Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
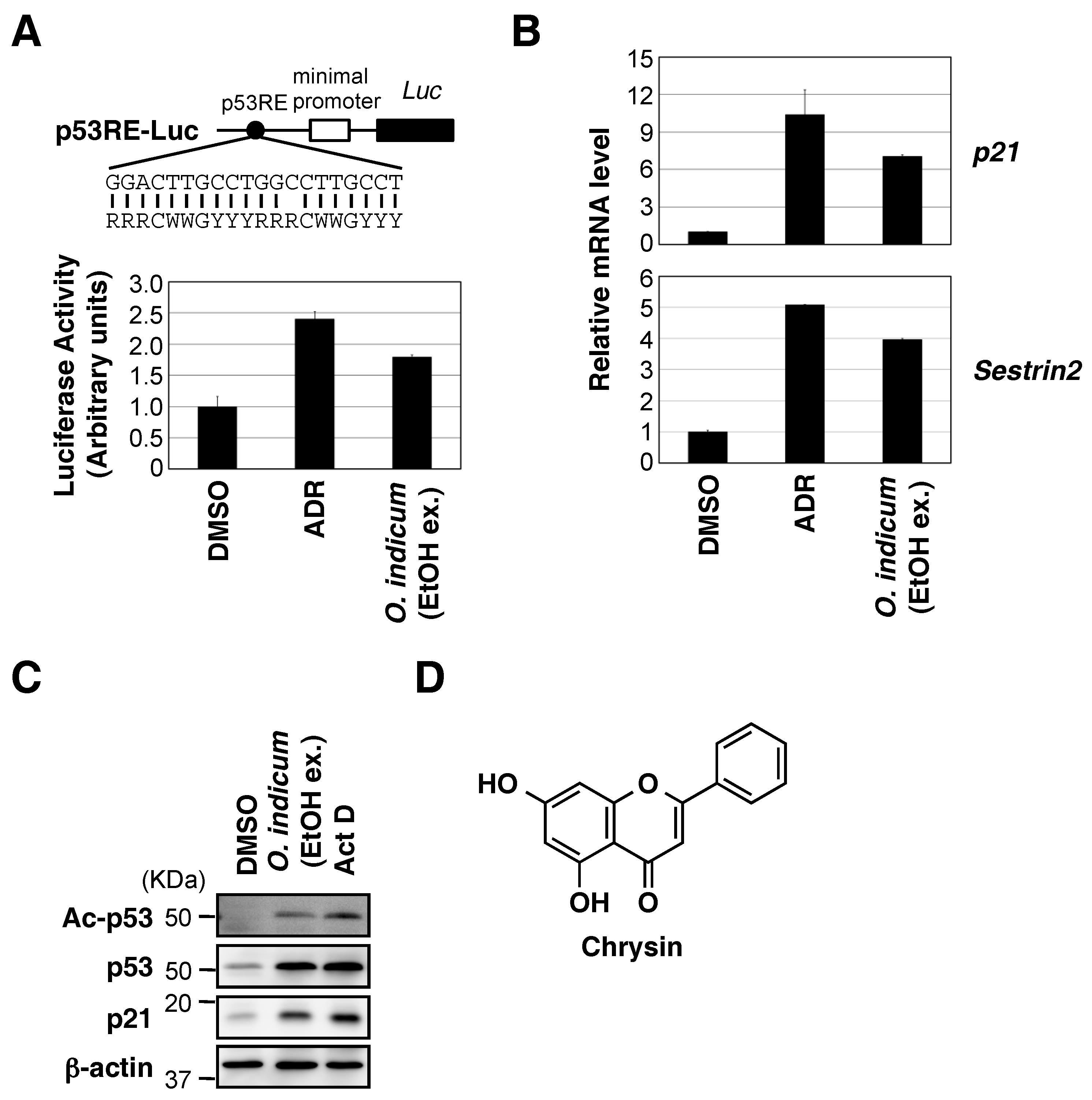
2.1. Ethanol Extract of O. indicum Bark Increased p53 Transcriptional Activity
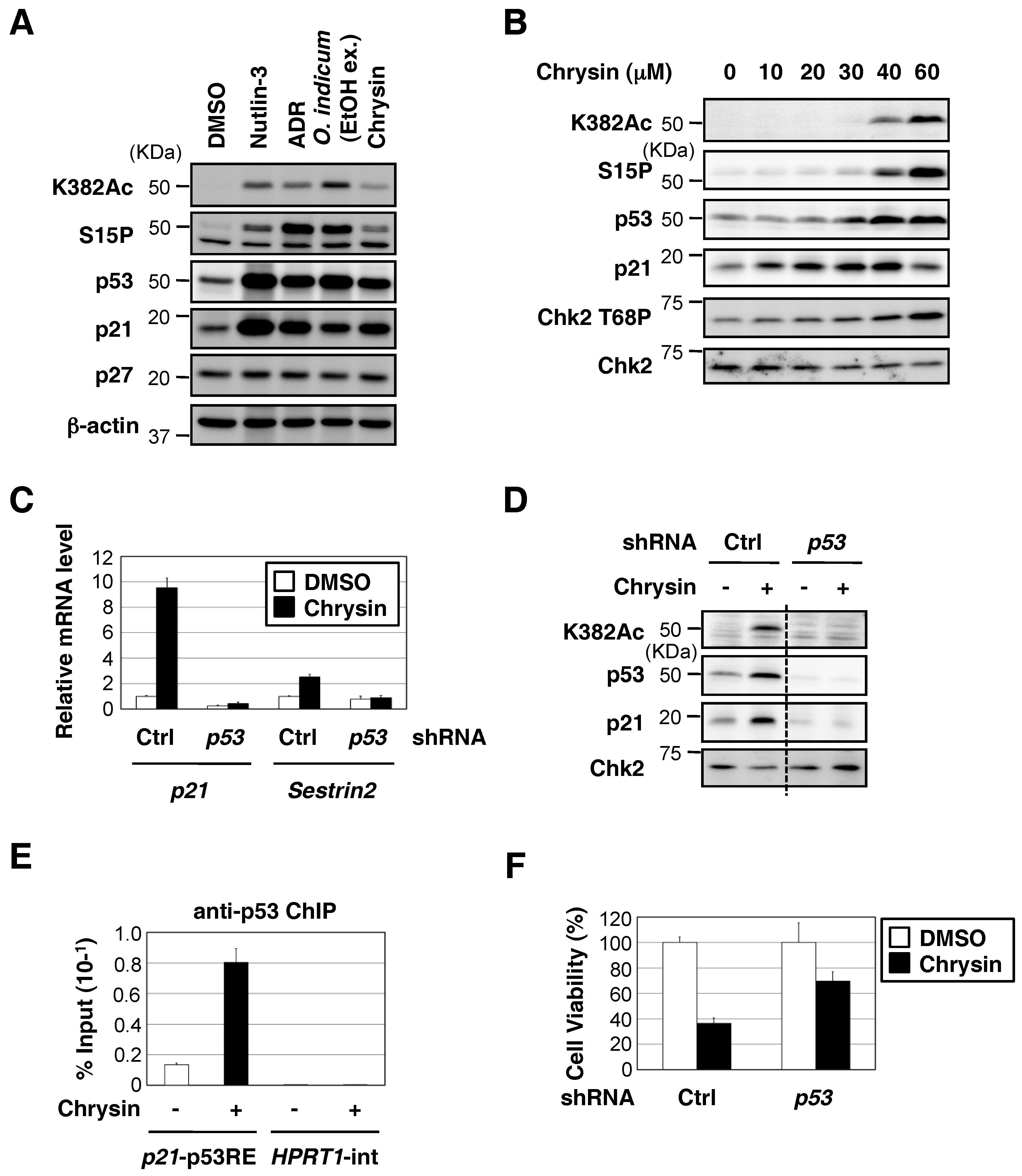
2.2. Chrysin Increased p53 Protein Expression and the p53-Mediated Expression of Downstream Target Genes, and Decreased Cell Viability in a p53-Dependent Manner
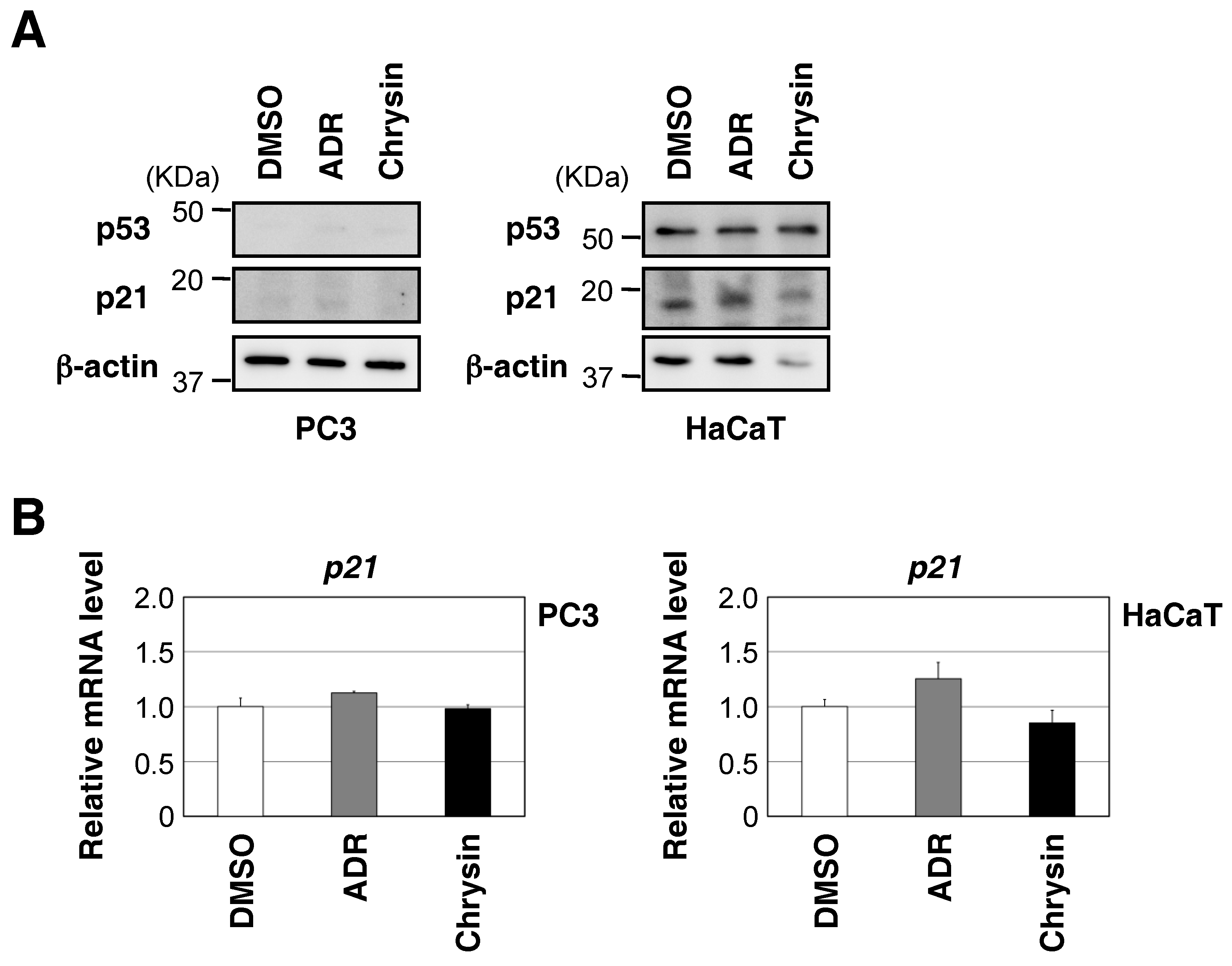
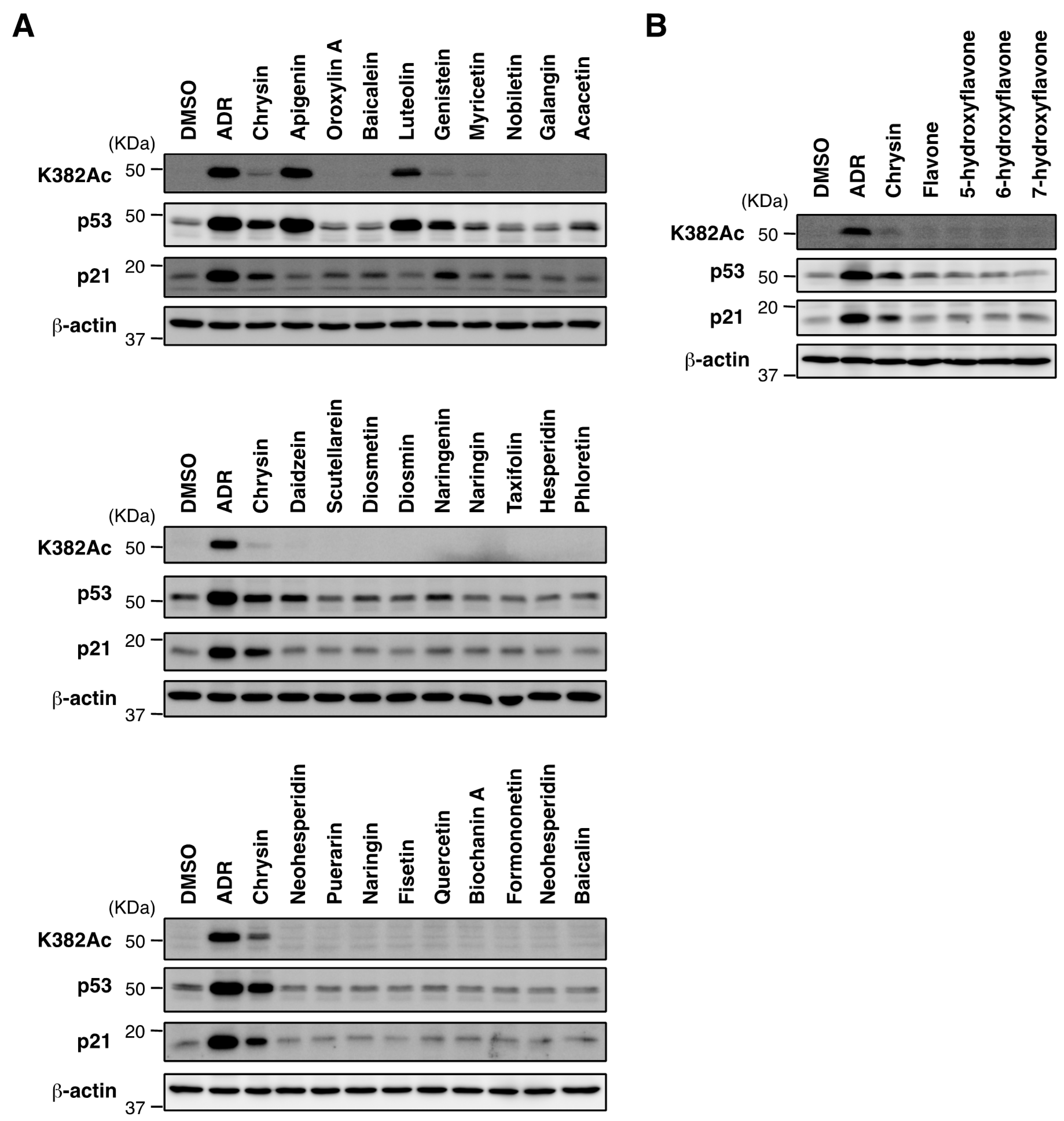
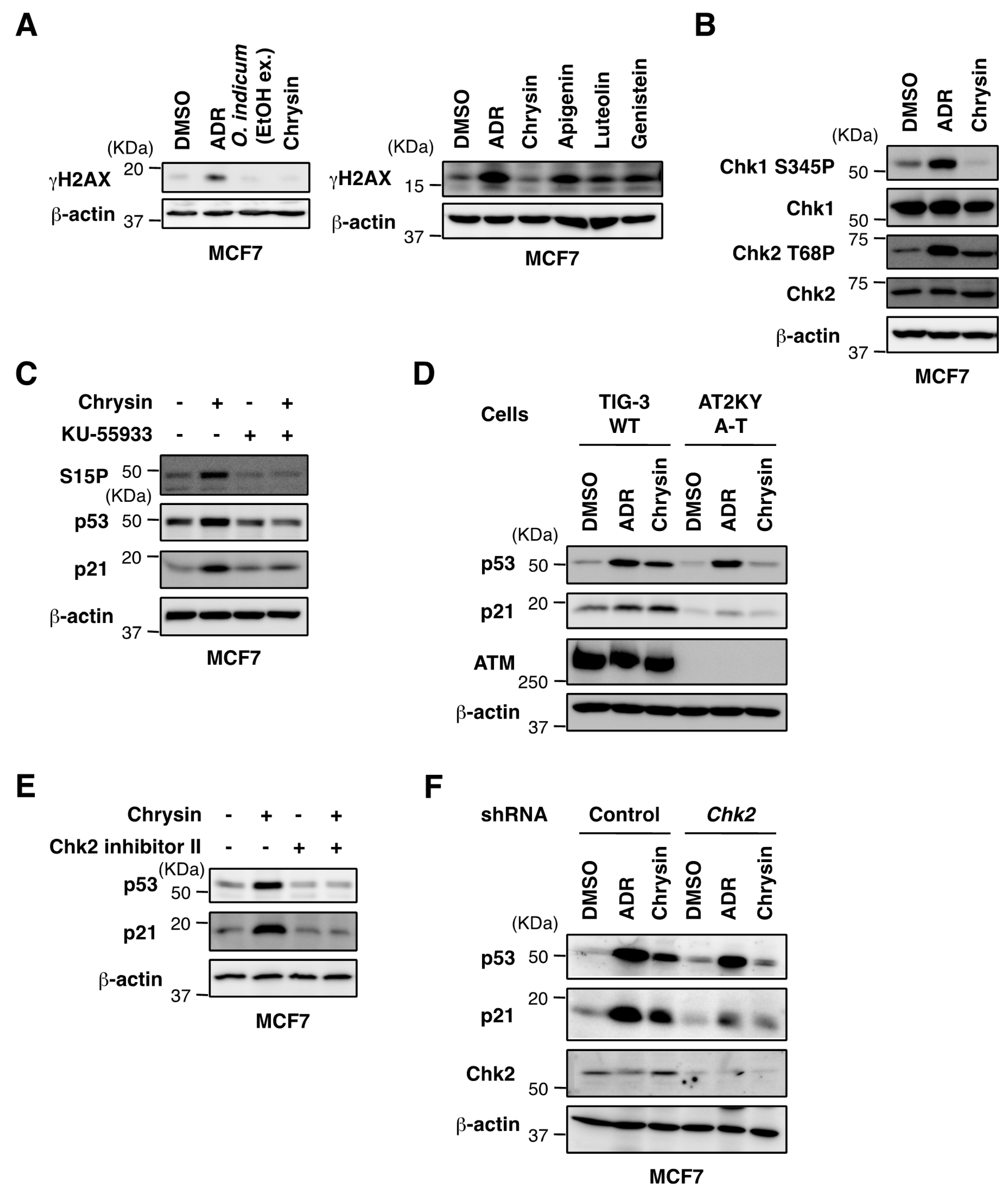
2.3. Biological Evaluation of p53 Activation by Flavonoids in MCF7 Cells
2.4. Chrysin Activated the ATM-Chk2 Pathway in the Absence of DNA Damage
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Plasmids, and RNA Interference
4.2. RNA Extraction, Reverse Transcription, and Quantitative PCR (qPCR)
4.3. Immunochemical Methods and Antibodies
4.4. Reporter Assay
4.5. Chromatin Immunoprecipitation Assay
4.6. Cell Viability Assay
4.7. Myanmar Natural Plant Extract Library
4.8. Plant Material
4.9. Spectroscopic Experimental Procedures
4.10. Extraction and Isolation of Chrysin from O. indicum Bark
4.11. Chemicals
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hainaut, P.; Hollstein, M. P53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Menedez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypothesis, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, V.; Murahari, M.; Khan, T.; Chaubey, P.; Sangave, P. Phytochemicals and PI3K inhibitors in cancer-an insight. Front. Pharmacol. 2017, 8, 916. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Natesan, V. Chrysin: Sources, beneficial pharmacological activities, and molecular mechanism of action. Phytochemistry 2018, 145, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.Y.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Sakamuru, S.; Huang, R.; Teneva, N.; Simmons, S.O.; Xia, M.; Tice, R.R.; Austin, C.P.; Myung, K. High-throughput genotoxicity assay identifies antioxidants as inducers of DNA damage response and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 5423–5428. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell. Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Arienti, K.L.; Brunmark, A.; Axe, F.U.; McClure, K.; Lee, A.; Blevitt, J.; Neff, D.K.; Huang, L.; Crawford, S.; Pandit, C.R.; et al. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J. Med. Chem. 2005, 48, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. DNA damage checkpoint: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. ATM activation and signaling under hypoxic conditions. Mol. Cell. Biol. 2009, 29, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Davatgaran-Taghipour, Y.; Masoomzadeh, S.; Farzaei, M.H.; Bahramsoltani, R.; Karimi-Soureh, Z.; Rahimi, R.; Abdollahi, M. Polyphenol nanoformulations for cancer therapy: Experimental evidence and clinical perspective. Int. J. Nanomed. 2017, 12, 2689–2702. [Google Scholar] [CrossRef] [PubMed]
- Khoo, B.Y.; Chua, S.L.; Balaram, P. Apoptotic effects of chrysin in human cancer cell lines. Int. J. Mol. Sci. 2010, 11, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Bahadori, M.; Baharara, J.; Amini, E. Anticancer properties of chrysin on colon cancer cells, in vitro and in vivo with modulation of Caspase-3, -9, Bax and Sall4. Iran. J. Biothechnol. 2016, 14, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Jin, H.; Pi, J.; Jiang, J.H.; Liu, L.; Bai, H.H.; Yang, P.H.; Cai, J.Y. Anti-tumor activity evaluation of novel chrysin-organogermanium(IV) complex in MCF-7 cells. Bioorg. Med. Chem. Lett. 2013, 23, 5544–5551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, S.; Liu, B.; Liu, J.; Zhu, R.; Li, M. Chrysin induces cell apoptosis via activation of the p53/Bcl-2/caspase-9 pathway in hepatocellular carcinoma cells. Exp. Ther. Med. 2016, 12, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Polier, G.; Köhler, R.; Giaisi, M.; Krammer, P.H.; Li-Weber, M. Wogonin and related natural flavones overcome tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein resistance of tumors by down-regulation of c-FLIP protein and up-regulation of TRAIL receptor 2 expression. J. Biol. Chem. 2012, 287, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, J.M.; Wang, J.N.; Xiong, X.K.; Yang, X.F.; Zou, F. Combination of chrysin and cisplatin promotes the apoptosis of HepG2 cells by up-regulating p53. Chem. Biol. Interact. 2015, 232, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Anari, E.; Akbarzadeh, A.; Zarghami, N. Chrysin-loaded PLGA-PEG nanoparticles designed for enhanced effect on the breast cancer cell line. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Mohammadinejad, S.; Akbarzadeh, A.; Rahmati-Yamchi, M.; Hatam, S.; Kachalaki, S.; Zohreh, S.; Zarghami, N. Preparation and evaluation of chrysin encapsulated in PLGA-PEG nanoparticles in the T47-D breast cancer cell line. Asian Pac. J. Cancer Prev. 2015, 16, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Li, S.; Pu, Y.; Lai, Y.; He, B.; Gu, Z. Nanoparticles generated by PEG-Chrysin conjugates for efficient anticancer drug delivery. Eur. J. Pharm. Biopharm. 2014, 87, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kitagawa, M.; Taya, Y. Phosphorylation of pRB at Ser612 by Chk1/2 leads to a complex between pRB and E2F-1 after DNA damage. EMBO J. 2007, 26, 2083–2093. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kawachi, C.; Ohkubo, T.; Nagasaka, M.; Ito, S.; Fukuura, K.; Itoh, Y.; Ohoka, N.; Morishita, D.; Hayashi, H. The CDK inhibitor p21 is a novel target gene of ATF4 and contributes to cell survival under ER stress. FEBS Lett. 2017, 591, 3682–3691. [Google Scholar] [CrossRef] [PubMed]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-beta induces p53/Smads complex formation in the PAI-1 promoter to active transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef] [PubMed]
- Kern, S.E.; Kinzler, K.W.; Bruskin, A.; Jarosz, D.; Friedman, P.; Prives, C.; Vogelstein, B. Identification of p53 as a sequence-specific DNA-binding protein. Science 1991, 252, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Abe, K.; Onozaki, K.; Hayashi, H. TGF-beta decreases the stability of IL-18-induced IFN-gamma mRNA through the expression of TGF-beta-induced tristetraprolin in KG-1 cells. Biol. Pharm. Bull. 2015, 38, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, C.; Inoue, Y.; Hayashi, H. Pseudokinase tribbles1 (TRB1) negatively regulates tumor-suppressor activity of p53 through p53 deacetylation. Biol. Pharm. Bull. 2015, 38, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Iemura, S.I.; Natsume, T.; Miyazawa, K.; Imamura, T. Suppression of p53 activity through the cooperative action of Ski and histone deacetylase SIRT1. J. Biol. Chem. 2011, 286, 6311–6320. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Parmigiani, A.; Divakaruni, A.S.; Archer, K.; Murphy, A.N.; Budanov, A.V. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci. Rep. 2016, 6, 22538. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Itoh, Y.; Abe, K.; Okamoto, T.; Daitoku, H.; Fukamizu, A.; Onozaki, K.; Hayashi, H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 2017, 26, 500–508. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagasaka, M.; Hashimoto, R.; Inoue, Y.; Ishiuchi, K.; Matsuno, M.; Itoh, Y.; Tokugawa, M.; Ohoka, N.; Morishita, D.; Mizukami, H.; et al. Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway. Molecules 2018, 23, 1394. https://doi.org/10.3390/molecules23061394
Nagasaka M, Hashimoto R, Inoue Y, Ishiuchi K, Matsuno M, Itoh Y, Tokugawa M, Ohoka N, Morishita D, Mizukami H, et al. Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway. Molecules. 2018; 23(6):1394. https://doi.org/10.3390/molecules23061394
Chicago/Turabian StyleNagasaka, Mai, Ryoko Hashimoto, Yasumichi Inoue, Kan’ichiro Ishiuchi, Michiyo Matsuno, Yuka Itoh, Muneshige Tokugawa, Nobumichi Ohoka, Daisuke Morishita, Hajime Mizukami, and et al. 2018. "Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway" Molecules 23, no. 6: 1394. https://doi.org/10.3390/molecules23061394
APA StyleNagasaka, M., Hashimoto, R., Inoue, Y., Ishiuchi, K., Matsuno, M., Itoh, Y., Tokugawa, M., Ohoka, N., Morishita, D., Mizukami, H., Makino, T., & Hayashi, H. (2018). Anti-Tumorigenic Activity of Chrysin from Oroxylum indicum via Non-Genotoxic p53 Activation through the ATM-Chk2 Pathway. Molecules, 23(6), 1394. https://doi.org/10.3390/molecules23061394