
O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
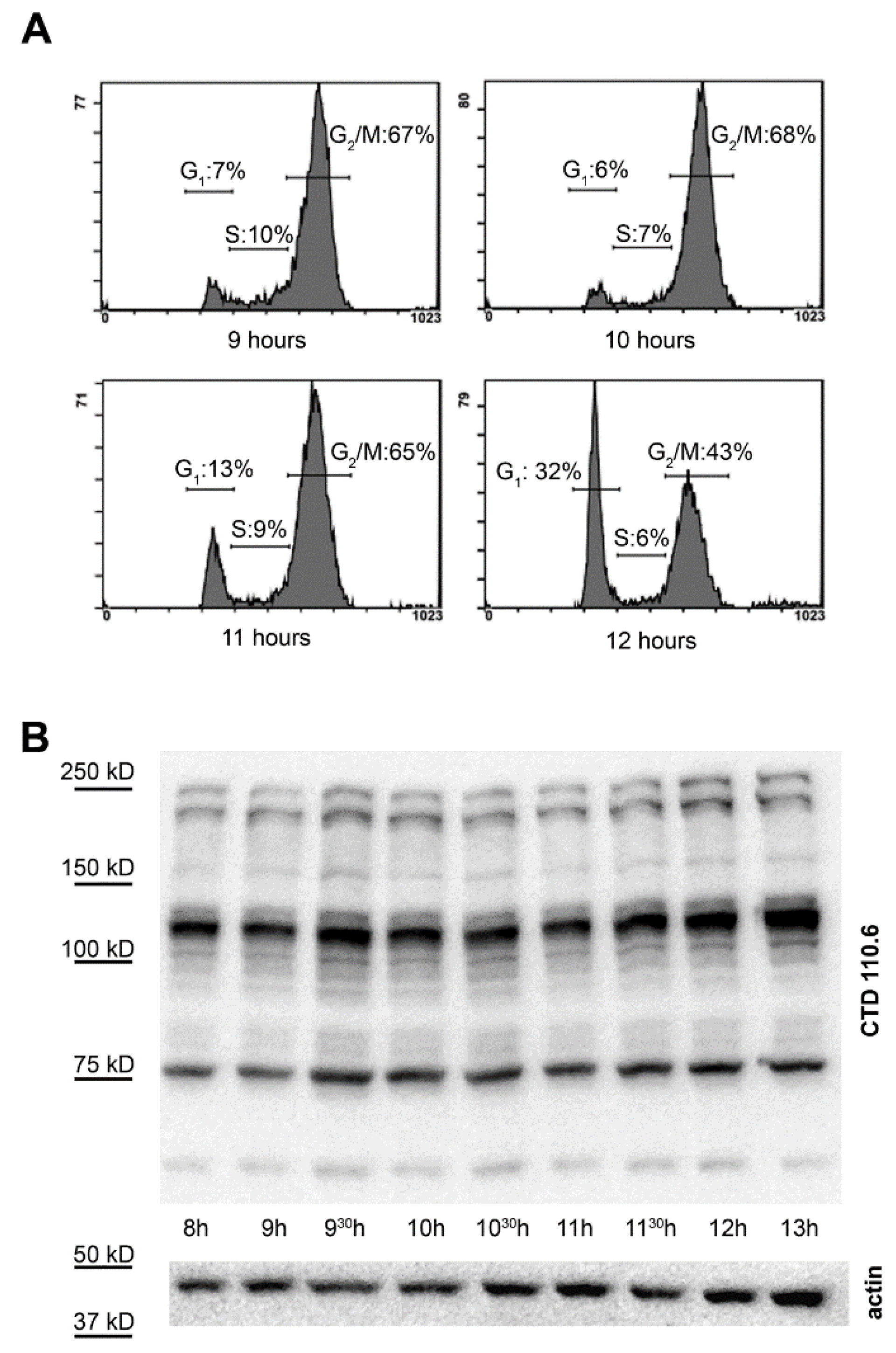
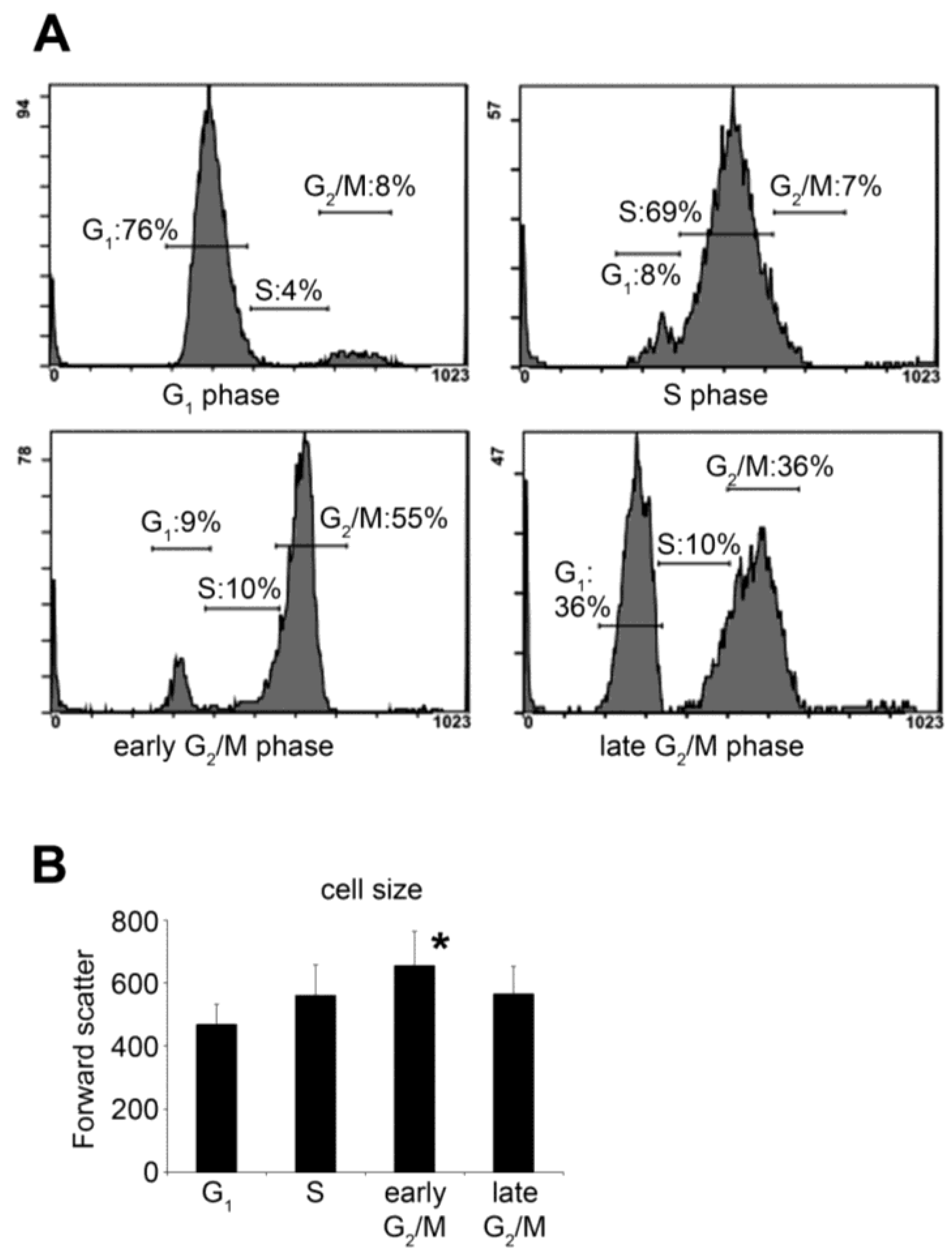
2.1. Synchronization of HeLa Cells by Double Thymidine Block
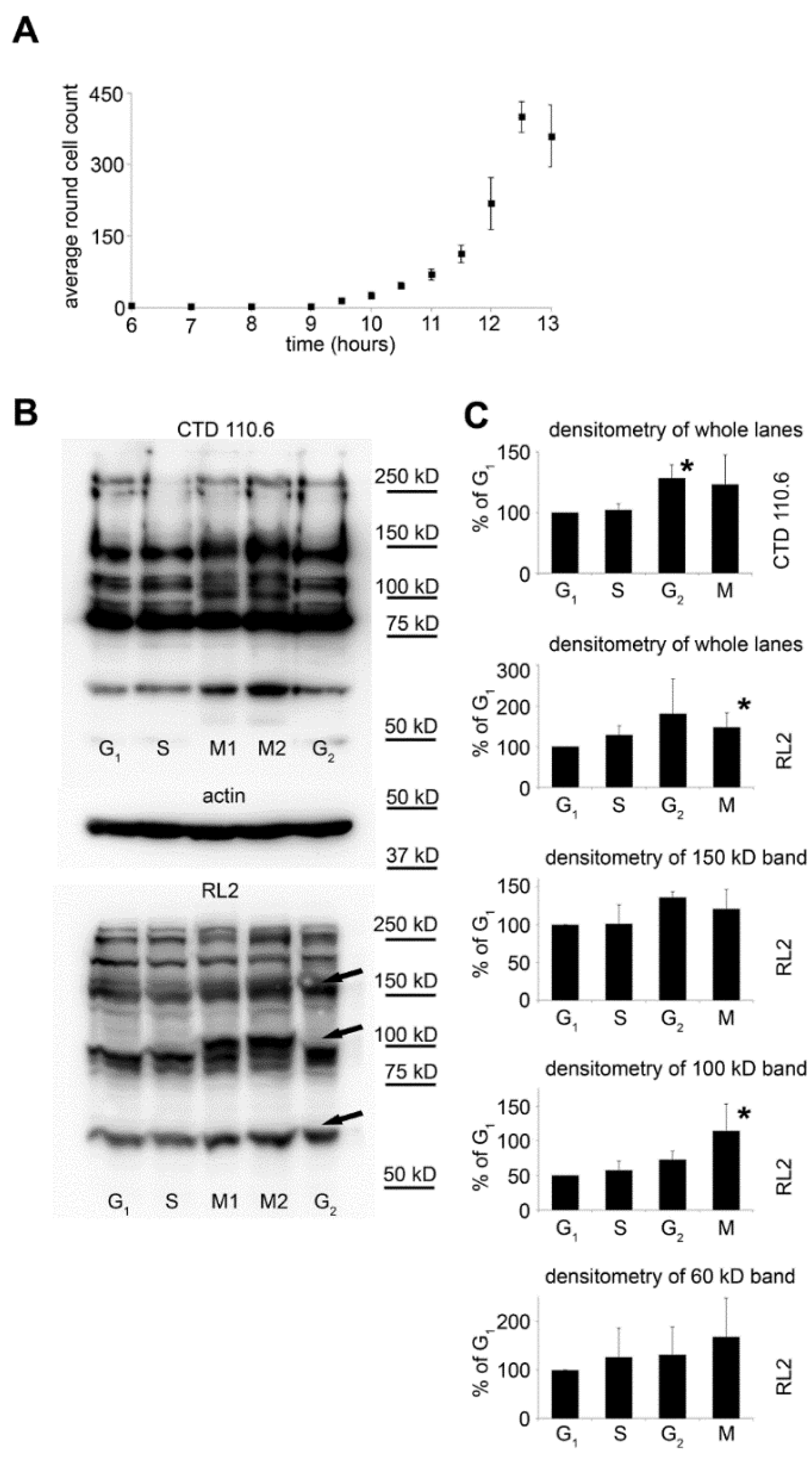
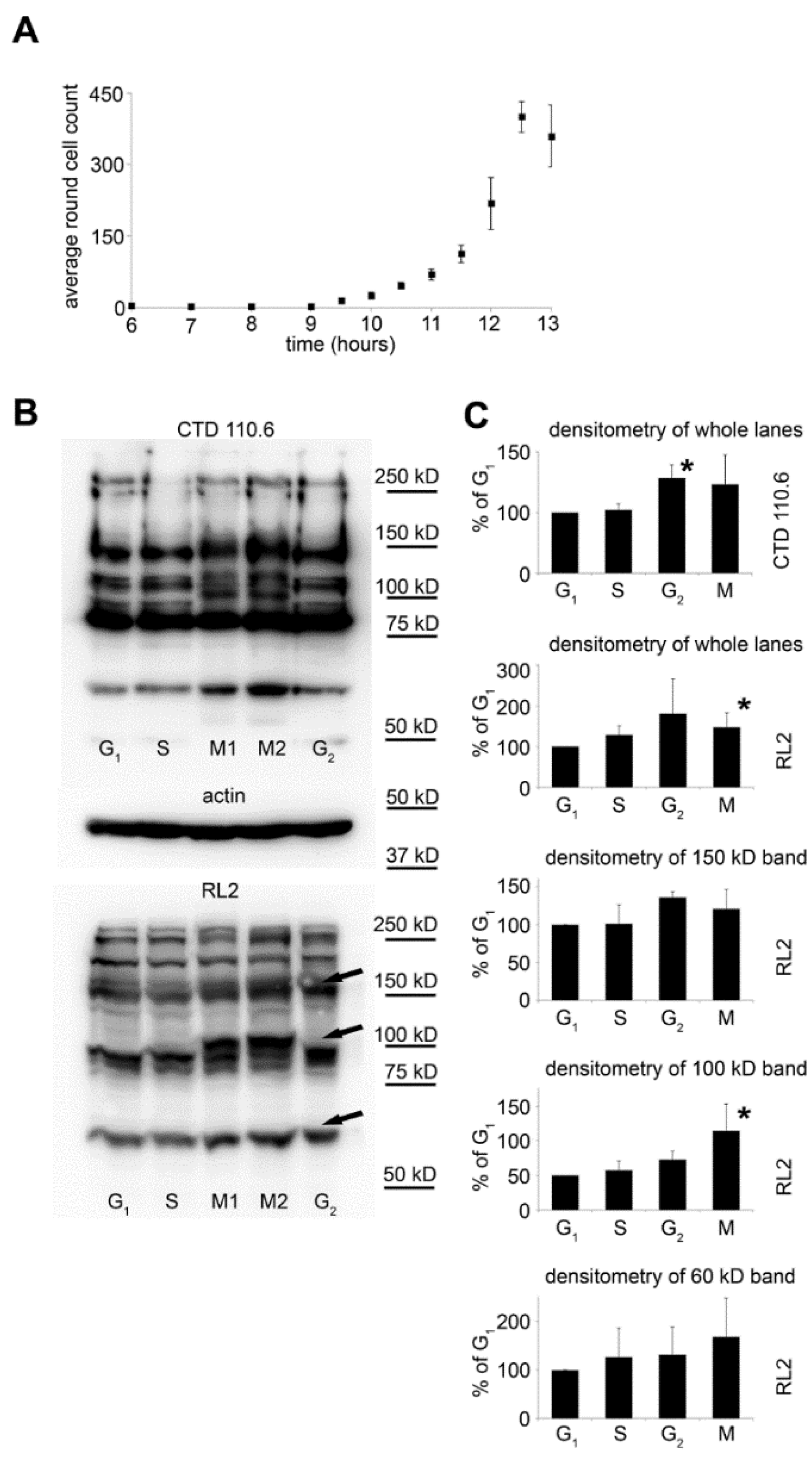
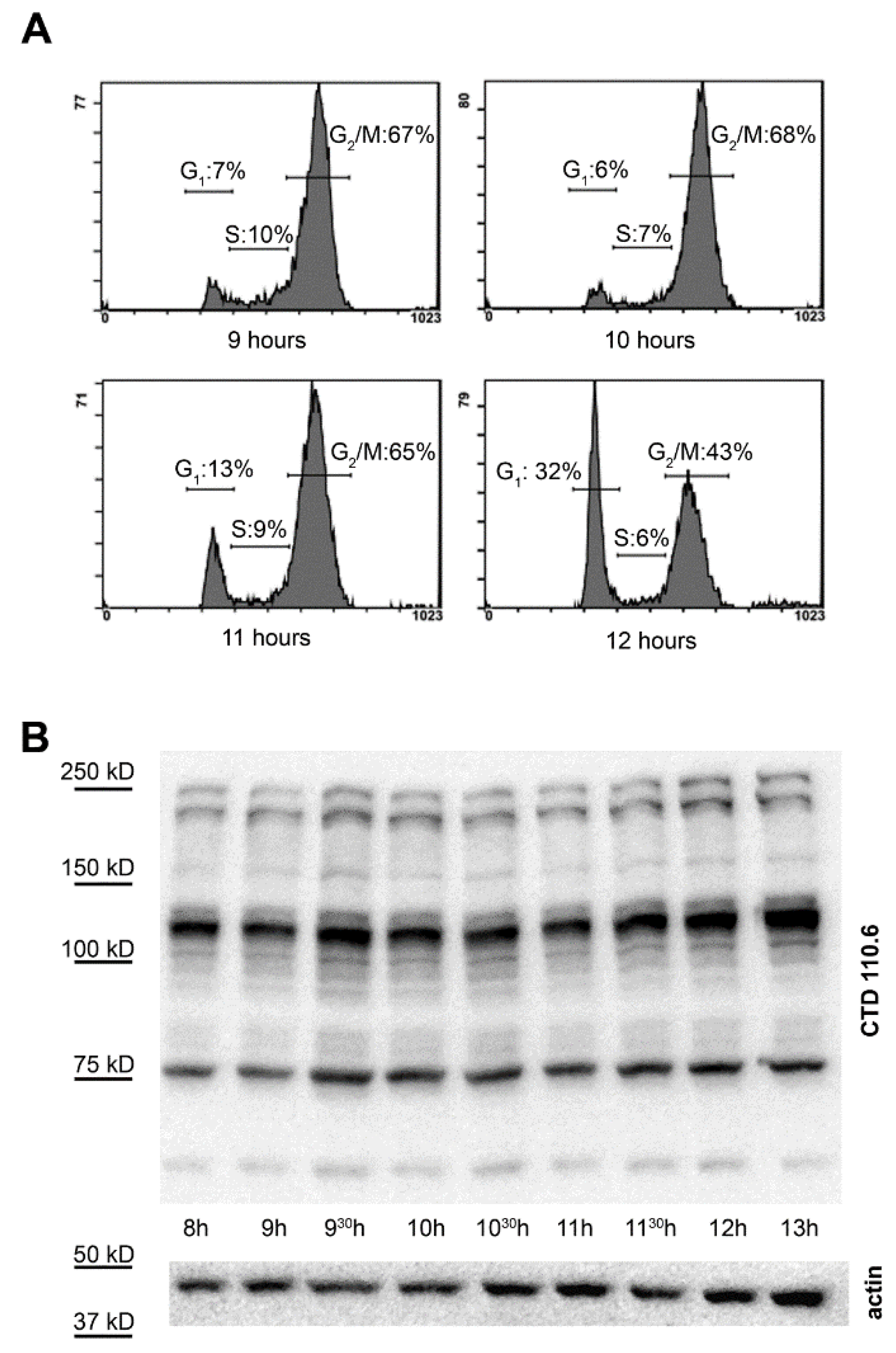
2.2. Selective Collection of Mitotic Cells Resulted in Detection of Distinct Changes in O-GlcNAc Pattern
2.3. Synchronization Alone Is Not Sufficient to Detect Changes in Overall O-GlcNAc Levels during G2/M–G1 Transition
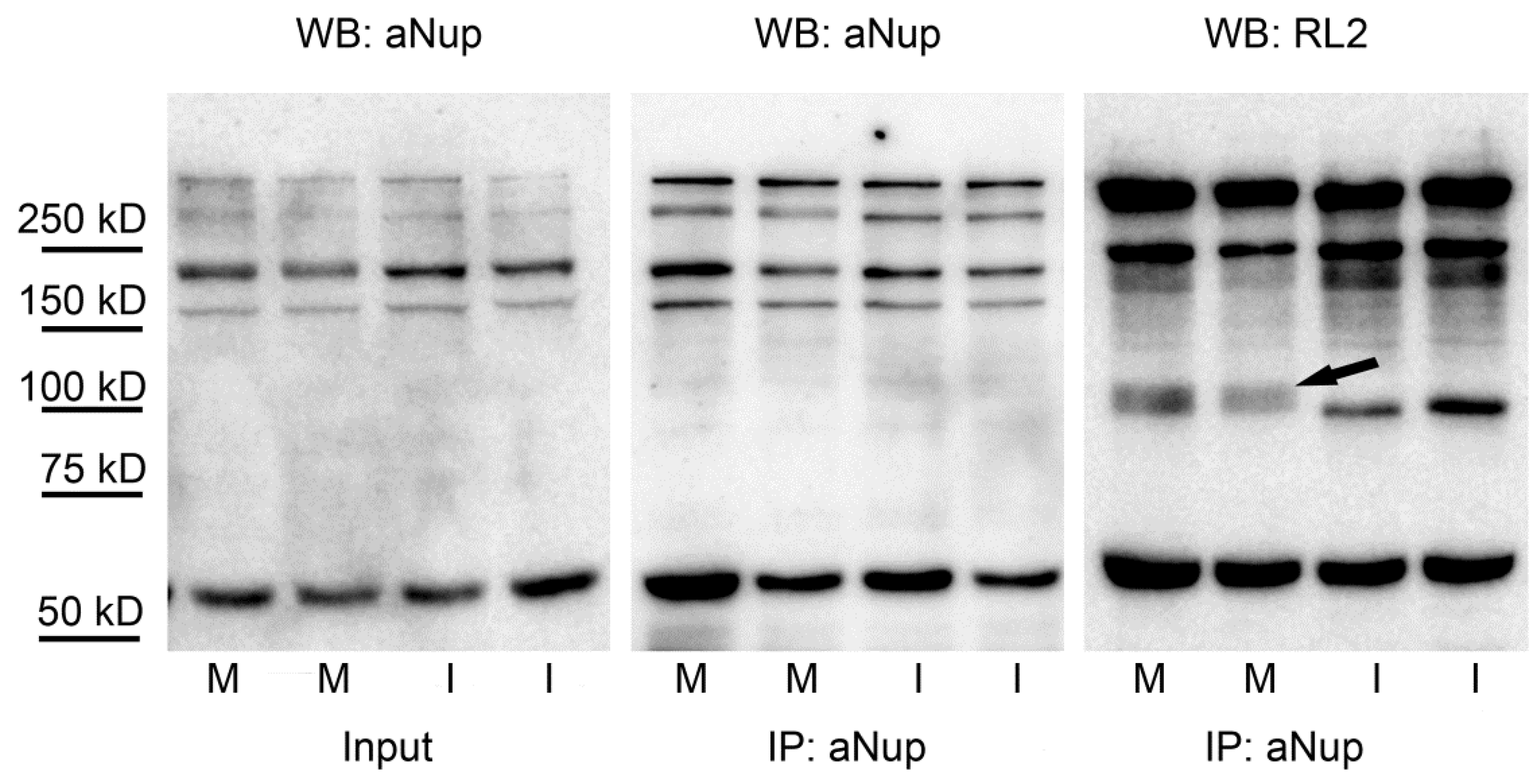
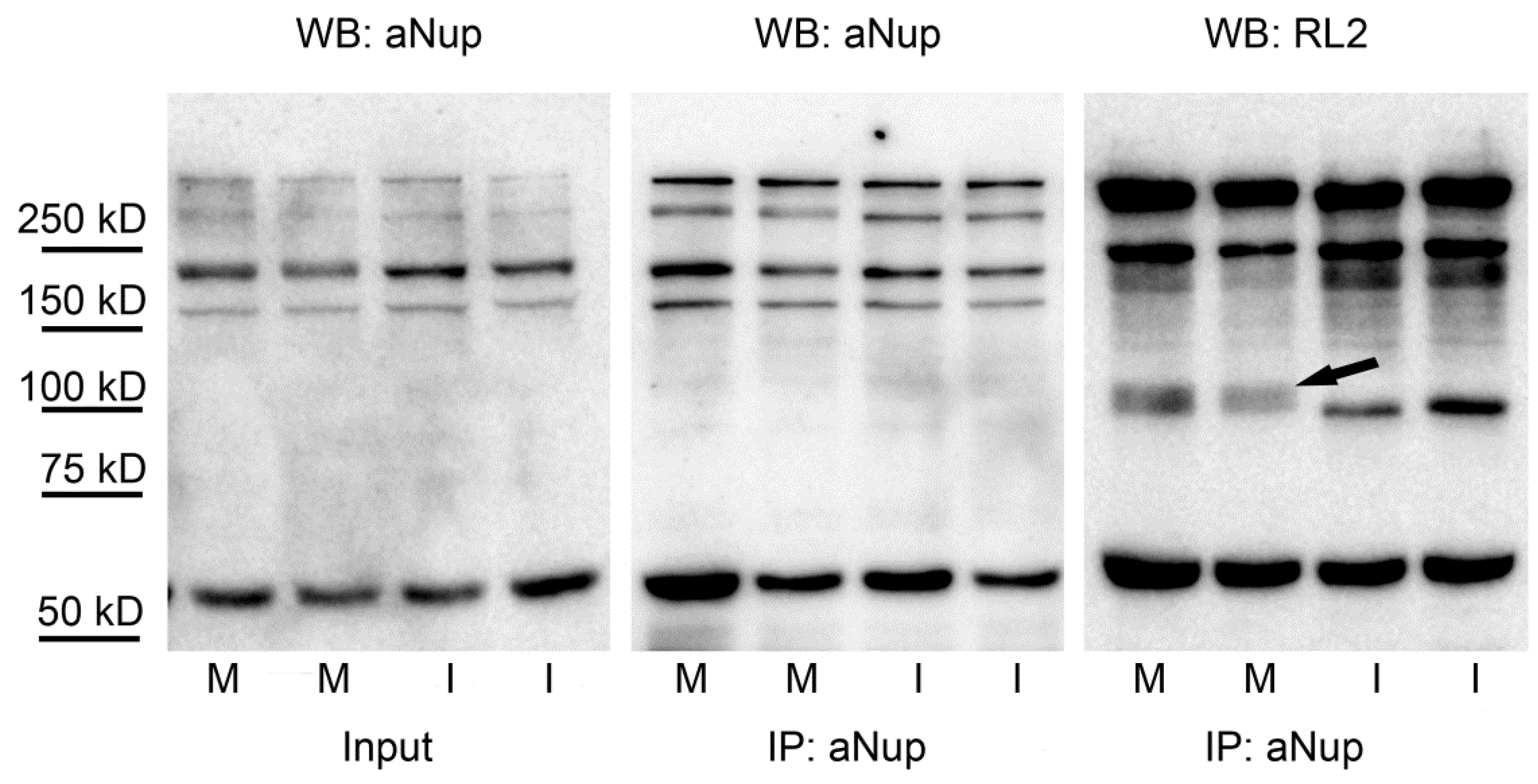
2.4. Nuclear Pore Proteins Are Associated with Variable O-GlcNAc Content during Cell Cycle
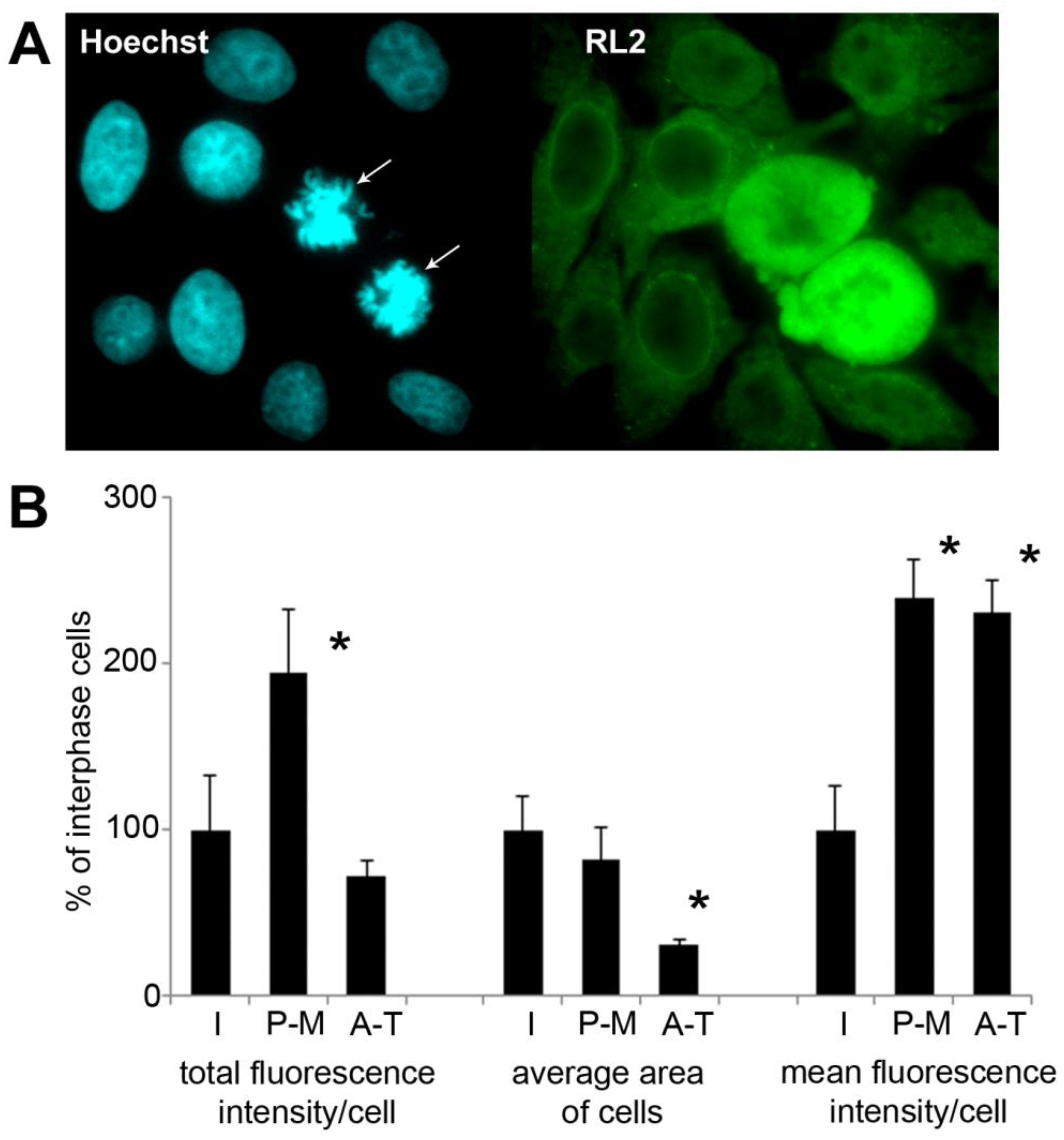
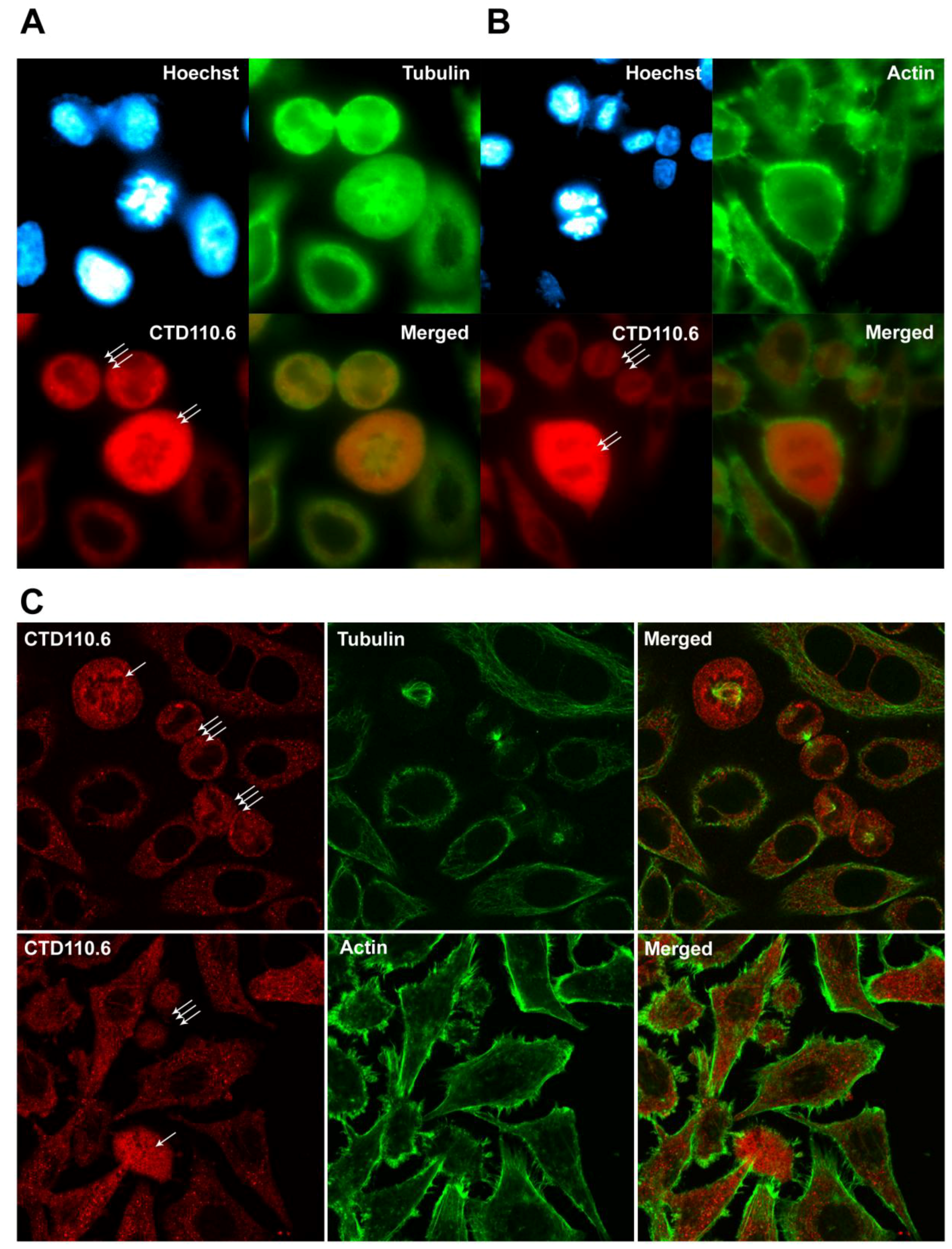
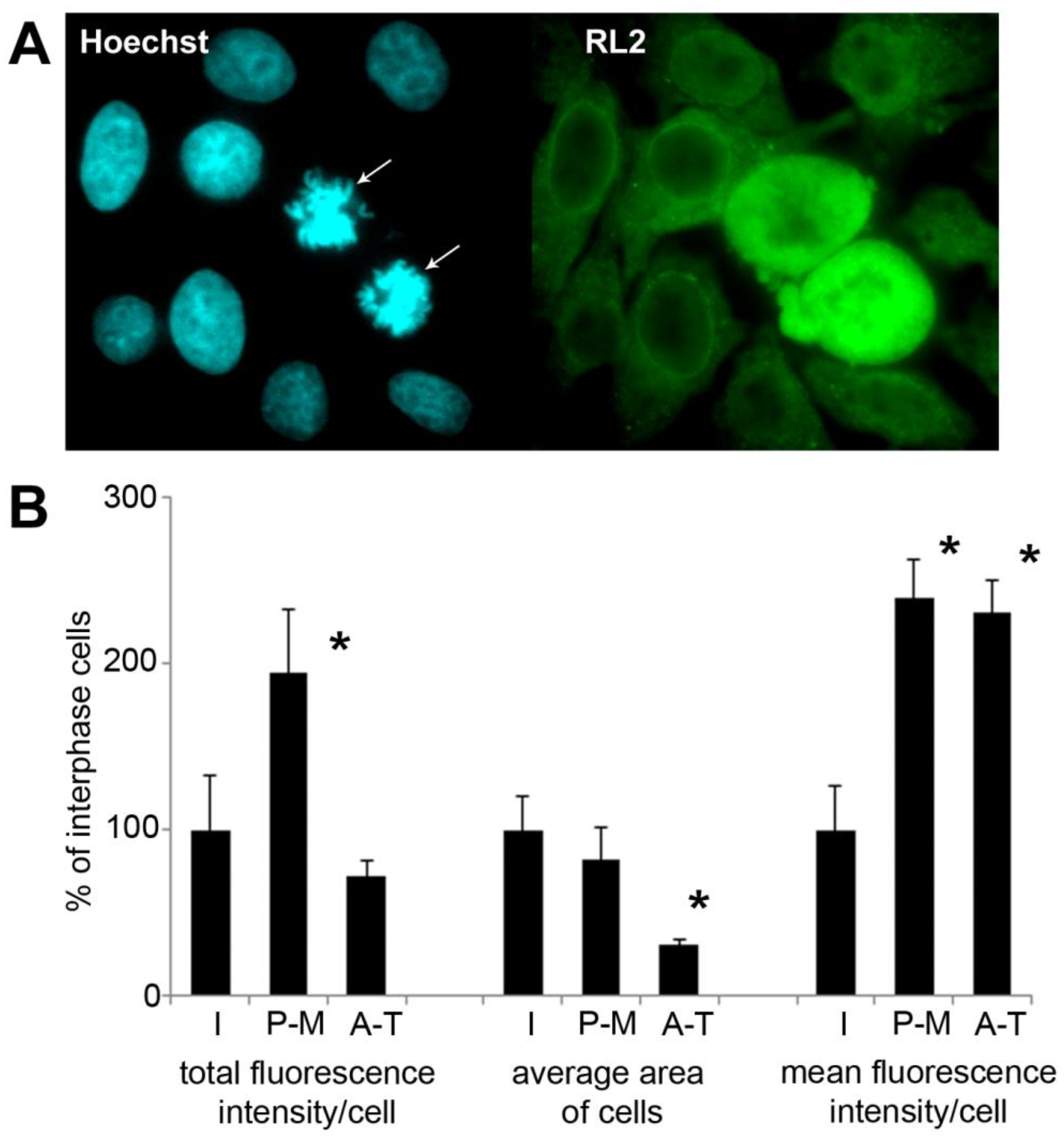
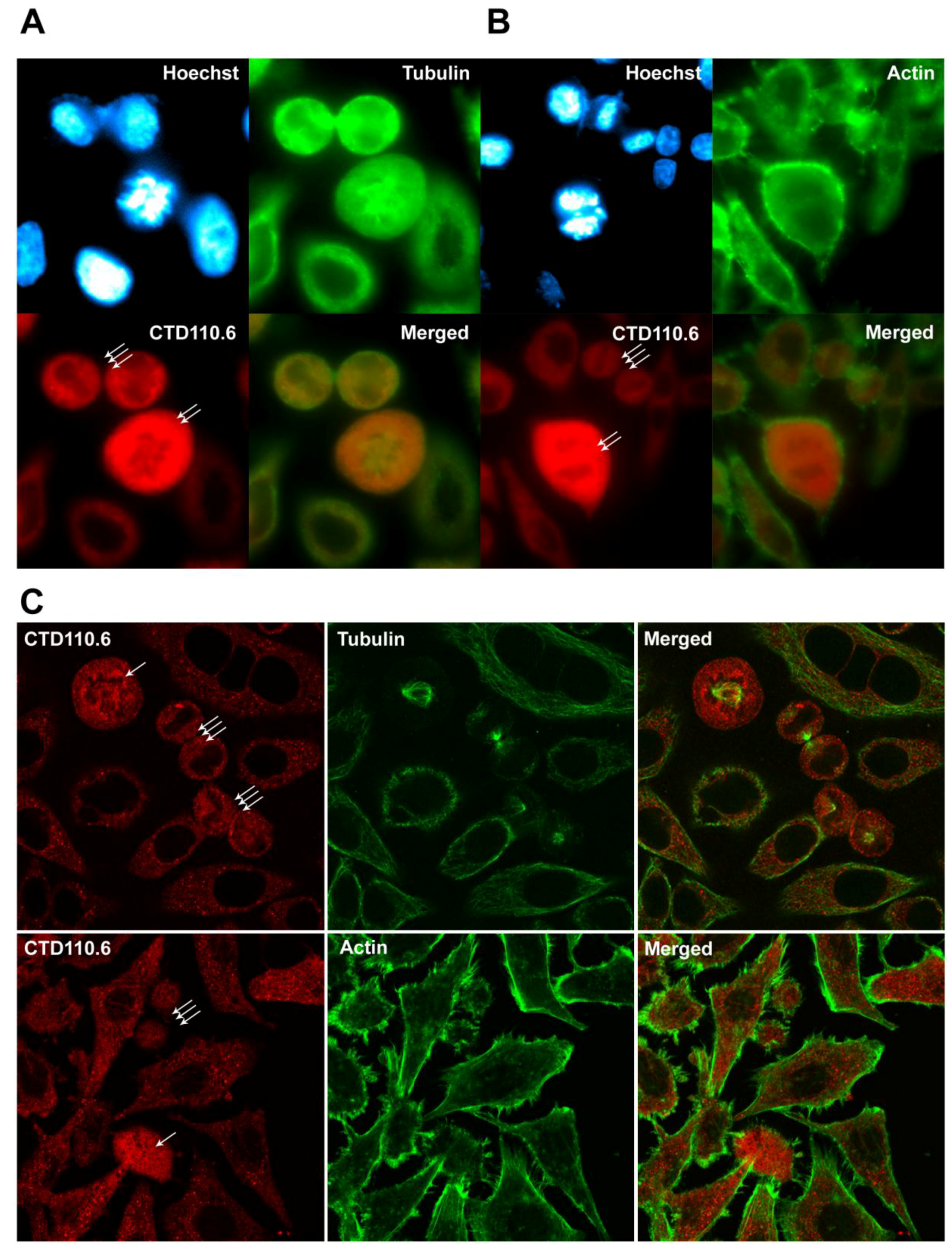
2.5. Immunofluorescence Detection Shows Increased Level of O-GlcNAc in Mitotic HeLa Cells
3. Discussion
4. Materials and Methods
4.1. Cell Line and Culture Conditions
4.2. Flow Cytometry
4.3. Cell Counting
4.4. Western Blot Analysis
4.5. Immunofluorescence Microscopy
4.6. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Wang, Z. Studying the relationship between cell cycle and Alzheimer’s disease by gold nanoparticle probes. Anal. Biochem. 2015, 489, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-GlcNAc signaling: A metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Balomenos, D.; Martínez-A, C. Cell-cycle regulation in immunity, tolerance and autoimmunity. Immunol. Today 2000, 21, 551–555. [Google Scholar] [CrossRef]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Revisiting the “Cdk-centric” view of the mammalian cell cycle. Cell Cycle 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Mocciaro, A.; Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 2012, 125, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.A.; Kaneko, T.; Li, S.S.C. Cell Regulation by Phosphotyrosine-Targeted Ubiquitin Ligases. Mol. Cell. Biol. 2015, 35, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.-H.; Abe, J.-I. SUMO—A post-translational modification with therapeutic potential? Curr. Opin. Pharmacol. 2010, 10, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, T.; Langelier, M.-F.; Pascal, J.M.; Schüler, H. Structural biology of the writers, readers, and erasers in mono- and poly(ADP-ribose) mediated signaling. Mol. Aspects Med. 2013, 34, 1088–1108. [Google Scholar] [CrossRef] [PubMed]
- Hahne, H.; Sobotzki, N.; Nyberg, T.; Helm, D.; Borodkin, V.S.; van Aalten, D.M.F.; Agnew, B.; Kuster, B. Proteome wide purification and identification of O-GlcNAc-modified proteins using click chemistry and mass spectrometry. J. Proteome Res. 2013, 12, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Butkinaree, C.; Park, K.; Hart, G.W. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta 2010, 1800, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.-B.; Nie, Y.; Yang, X. Regulation of protein degradation by O-GlcNAcylation: Crosstalk with ubiquitination. Mol. Cell. Proteom. 2013, 12, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Keembiyehetty, C.N.; Krzeslak, A.; Love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 124, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Guinez, C.; Mir, A.-M.; Dehennaut, V.; Cacan, R.; Harduin-Lepers, A.; Michalski, J.-C.; Lefebvre, T. Protein ubiquitination is modulated by O-GlcNAc glycosylation. FASEB J. 2008, 22, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Su, K.; Yang, X.; Bowe, D.B.; Paterson, A.J.; Kudlow, J.E. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 2003, 115, 715–725. [Google Scholar] [CrossRef]
- Sümegi, M.; Hunyadi-Gulyás, E.; Medzihradszky, K.F.; Udvardy, A. 26S proteasome subunits are O-linked N-acetylglucosamine-modified in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2003, 312, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Hart, G.W. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell. Mol. Life Sci. 2003, 60, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Drougat, L.; Olivier-Van Stichelen, S.; Mortuaire, M.; Foulquier, F.; Lacoste, A.-S.; Michalski, J.-C.; Lefebvre, T.; Vercoutter-Edouart, A.-S. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim. Biophys. Acta 2012, 1820, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; Hart, G.W. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 2006, 1761, 599–617. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010, 3, ra2. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Hanoulle, X.; Bodart, J.-F.; Vilain, J.-P.; Michalski, J.-C.; Landrieu, I.; Lippens, G.; Lefebvre, T. Microinjection of recombinant O-GlcNAc transferase potentiates Xenopus oocytes M-phase entry. Biochem. Biophys. Res. Commun. 2008, 369, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.J.; Nguyen, B.L.; Bridger, R.; Medrano, E.E.; Wells, L.; Pan, S.; Sifers, R.N. β-N-acetylglucosamine (O-GlcNAc) is a novel regulator of mitosis-specific phosphorylations on histone H3. J. Biol. Chem. 2012, 287, 12195–12203. [Google Scholar] [CrossRef] [PubMed]
- Delporte, A.; De Zaeytijd, J.; De Storme, N.; Azmi, A.; Geelen, D.; Smagghe, G.; Guisez, Y.; Van Damme, E.J.M. Cell cycle-dependent O-GlcNAc modification of tobacco histones and their interaction with the tobacco lectin. Plant Physiol. Biochem. 2014, 83, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.P.; Caro, S.; Potnis, A.; Lanza, C.; Slawson, C. O-linked N-acetylglucosamine cycling regulates mitotic spindle organization. J. Biol. Chem. 2013, 288, 27085–27099. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.M.; Minner, S.; Guldvik, I.J.J.; Sandmann, M.J.J.; Tsourlakis, M.C.C.; Berge, V.; Svindland, A.; Schlomm, T.; Mills, I.G.G. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Cancer Res. 2013, 73, 5277–5287. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, K.; Wang, Z.; Hart, G.W. Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl. Acad. Sci. USA 2010, 107, 19915–19920. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.; Daou, S.; Zamorano, N.; Iannantuono, N.V.G.; Hammond-Martel, I.; Mashtalir, N.; Bonneil, E.; Wurtele, H.; Thibault, P.; Affar, E.B. Undetectable histone O-GlcNAcylation in mammalian cells. Epigenetics 2015, 10, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Baert, F.; Bodart, J.F.; Flament, S.; Michalski, J.C.; Vilain, J.P. Modulation of O-GlcNAc glycosylation during xenopus oocyte maturation. J. Cell. Biochem. 2004, 93, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Lefebvre, T.; Sellier, C.; Leroy, Y.; Gross, B.; Walker, S.; Cacan, R.; Michalski, J.-C.; Vilain, J.-P.; Bodart, J.-F. O-linked N-acetylglucosaminyltransferase inhibition prevents G2/M transition in Xenopus laevis oocytes. J. Biol. Chem. 2007, 282, 12527–12536. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.J.; Jang, H.J.; Moon, H.Y.; Byun, H.Y.; Kim, E.K.; Kim, D.H.; et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, K.; Hart, G.W. O-GlcNAc transferase regulates mitotic chromatin dynamics. J. Biol. Chem. 2010, 285, 34460–34468. [Google Scholar] [CrossRef] [PubMed]
- Pederson, T.; Robbins, E. A method for improving synchrony in the G2 phase of the cell cycle. J. Cell Biol. 1971, 49, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Bostock, C.J.; Prescott, D.M.; Kirkpatrick, J.B. An evaluation of the double thymidine block for synchronizing mammalian cells at the G1-S border. Exp. Cell Res. 1971, 68, 163–168. [Google Scholar] [CrossRef]
- Fisi, V.; Kátai, E.; Bogner, P.; Miseta, A.; Nagy, T. Timed, sequential administration of paclitaxel improves its cytotoxic effectiveness in a cell culture model. Cell Cycle 2016, 15, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Cohen, C.K.; Willingham, M.C.; Park, M.K. O-linked N-acetylglucosamine is attached to proteins of the nuclear pore. Evidence for cytoplasmic and nucleoplasmic glycoproteins. J. Biol. Chem. 1987, 262, 9887–9894. [Google Scholar] [PubMed]
- Zhu, Y.; Liu, T.-W.; Madden, Z.; Yuzwa, S.A.; Murray, K.; Cecioni, S.; Zachara, N.; Vocadlo, D.J. Post-translational O-GlcNAcylation is essential for nuclear pore integrity and maintenance of the pore selectivity filter. J. Mol. Cell Biol. 2016, 8, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Fisi, V.; Miseta, A.; Nagy, T. The Role of Stress-Induced O-GlcNAc Protein Modification in the Regulation of Membrane Transport. Oxid. Med. Cell. Longev. 2017, 2017, 1308692. [Google Scholar] [CrossRef] [PubMed]
- Walgren, J.L.E.; Vincent, T.S.; Schey, K.L.; Buse, M.G. High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E424–E434. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Slomianny, M.-C.; Page, A.; Vercoutter-Edouart, A.-S.; Jessus, C.; Michalski, J.-C.; Vilain, J.-P.; Bodart, J.-F.; Lefebvre, T. Identification of structural and functional O-linked N-acetylglucosamine-bearing proteins in Xenopus laevis oocyte. Mol. Cell. Proteom. 2008, 7, 2229–2245. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Hengst, L.; Slingerland, J.M. The Cdk inhibitor p27 in human cancer: Prognostic potential and relevance to anticancer therapy. Nat. Rev. Cancer 2008, 8, 253–267. [Google Scholar] [CrossRef]
- Boehmelt, G.; Wakeham, A.; Elia, A.; Sasaki, T.; Plyte, S.; Potter, J.; Yang, Y.; Tsang, E.; Ruland, J.; Iscove, N.N.; et al. Decreased UDP-GlcNAc levels abrogate proliferation control in EMeg32-deficient cells. EMBO J. 2000, 19, 5092–5104. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Drougat, L.; Dehennaut, V.; El Yazidi-Belkoura, I.; Guinez, C.; Mir, A.-M.; Michalski, J.-C.; Vercoutter-Edouart, A.-S.; Lefebvre, T. Serum-stimulated cell cycle entry promotes ncOGT synthesis required for cyclin D expression. Oncogenesis 2012, 1, e36. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.A.; Baker, J.B.; Pelham, R.J. Modulators of sensitivity and resistance to inhibition of PI3K identified in a pharmacogenomic screen of the NCI-60 human tumor cell line collection. PLoS ONE 2012, 7, e46518. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Fardini, Y.; Dehennaut, V.; Lefebvre, T.; Issad, T. O-GlcNAcylation: A New Cancer Hallmark? Front. Endocrinol. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Lanza, C.; Tan, E.P.; Zhang, Z.; Machacek, M.; Brinker, A.E.; Azuma, M.; Slawson, C. Reduced O-GlcNAcase expression promotes mitotic errors and spindle defects. Cell Cycle 2016, 15, 1363–1375. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Lakshmanan, T.; Knapp, S.; Hart, G.W. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol. Biol. Cell 2008, 19, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Geng, Q.; Ding, Y.; Liao, J.; Dong, M.-Q.; Xu, X.; Li, J. O-GlcNAcylation Antagonizes Phosphorylation of CDH1 (CDC20 Homologue 1). J. Biol. Chem. 2016, 291, 12136–12144. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Lefebvre, T.; Leroy, Y.; Vilain, J.-P.; Michalski, J.-C.; Bodart, J.-F. Survey of O-GlcNAc level variations in Xenopus laevis from oogenesis to early development. Glycoconj. J. 2009, 26, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Haltiwanger, R.S.; Philipsberg, G.A. Mitotic arrest with nocodazole induces selective changes in the level of O-linked N-acetylglucosamine and accumulation of incompletely processed N-glycans on proteins from HT29 cells. J. Biol. Chem. 1997, 272, 8752–8758. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.; O’Connor, P.M. Methods for synchronizing cells at specific stages of the cell cycle. Curr. Protoc. Cell Biol. 2001. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Betzendahl, I.; Sun, F.; Tinneberg, H.-R.; Eichenlaub-Ritter, U. Non-invasive method to assess genotoxicity of nocodazole interfering with spindle formation in mammalian oocytes. Reprod. Toxicol. 2005, 19, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Steenackers, A.; Olivier-Van Stichelen, S.; Baldini, S.F.; Dehennaut, V.; Toillon, R.-A.; Le Bourhis, X.; El Yazidi-Belkoura, I.; Lefebvre, T. Silencing the Nucleocytoplasmic O-GlcNAc Transferase Reduces Proliferation, Adhesion, and Migration of Cancer and Fetal Human Colon Cell Lines. Front. Endocrinol. 2016, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Tarbet, H.J.; Dolat, L.; Smith, T.J.; Condon, B.M.; O’Brien, E.T.; Valdivia, R.H.; Boyce, M. Site-specific glycosylation regulates the form and function of the intermediate filament cytoskeleton. eLife 2018, 7, e31807. [Google Scholar] [CrossRef] [PubMed]
- Theisen, U.; Straube, A.; Steinberg, G. Dynamic rearrangement of nucleoporins during fungal “open” mitosis. Mol. Biol. Cell 2008, 19, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Fahrenkrog, B. Nucleoporins: Leaving the nuclear pore complex for a successful mitosis. Cell. Signal. 2011, 23, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Dossus, L.; Kaaks, R. Nutrition, metabolic factors and cancer risk. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 551–571. [Google Scholar] [CrossRef] [PubMed]
- Rozanski, W.; Krzeslak, A.; Forma, E.; Brys, M.; Blewniewski, M.; Wozniak, P.; Lipinski, M. Prediction of bladder cancer based on urinary content of MGEA5 and OGT mRNA level. Clin. Lab. 2012, 58, 579–583. [Google Scholar] [PubMed]
- Lefebvre, T. O-GlcNAcylation: A sweet thorn in the spindle! Cell Cycle 2016, 15, 1954–1955. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.T.; Poon, R.Y.C. Synchronization of HeLa cells. Methods Mol. Biol. 2011, 761, 151–161. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisi, V.; Kátai, E.; Orbán, J.; Dossena, S.; Miseta, A.; Nagy, T. O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis. Molecules 2018, 23, 1275. https://doi.org/10.3390/molecules23061275
Fisi V, Kátai E, Orbán J, Dossena S, Miseta A, Nagy T. O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis. Molecules. 2018; 23(6):1275. https://doi.org/10.3390/molecules23061275
Chicago/Turabian StyleFisi, Viktória, Emese Kátai, József Orbán, Silvia Dossena, Attila Miseta, and Tamás Nagy. 2018. "O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis" Molecules 23, no. 6: 1275. https://doi.org/10.3390/molecules23061275
APA StyleFisi, V., Kátai, E., Orbán, J., Dossena, S., Miseta, A., & Nagy, T. (2018). O-Linked N-Acetylglucosamine Transiently Elevates in HeLa Cells during Mitosis. Molecules, 23(6), 1275. https://doi.org/10.3390/molecules23061275