
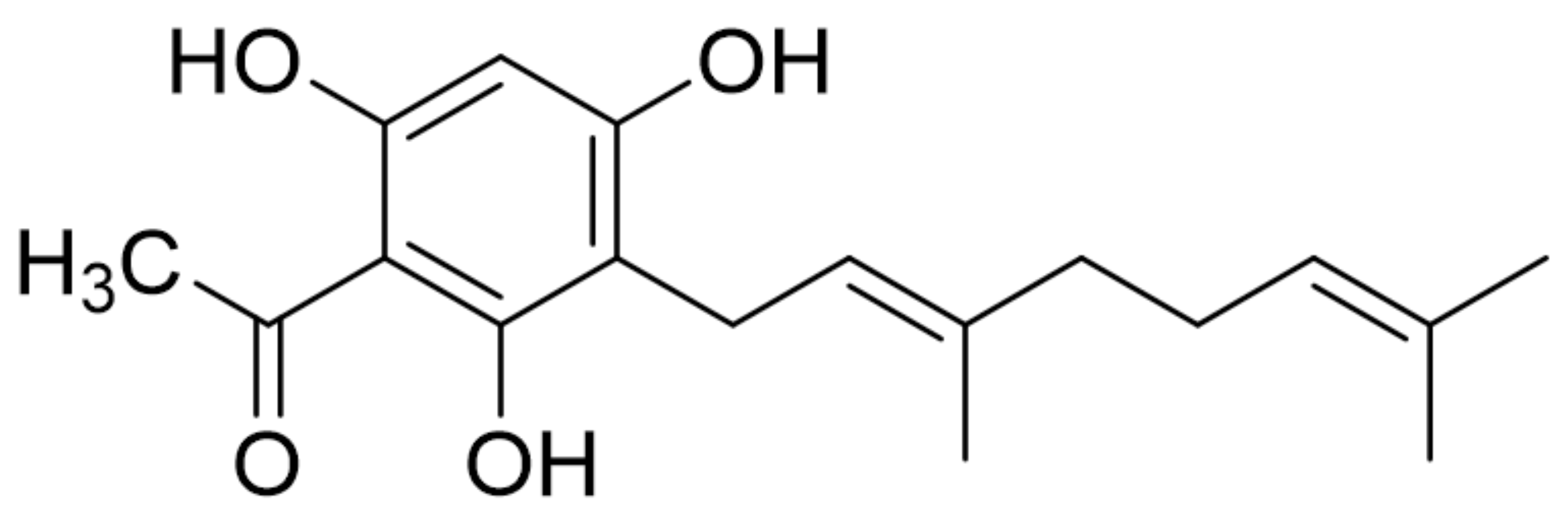
The Protective Effects of a Synthetic Geranyl Acetophenone in a Cellular Model of TNF-α-Induced Pulmonary Epithelial Barrier Dysfunction

 ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. Cytotoxicity
2.2. tHGA Inhibited TNF-α-Induced Monocyte Adhesion and Transepithelial Migration (TEM)
2.3. tHGA Inhibited TNF-α Induced MCP-1 and ICAM-1 Expression and Secretion
2.4. tHGA Reduced Hyperpermeability and Increased Trans-Epithelial Electrical Resistance (TEER) of Epithelium Monolayers
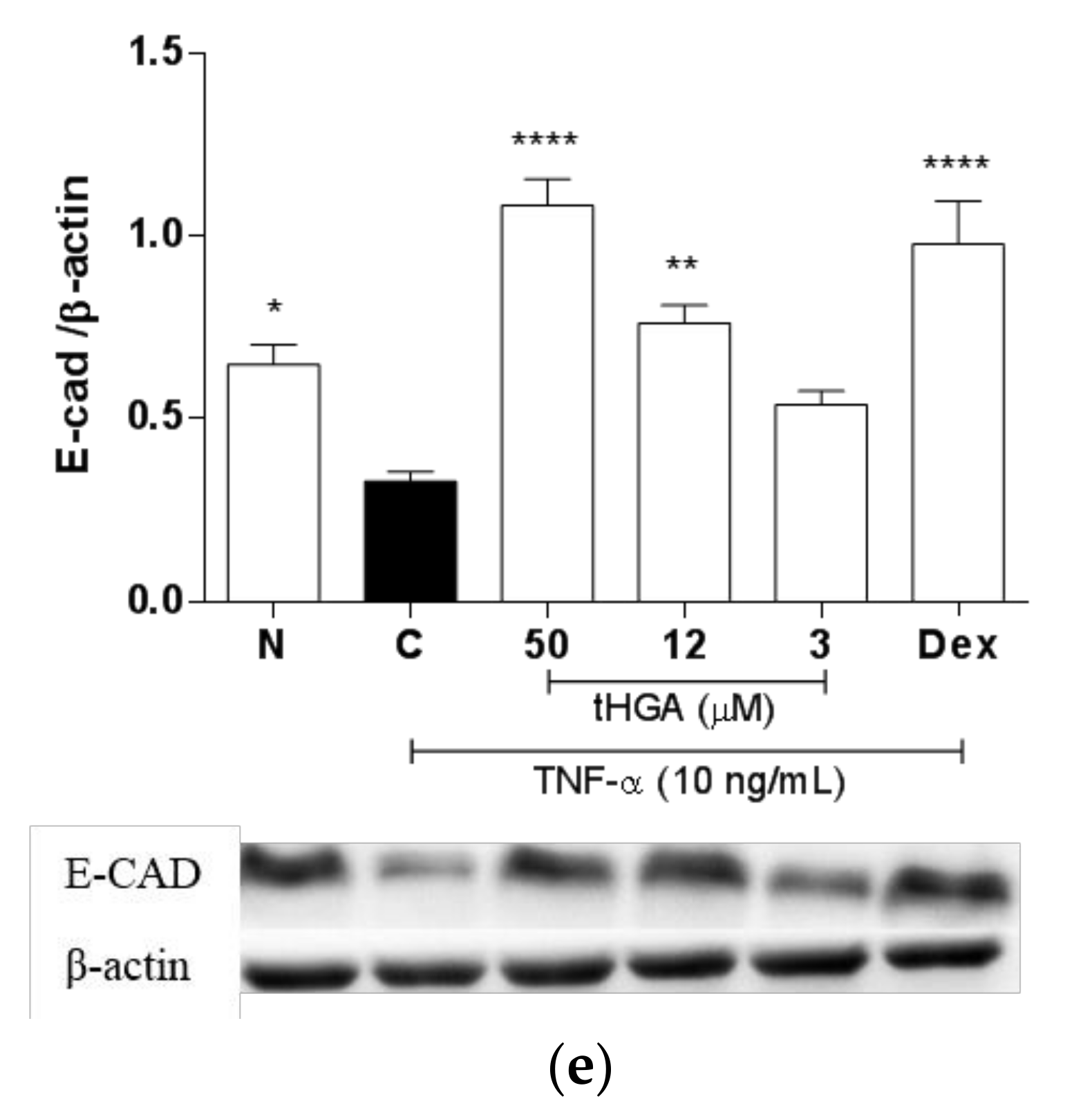
2.5. tHGA Prevented Disruption of Junctional Complexes Molecules
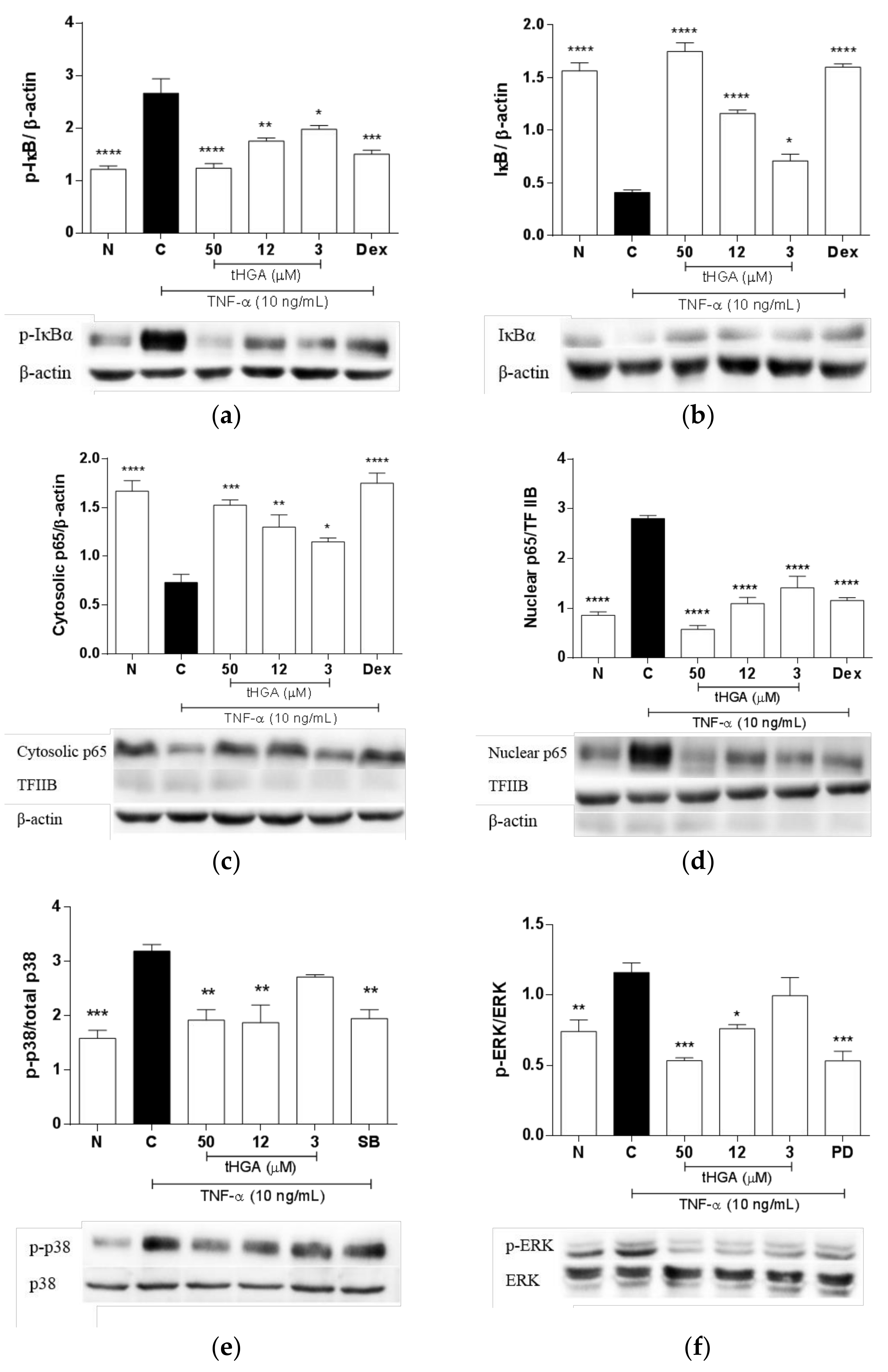
2.6. tHGA Inhibited the Activation of NFκB and MAPK Pathways
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Monocyte Adhesion Assay
4.5. Transepithelial Migration (TEM) Assay
4.6. Cytokine Immunoassay
4.7. Reverse-Transcriptase-Polymerase Chain Reaction (RT-PCR)
4.8. Western Blot Analysis
4.9. Paracellular Permeability Assay
4.10. Transepithelial Electrical Resistance
4.11. Preparation of Whole Cell Lysates and Subcellular Fractions
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wittekindt, O.H. Tight junctions in pulmonary epithelia during lung inflammation. Pflüg. Arch.-Eur. J. Physiol. 2017, 469, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Susanne, H.; Nieves, M.G.; István, V. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L665–L681. [Google Scholar]
- Kieran, B.; James, F.; Andreas, S.; James, F.; Venkataramana, K.S. Pulmonary epithelial barrier function: Some new players and mechanisms. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L731–L745. [Google Scholar]
- Vogelmann, R.; Amieva, M.R.; Falkow, S.; Nelson, W.J. Breaking into the epithelial apical–junctional complex—News from pathogen hackers. Curr. Opin. Cell Biol. 2004, 16, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Manicone, A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2009, 5, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Barravecchia, M.; Kothari, P.; Young, J.L.; Dean, D.A. B1-Na+, k+-ATPase gene therapy upregulates tight junctions to rescue lipopolysaccharide-induced acute lung injury. Gene Ther. 2016, 23, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Rokkam, D.; LaFemina, M.J.; Lee, J.W.; Matthay, M.A.; Frank, J.A. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am. J. Pathol. 2011, 179, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Shaari, K.; Safri, S.; Abas, F.; Lajis, N.H.J.; Israf, D.A. A geranylacetophenone from the leaves of Melicope ptelefolia. Nat. Prod. Res. 2006, 20, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Abas, F.; Lajis, N.H.; Israf, D.A.; Khozirah, S.; Umi Kalsom, Y. Antioxidant and nitric oxide inhibition activities of selected Malay traditional vegetables. Food Chem. 2006, 95, 566–573. [Google Scholar] [CrossRef]
- Shaari, K.; Suppaiah, V.; Wai, L.K.; Stanslas, J.; Tejo, B.A.; Israf, D.A.; Abas, F.; Ismail, I.S.; Shuaib, N.H.; Zareen, S.; et al. Bioassay-guided identification of an anti-inflammatory prenylated acylphloroglucinol from melicope ptelefolia and molecular insights into its interaction with 5-lipoxygenase. Bioorg. Med. Chem. 2011, 19, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Jambari, N.N.; Zareen, S.; Akhtar, M.N.; Shaari, K.; Zamri-Saad, M.; Tham, C.L.; Sulaiman, M.R.; Lajis, N.H.; Israf, D.A. A geranyl acetophenone targeting cysteinyl leukotriene synthesis prevents allergic airway inflammation in ovalbumin-sensitized mice. Toxicol. Appl. Pharmacol. 2012, 259, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.J.; Musa, N.F.; Ng, C.H.; Shaari, K.; Israf, D.A.; Tham, C.L. Barrier protective effects of 2,4,6-trihydroxy-3-geranyl acetophenone on lipopolysaccharides-stimulated inflammatory responses in human umbilical vein endothelial cells. J. Ethnopharmacol. 2016, 192, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Z.; Yap, H.M.; Shaari, K.; Tham, C.L.; Sulaiman, M.R.; Israf, D.A. Blockade of eosinophil-induced bronchial epithelial-mesenchymal transition with a geranyl acetophenone in a coculture model. Front. Pharmacol. 2017, 8, 837. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Israf, D.A.; Harith, H.H.; Md Hashim, N.F.; Ng, C.H.; Shaari, K.; Tham, C.L. Anti-allergic activity of 2,4,6-trihydroxy-3-geranylacetophenone (tHGA) via attenuation of IgE-mediated mast cell activation and inhibition of passive systemic anaphylaxis. Toxicol. Appl. Pharmacol. 2017, 319, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Israf, D.A.; Md Hashim, N.F.; Cheah, Y.K.; Harith, H.H.; Shaari, K.; Tham, C.L. LAT is essential for the mast cell stabilising effect of tHGA in IgE-mediated mast cell activation. Biochem. Pharmacol. 2017, 144, 132–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Z.; Shaari, K.; Cheema, M.S.; Tham, C.L.; Sulaiman, M.R.; Israf, D.A. An orally active geranyl acetophenone attenuates airway remodeling in a murine model of chronic asthma. Eur. J. Pharmacol. 2017, 797, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Rosseau, S.; Selhorst, J.; Wiechmann, K.; Leissner, K.; Maus, U.; Mayer, K.; Grimminger, F.; Seeger, W.; Lohmeyer, J. Monocyte migration through the alveolar epithelial barrier: Adhesion molecule mechanisms and impact of chemokines. J. Immunol. 2000, 164, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chan, S.T.; Yang, T.L.; Tzeng, C.C.; Chen, C.C. Inhibition of ICAM-1 gene expression, monocyte adhesion and cancer cell invasion by targeting IKK complex: Molecular and functional study of novel α-methylene-γ-butyrolactone derivatives. Carcinogenesis 2004, 25, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hwang, J.S.; Woo, C.H.; Kim, E.Y.; Kim, T.H.; Cho, K.J.; Seo, J.M.; Lee, S.S.; Kim, J.H. TNF-α-induced up-regulation of intercellular adhesion molecule-1 is regulated by a rac-ros-dependent cascade in human airway epithelial cells. Exp. Mol. Med. 2008, 40, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–1119. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guan, W.J.; Huang, R.Q.; Xie, Y.Q.; Zheng, J.P.; Zhu, S.X.; Chen, M.; Zhong, N.S. Carbocisteine attenuates TNF-α-induced inflammation in human alveolar epithelial cells in vitro through suppressing NF-κB and ERK1/2 MAPK signaling pathways. Acta Pharmacol. Sin. 2016, 37, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Brazee, P.L.; Soni, P.N.; Tokhtaeva, E.; Magnani, N.; Yemelyanov, A.; Perlman, H.R.; Ridge, K.M.; Sznajder, J.I.; Vagin, O.; Dada, L.A. FXYD5 is an essential mediator of the inflammatory response during lung injury. Front. Immunol. 2017, 8, 623. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Neumann, M.; König, W. Peroxisome proliferator-activated receptor-γ agonists inhibit respiratory syncytial virus-induced expression of intercellular adhesion molecule-1 in human lung epithelial cells. Immunology 2007, 121, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Shi, Y.F.; Yang, J.J.; Hsiao, Y.C.; Tzang, S.; Hsu, T.C. Effects of human parvovirus B19 and Bocavirus VP1 unique region on tight junction of human airway epithelial A549 cells. PLoS ONE 2014, 9, e107970. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Cuzzocrea, S. Role of TNF-α in lung tight junction alteration in mouse model of acute lung inflammation. Respir. Res. 2007, 8, 75–75. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.M.; Williams, A.; Yauk, C.L.; Vincent, R. Overexpression of tumor necrosis factor-α in the lungs alters immune response, matrix remodeling, and repair and maintenance pathways. Am. J. Pathol. 2012, 180, 1413–1430. [Google Scholar] [CrossRef] [PubMed]
- Weifeng, Y.; Li, L.; Yujie, H.; Weifeng, L.; Zhenhui, G.; Wenjie, H. Inhibition of acute lung injury by TNFR-Fc through regulation of an inflammation-oxidative stress pathway. PLoS ONE 2016, 11, e0151672. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; von Wulffen, W.; Steinmueller, M.; Pleschka, S.; Kuziel, W.A.; Mack, M.; Srivastava, M.; Seeger, W.; Maus, U.A.; Lohmeyer, J. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: Impact of chemokines and adhesion molecules. J. Immunol. 2006, 177, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Yen, F.L.; Tsai, M.H.; Yang, C.M.; Liang, C.J.; Lin, C.C.; Chiang, Y.C.; Lee, H.C.; Ko, H.H.; Lee, C.W. Curcumin nanoparticles ameliorate ICAM-1 expression in TNF-α-treated lung epithelial cells through p47 phox and MAPK/AP-1 pathways. PLoS ONE 2013, 8, e63845. [Google Scholar] [CrossRef] [PubMed]
- Yael, G.; Nga Vu, T.Q.; Raphaël, C.; Valérie, U. Lxa4 stimulates ZO-1 expression and transepithelial electrical resistance in human airway epithelial (16hbe14o-) cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L101–L108. [Google Scholar]
- Hardyman, M.A.; Wilkinson, E.; Martin, E.; Jayasekera, N.P.; Blume, C.; Swindle, E.J.; Gozzard, N.; Holgate, S.T.; Howarth, P.H.; Davies, D.E.; et al. TNF-α–mediated bronchial barrier disruption and regulation by src-family kinase activation. J. Allergy Clin. Immunol. 2013, 132, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.C.; Mise, N.; Jia, J.; Genoyer, E.; Yildirim, A.O.; Meiners, S.; Eickelberg, O. Cigarette smoke-induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-beta. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Shang, V.C.M.; Kendall, D.A.; Roberts, R.E. Δ9-tetrahydrocannabinol reverses TNF α-induced increase in airway epithelial cell permeability through cb2 receptors. Biochem. Pharmacol. 2016, 120, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kenneth, J.C.J.; Jane, O.; Susan, S.M. Role of stretch on tight junction structure in alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol. 2001, 25, 584–591. [Google Scholar]
- Hermanns, M.I.; Unger, R.E.; Kehe, K.; Peters, K.; Kirkpatrick, C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: Development of an alveolo-capillary barrier in vitro. Lab. Investig. 2004, 84, 736–752. [Google Scholar] [CrossRef] [PubMed]
- Felix, K.; Hanna, S.; Peter, B.; Veronika, E.W.; Kristin, E.T.; Manfred, F.; Paul, D.; Oliver, H.W. Glucocorticoids regulate tight junction permeability of lung epithelia by modulating claudin 8. Am. J. Respir. Cell Mol. Biol. 2016, 54, 707–717. [Google Scholar]
- Yang, J.; Wang, Y.; Liu, H.; Bi, J.; Lu, Y. C2-ceramide influences alveolar epithelial barrier function by downregulating ZO-1, occludin and claudin-4 expression. Toxicol. Mech. Meth. 2017, 27, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Nam, M.H.; Lee, H.R.; Hong, C.O.; Lee, K.W. Protective effects of chebulic acid on alveolar epithelial damage induced by urban particulate matter. BMC Compl. Altern. Med. 2017, 17, 373. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K.; Smink, J.J.; Leutz, A. Upstream open reading frames: Molecular switches in (patho)physiology. Bioessays 2010, 32, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.W.; Fletcher, S.; Wilton, S.D. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell. Mol. Life Sci. 2012, 69, 3613–3634. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Amon, A. Gene copy-number alterations: A cost-benefit analysis. Cell 2013, 152, 394–405. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.; Cheng, Z.; Vogel, C. Next-generation analysis of gene expression regulation—Comparing the roles of synthesis and degradation. Mol. BioSyst. 2015, 11, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mRNA abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Xu, X.; Fu, J.; Xu, S.; Yue, X.; Yu, Z.; Xue, X. Hyperoxia disrupts pulmonary epithelial barrier in newborn rats via the deterioration of occludin and ZO-1. Respir. Res. 2012, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wang, H.; Wang, L.; Yao, C.; Yuan, R.; Wu, Q. Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab. Investig. 2013, 93, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gu, C.; Wang, Y. Upregulation of the tight junction protein occludin: Effects on ventilation-induced lung injury and mechanisms of action. BMC Pulmonary Med. 2014, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Englert, J.A.; Macias, A.A.; Amador-Munoz, D.; Pinilla Vera, M.; Isabelle, C.; Guan, J.; Magaoay, B.; Suarez Velandia, M.; Coronata, A.; Lee, A. Isoflurane ameliorates acute lung injury by preserving epithelial tight junction integrity. Anesthesiology 2015, 123, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.Y.; Liu, T.J.; Fu, J.H.; Xu, W.; Wu, L.L.; Hou, A.N.; Xue, X.D. Vitamin D/VDR signaling attenuates lipopolysaccharide-induced acute lung injury by maintaining the integrity of the pulmonary epithelial barrier. Mol. Med. Rep. 2016, 13, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Nathalie, C.; Alison, C.; Isabelle, V.; Brigitte, M.; Jean, B.; Philippe, G.; Pascal, C. Modulation of cadherin and catenins expression by tumor necrosis factor- α and dexamethasone in human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2002, 26, 341–347. [Google Scholar]
- Heijink, I.; Brandenburg, S.; Noordhoek, J.; Postma, D.S.; Slebos, D.J.; van Oosterhout, A.J.M. Characterisation of cell adhesion in airway epithelial cell types using electric cell-substrate impedance sensing. Eur. Respir. J. 2009, 35, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Steven, M.E.; David, I.B.; Tony, W.; Shahin, S.; Michael, R.W. Decreased distribution of lung epithelial junction proteins after intratracheal antigen or lipopolysaccharide challenge. Am. J. Respir. Cell Mol. Biol. 2002, 27, 446–454. [Google Scholar]
- Chignalia, A.Z.; Vogel, S.M.; Reynolds, A.B.; Mehta, D.; Dull, R.O.; Minshall, R.D.; Malik, A.B.; Liu, Y. P120-catenin expressed in alveolar type II cells is essential for the regulation of lung innate immune response. Am. J. Pathol. 2015, 185, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—From molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 2005, 1754, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Deng, J.; Gantner, B.N.; Flavell, R.A.; Dong, C.; Christman, J.W.; Ye, R.D. MAP kinase phosphatase 5 protects against sepsis-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L866–L874. [Google Scholar] [CrossRef] [PubMed]
- Yunhe, F.; Bo, L.; Xiaosheng, F.; Fengyang, L.; Dejie, L.; Zhicheng, L.; Depeng, L.; Yongguo, C.; Xichen, Z.; Naisheng, Z. The effect of magnolol on the toll-like receptor 4/nuclear factor kappa b signaling pathway in lipopolysaccharide-induced acute lung injury in mice. Eur. J. Pharmacol. 2012, 689, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Song, Y.; Zhao, W.; Han, T.; Lin, S.; Ramirez, O.; Liang, L. Small interfering RNA targeting NF-κB attenuates lipopolysaccharide-induced acute lung injury in rats. BMC Physiol. 2016, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci. Rep. 2016, 6, 27911. [Google Scholar] [CrossRef] [PubMed]
- Holden, N.S.; Catley, M.C.; Cambridge, L.M.; Barnes, P.J.; Newton, R. ICAM-1 expression is highly NF-kappaB-dependent in A549 cells. No role for ERK and p38 MAPK. Eur. J. Biochem. 2004, 271, 785–791. [Google Scholar] [CrossRef] [PubMed]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 729–756. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Phillips, O.; Fukumoto, J.; Fukumoto, I.; Tamarapu, P.P.; Mandry, M.; Cho, Y.; Lockey, R.; Kolliputi, N. Resolvins decrease oxidative stress mediated macrophage and epithelial cell interaction through decreased cytokine secretion. PLoS ONE 2015, 10, e0136755. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Wang, Y.; Thorlacius, H. Critical role of p38 mitogen-activated protein kinase signaling in septic lung injury. Crit. Care Med. 2008, 36, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Feng, G.; Wang, G.L.; Liu, G.J. P38 MAPK inhibition attenuates LPS-induced acute lung injury involvement of NF-κB pathway. Eur. J. Pharmacol. 2008, 584, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Schuh, K.; Pahl, A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem. Pharmacol. 2009, 77, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Ngamsri, K.C.; Müller, A.; Bösmüller, H.; Gamper-Tsigaras, J.; Reutershan, J.; Konrad, F.M. The pivotal role of CXCR7 in stabilization of the pulmonary epithelial barrier in acute pulmonary inflammation. J. Immunol. 2017, 198, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Nurit, D.; Brian, C.D.; Gladys, G.L.; Peter, C.; Nadir, Y.; Susan, S.M. Cyclic stretch–induced oxidative stress increases pulmonary alveolar epithelial permeability. Am. J. Respir. Cell Mol. Biol. 2013, 49, 156–164. [Google Scholar]
- Ward, C.; Schlingmann, B.; Stecenko, A.A.; Guidot, D.M.; Koval, M. NF-κB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 2015, 3, e982424. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.S.; Gray, L.G.; Margulies, S.S. Cultured alveolar epithelial cells from septic rats mimic in vivo septic lung. PLoS ONE 2010, 5, e11322. [Google Scholar] [CrossRef] [PubMed]
- Petecchia, L.; Sabatini, F.; Usai, C.; Caci, E.; Varesio, L.; Rossi, G.A. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Investig. 2012, 92, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Peerapen, P.; Thongboonkerd, V. p38 MAPK mediates calcium oxalate crystal-induced tight junction disruption in distal renal tubular epithelial cells. Sci. Rep. 2013, 3, 1041. [Google Scholar] [CrossRef] [PubMed]
- Enjoji, S.; Ohama, T.; Sato, K. Regulation of epithelial cell tight junctions by protease-activated receptor 2. J. Vet. Med. Sci. 2014, 76, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Qing, Q.; Zhang, S.; Chen, Y.; Li, R.; Mao, H.; Chen, Q. High glucose-induced intestinal epithelial barrier damage is aggravated by syndecan-1 destruction and heparanase overexpression. J. Cell. Mol. Med. 2015, 19, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Jiao, L.; Cao, S.; Song, Z.; Hu, C.; Han, X. Whey protein concentrate enhances intestinal integrity and influences transforming growth factor-beta1 and mitogen-activated protein kinase signalling pathways in piglets after lipopolysaccharide challenge. Br. J. Nutr. 2016, 115, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.J.; Lee, Y.L.; Shih, N.Y.; Yang, Y.Y.; Charoenfuprasert, S.; Dai, Y.S.; Chang, S.M.; Tsai, Y.H.; Tseng, H.; Liu, C.Y. Reduction of monocyte chemoattractant protein-1 and interleukin-8 levels by ticlopidine in TNF-alpha stimulated human umbilical vein endothelial cells. J. Biomed. Biotechnol. 2009, 2009, 917837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Fisher, K. Tight junction-related barrier contributes to the electrophysiological asymmetry across vocal fold epithelium. PLoS ONE 2012, 7, e34017. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound are available from Daud Ahmad Israf. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequences (5′-3′) | Product Size (bp) |
|---|---|---|
| MCP-1 | forward-GCTCATAGCAGCCACCTTCATTC | 297 |
| reverse-TGCAGATTCTTGGGTTGTGGAG | ||
| ZO-1 | forward-AGAAGATAGCCCTGCAGC | 252 |
| reverse–AGTCCATAGGGAGATTCC | ||
| Occludin | forward–GATCAGGAATATCCACC | 199 |
| reverse–ATTGTACTCGTCAGCAGC | ||
| GAPDH | forward-GCCATCGACCCCTTCATTGAC | 606 |
| reverse-ACGGAAGGACATGCCAGTGAGCTT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, T.Y.; Harith, H.H.; Tham, C.L.; Md Hashim, N.F.; Shaari, K.; Sulaiman, M.R.; Israf, D.A. The Protective Effects of a Synthetic Geranyl Acetophenone in a Cellular Model of TNF-α-Induced Pulmonary Epithelial Barrier Dysfunction. Molecules 2018, 23, 1355. https://doi.org/10.3390/molecules23061355
Sim TY, Harith HH, Tham CL, Md Hashim NF, Shaari K, Sulaiman MR, Israf DA. The Protective Effects of a Synthetic Geranyl Acetophenone in a Cellular Model of TNF-α-Induced Pulmonary Epithelial Barrier Dysfunction. Molecules. 2018; 23(6):1355. https://doi.org/10.3390/molecules23061355
Chicago/Turabian StyleSim, Tee Yee, Hanis Hazeera Harith, Chau Ling Tham, Nur Fariesha Md Hashim, Khozirah Shaari, Mohd Roslan Sulaiman, and Daud Ahmad Israf. 2018. "The Protective Effects of a Synthetic Geranyl Acetophenone in a Cellular Model of TNF-α-Induced Pulmonary Epithelial Barrier Dysfunction" Molecules 23, no. 6: 1355. https://doi.org/10.3390/molecules23061355
APA StyleSim, T. Y., Harith, H. H., Tham, C. L., Md Hashim, N. F., Shaari, K., Sulaiman, M. R., & Israf, D. A. (2018). The Protective Effects of a Synthetic Geranyl Acetophenone in a Cellular Model of TNF-α-Induced Pulmonary Epithelial Barrier Dysfunction. Molecules, 23(6), 1355. https://doi.org/10.3390/molecules23061355