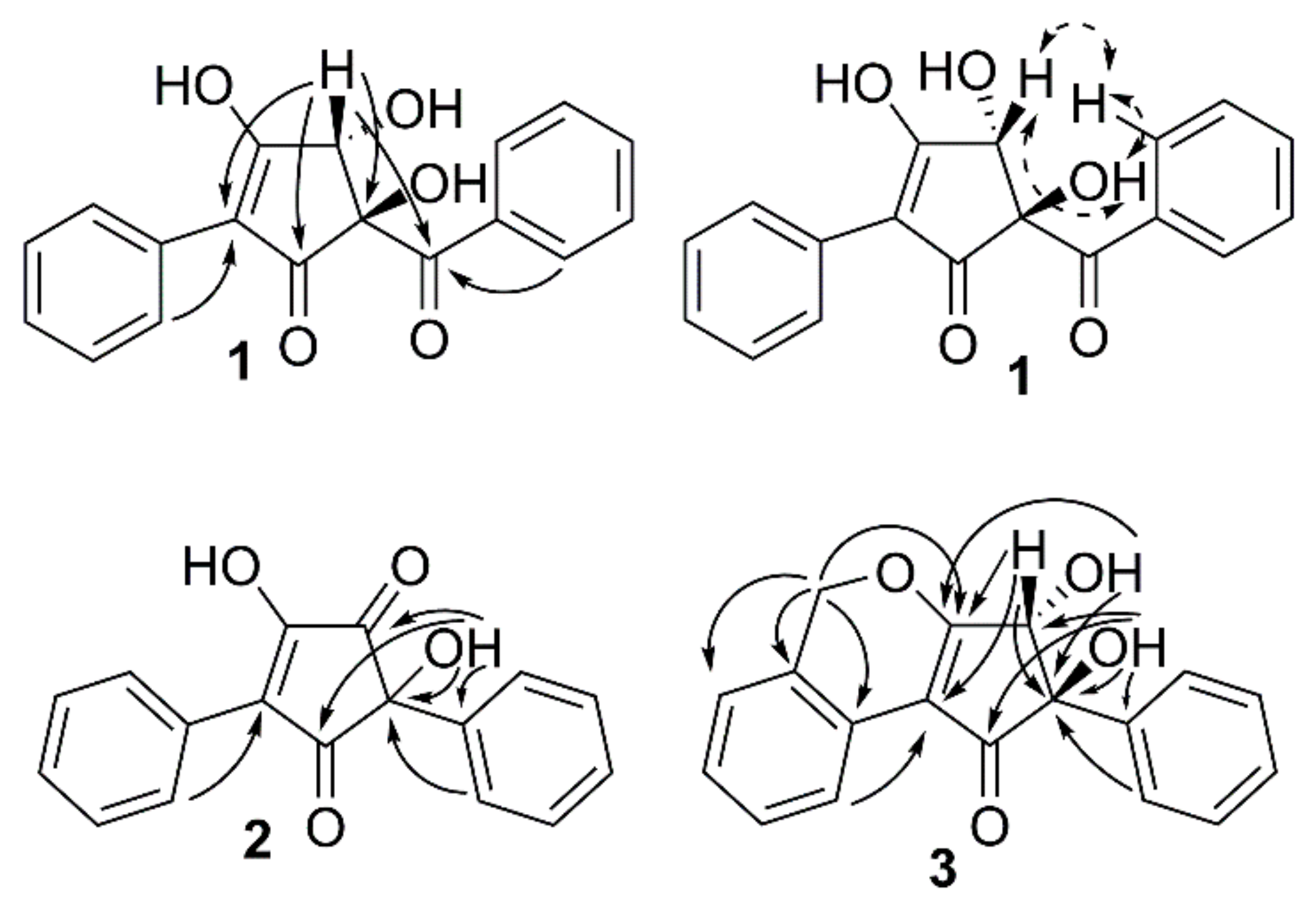
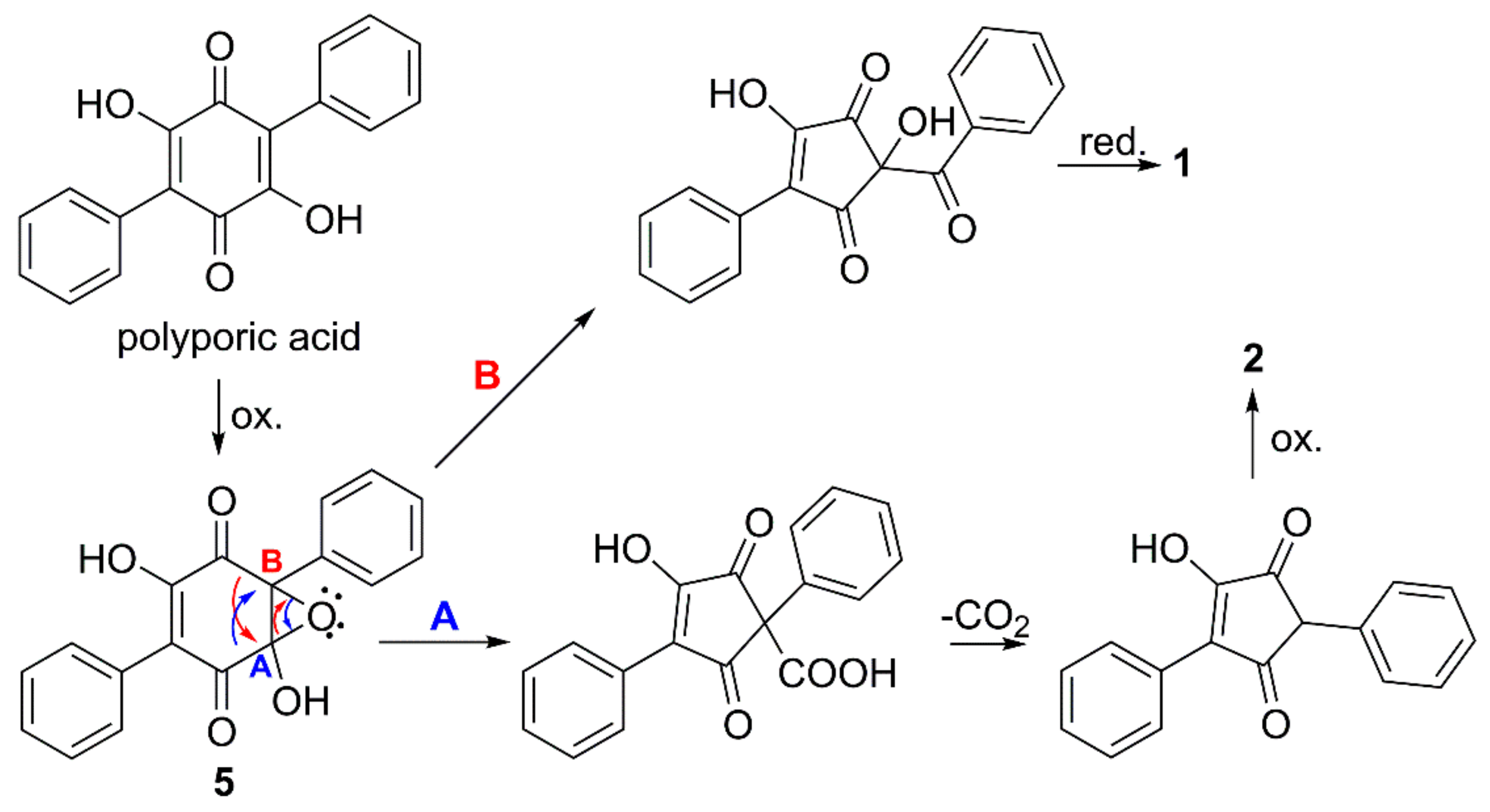
2.2. Structure Determination
2.3. Biological Activities
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Production of Fungal Cultures for Metabolite Isolation
3.3. Production of Microbial Inoculums for Bioassay
3.4. Bioassay Procedure
3.5. Extraction and Isolation
3.6. Relative Configuration of Compound 1
3.7. Absolute Configuration of Compound 4
3.8. Phlebiopsin A (1)
3.9. Phlebiopsin B (2)
3.10. Phlebiopsin C (3)
3.11. Methyl-Terfestatin A (4)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dalman, K.; Olson, Å.; Stenlid, J. Evolutionary history of the conifer root rot fungus Heterobasidion annosum sensu lato. Mol. Ecol. 2010, 19, 4979–4993. [Google Scholar] [CrossRef] [PubMed]
- Redfern, D.B.; Stenlid, J. Spore dispersal and infection. In Heterobasidion annosum, Biology, Ecology, Impact and Control; Woodward, S., Stenlid, J., Karjalainen, R., Hüttermann, A., Eds.; CAB International: Wallingford, UK, 1998; pp. 105–124. [Google Scholar]
- Thor, M.; Stenlid, J. Heterobasidion annosum infection of Picea abies following manual or mechanized stump treatment. Scand. J. For. Res. 2005, 20, 154–164. [Google Scholar] [CrossRef]
- Sun, H.; Paulin, L.; Alatalo, E.; Asiegbu, F.O. Response of living tissues of Pinus sylvestris to the saprotrophic biocontrol fungus Phlebiopsis gigantea. Tree Physiol. 2011, 31, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Adomas, A.; Eklund, M.; Johansson, M.; Asiegbu, F.O. Identification and analysis of differentially expressed cDNAs during nonself-competitive interaction between Phlebiopsis gigantea and Heterobasidion parviporum. FEMS Microbiol. Ecol. 2006, 57, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Briggs, L.H.; Cambie, R.C.; Dean, I.C.; Dromgoole, S.H.; Fergus, B.J.; Ingram, W.B.; Lewis, K.G.; Small, C.W.; Thomas, R.; Walker, D.A. Chemistry of fungi. 10. Metabolites of some fungal species. N. Z. J. Sci. 1975, 18, 565–576. [Google Scholar]
- Liu, J.-K. Natural terphenyls: Developments since 1877. Chem. Rev. 2006, 106, 2209–2223. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.-Z.; Pu, X.; Luo, G.; Zhao, L.-X.; Xu, L.-H.; Li, W.-J.; Luo, Y. Isolation and characterization of new p-terphenyls with antifungal, antibacterial and antioxidant activities from halophilic actinomycete Nocardiopsis gilva YIM 90087. J. Agric. Food Chem. 2013, 61, 3006–3012. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Christensen, S.B.; Cruciani, G.; Kharazmi, A.; Liljefors, T. Antileishmanial chalcones: Statistical design, synthesis and three-dimensional quantitative structure—Activity relationship analysis. J. Med. Chem. 1998, 41, 4819–4832. [Google Scholar] [CrossRef] [PubMed]
- Besl, H.; Bresinsky, A.; Steglich, W.; Zipfel, K. Pigments of fungi. 17. Gyrocyanin, the blueing principle of Gyroporus cyanescens, and an oxidative ring contraction of atromentin. Chem. Ber. 1973, 106, 3223–3229. [Google Scholar] [CrossRef]
- Steglich, W.; Thilmann, A.; Besl, H.; Bresinsky, A. Pigments of fungi. 29. 2,5-diarylcyclopentane-1,3-diones from Chamonixia caespitosa (Basidiomycetes). Zeitschrift für Naturforschung C 1977, 32c, 46–48. [Google Scholar]
- Besl, H.; Bresinsky, A.; Herrmann, R.; Steglich, W. Chamonixin and involutin, two chemosystematically interesting cyclopentanediones from Gyrodon lividus (Boletales). Zeitschrift für Naturforschung C 1980, 35c, 824–825. [Google Scholar]
- Edwards, R.L.; Elsworthy, G.C.; Kale, N. Constituents of the higher fungi. Part IV. Involutin, a diphenylcyclopenteneone from Paxillus involutus (Oeder ex Fries). J. Chem. Soc. 1967, 405–409. [Google Scholar] [CrossRef]
- Edwards, R.L.; Gill, M. Constituents of the higher fungi. Part XII. Identification of involutin as (-)-cis-5-(3,4-dihydroxyphenyl)-3,4-dihydroxy-2-(4-hydroxyphenyl)-cyclopent-2-enone and synthesis of (±)-cis-involutin trimethyl ether from isoxerocomic acid derivatives. J. Chem. Soc. Perkin Trans. 1973, 1, 1529–1537. [Google Scholar] [CrossRef]
- Antkowiak, R.; Antkowiak, W.Z.; Banczyk, I.; Mikolajczyk, L. A new phenolic metabolite, involutone, isolated from the mushroom Paxillus involutus. Can. J. Chem. 2003, 81, 118–124. [Google Scholar] [CrossRef]
- Teuscher, F.; Lin, W.; Wray, V.; Edrada, R.; Padmakumar, K.; Proksch, P.; Ebel, R. Two new cyclopentanoids from the endophytic fungus Aspergillus sydowii associated with the marine alga Acanthophora spicifera. Nat. Prod. Commun. 2006, 1, 927–933. [Google Scholar]
- Yamazoe, A.; Hayashi, K.; Kuboki, A.; Ohira, S.; Nozaki, H. The isolation, structural determination, and total synthesis of terfestatin A, a novel auxin signaling inhibitor from Streptomyces sp. Tetrahedron Lett. 2004, 45, 8359–8362. [Google Scholar] [CrossRef]
- Hayashi, K.; Yamazoe, A.; Ishibashi, Y.; Kusaka, N.; Oono, Y.; Nozaki, H. Active core structure of terfestatin A, a new specific inhibitor of auxin signaling. Bioorg. Med. Chem. 2008, 16, 5331–5344. [Google Scholar] [CrossRef] [PubMed]
- Read, G.; Vining, L.C.; Haskins, R.H. Biogenetic Studies on Volucrisporin. Can. J. Chem. 1962, 40, 2357–2361. [Google Scholar] [CrossRef]
- Braesel, J.; Götze, S.; Shah, F.; Heine, D.; Tauber, J.; Hertweck, C.; Tunlid, A.; Stallforth, P.; Hoffmeister, D. Three Redundant Synthetases Secure Redox-Active Pigment Production in the Basidiomycete Paxillus involutus. Chem. Biol. 2015, 22, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Feling, R.; Polborn, K.; Steglich, W.; Muhlbacher, J.; Bringmann, G. The absolute configuration of the mushroom metabolites involutin and chamonixin. Tetrahedron 2001, 57, 7857–7863. [Google Scholar] [CrossRef]
- Yamazoe, A.; Hayashi, K.; Kepinski, S.; Leyser, O.; Nozaki, H. Characterization of terfestatin A, a new specific inhibitor for auxin signaling. Plant Physiol. 2005, 139, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Hu, H.; Liu, S.; Liu, X.; Zhou, Y.; Che, Y. Bioactive p-terphenyl derivatives from cordysceps-colonizing isolate of Gliocladium sp. J. Nat. Prod. 2007, 70, 1519–1521. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-T.; Liu, W.-H. O-Orsellinaldehyde from the submerged culture of the edible mushroom Grifola frondosa exhibits selective cytotoxic effect against Hep 3B cells through apoptosis. J. Agric. Food Chem. 2006, 54, 7564–7569. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.H. Influence of ectotrophic mycorrhizal fungi on resistance of pine roots to pathogeninc infections. I. Antagonism of mycorrhizal fungi to root pathogenic fungi and soil bacteria. Phytopathology 1969, 59, 153–163. [Google Scholar]
- Gerwig, G.J.; Kamerling, J.P.; Vliegenthart, J.F.G. Determination of the D and L configuration of neutral monosaccharides by high-resolution capillary G.L.C. Carbohydr. Res. 1978, 62, 349–357. [Google Scholar] [CrossRef]
Sample Availability:P. gigantea is available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| pos. | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) |
| 1 | a 190.9, C | - | b 199.8, C | - | 198.7, C | |
| 2 | 117.5, C | - | c 130.6, C | - | 114.1, C | |
| 3 | d n.d. | - | 167.6, C | - | 182.4, C | - |
| 4 | 80.8, CH | 4.76, s | b 199.0, C | - | 78.0, CH | 4.69, d (6.6) |
| 5 | 89.9, C | - | 77.9, C | - | 86.0, C | - |
| 6 | 201.7, C | - | - | - | - | - |
| 1′ | 131.6, C | - | c 130.37, C | - | 127.1, C | - |
| 2′ | 129.3, CH | 7.77, d (7.7) | 130.41, CH | 8.14, d (7.7) | 127.7, C | - |
| 3′ | 129.1, CH | 7.37, t (7.7) | 129.5, CH | 7.47, t (7.6) | 125.0, CH | 7.13, d (7.5) |
| 4′ | 128.4, CH | 7.27, t (7.7) | 130.7, CH | 7.41, t (7.5) | 128.9, CH | 7.24, obsc. |
| 5′ | 129.1, CH | 7.37, t (7.7) | 129.5, CH | 7.47, t (7.6) | 129.3, CH | 7.28, t (7.5) |
| 6′ | 129.3, CH | 7.77, d (7.7) | 130.41, CH | 8.14, d (7.7) | 123.3, CH | 8.11, d (7.5) |
| 1′′ | 138.1, C | - | 138.8, C | - | 141.6, C | - |
| 2′′ | 131.0, CH | 8.01, d (7.8) | 127.1, CH | 7.45, d (7.6) | 128.6, CH | 7.42, d (7.7) |
| 3′′ | 129.0, CH | 7.43, t (7.8) | 129.8, CH | 7.37, t (7.5) | 128.0, CH | 7.23, obsc. |
| 4′′ | 133.7, CH | 7.54, t (7.8) | 129.7, CH | 7.33, t (7.5) | 127.72, CH | 7.17, t (7.3) |
| 5′′ | 129.0, CH | 7.43, t (7.8) | 129.8, CH | 7.37, t (7.5) | 128.0, CH | 7.23, obsc. |
| 6′′ | 131.0, CH | 8.01, d (7.8) | 127.1, CH | 7.45, d (7.6) | 128.6, CH | 7.42, d (7.7) |
| 2′-CH2 | - | - | - | - | 72.6, CH2 | 5.61, d (13.6) |
| 5.50, d (13.6) | ||||||
| 5-OH | e 6.02, br s | e 5.99, br s | 5.45, s | |||
| 4-OH | e 5.18, br s | 4.61, d (6.6) | ||||
| pos. | δC, mult. | δH (J in Hz) | pos. | δC, mult. | δH (J in Hz) |
|---|---|---|---|---|---|
| 1 | 140.0, C | 5′′ | 128.4, CH | 7.43, t (7.5) | |
| 2 | 130.3, CH | 7.62, d (7.5) | 6′′ | 131.5, CH | 7.46, d (7.5) |
| 3 | 128.4, CH | 7.40, t (7.5) | 1′′′ | 104.6, CH | 4.81, d (7.7) |
| 4 | 127.6, CH | 7.32, t (7.5) | 2′′′ | 75.3, CH | 3.24, obsc. |
| 5 | 128.4, CH | 7.40, t (7.5) | 3′′′ | 77.5, CH | 3.33, td (8.8, 3.3) |
| 6 | 130.3, CH | 7.62, d (7.5) | 4′′′ | 71.0, CH | 3.24, obsc. |
| 1′ | 137.3, C | 5′′′ | 76.9, CH | 3.05 , ddd (9.5, 4.7, 3.1) | |
| 2′ | 140.7, C | 6′′′ | 62.3, CH2 | 3.53 , ddd (11.4, 5.8, 3.1) | |
| 3′ | 152.3, C | 3.43, ddd (11.4, 7.3, 4.7) | |||
| 4′ | 124.0, C | 3′-OCH3 | 61.3, CH3 | 3.59, s | |
| 5′ | 151.8, C | 5′-OH | 7.94, s | ||
| 6′ | 112.9, CH | 6.76, s | 2′′′-OH | 4.10, obsc. | |
| 1′′ | 135.0, C | 3′′′-OH | 4.10, obsc. | ||
| 2′′ | 131.5, CH | 7.46, d (7.5) | 4′′′-OH | 4.04, d (3.3) | |
| 3′′ | 128.4, CH | 7.43, t (7.5) | 6′′′-OH | 2.19, dd (7.3, 5.8) | |
| 4′′ | 127.6, CH | 7.34, t (7.5) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kälvö, D.; Menkis, A.; Broberg, A. Secondary Metabolites from the Root Rot Biocontrol Fungus Phlebiopsis gigantea. Molecules 2018, 23, 1417. https://doi.org/10.3390/molecules23061417
Kälvö D, Menkis A, Broberg A. Secondary Metabolites from the Root Rot Biocontrol Fungus Phlebiopsis gigantea. Molecules. 2018; 23(6):1417. https://doi.org/10.3390/molecules23061417
Chicago/Turabian StyleKälvö, David, Audrius Menkis, and Anders Broberg. 2018. "Secondary Metabolites from the Root Rot Biocontrol Fungus Phlebiopsis gigantea" Molecules 23, no. 6: 1417. https://doi.org/10.3390/molecules23061417
APA StyleKälvö, D., Menkis, A., & Broberg, A. (2018). Secondary Metabolites from the Root Rot Biocontrol Fungus Phlebiopsis gigantea. Molecules, 23(6), 1417. https://doi.org/10.3390/molecules23061417