
Kinetic Basis of the Bifunctionality of SsoII DNA Methyltransferase

 and
and
Abstract
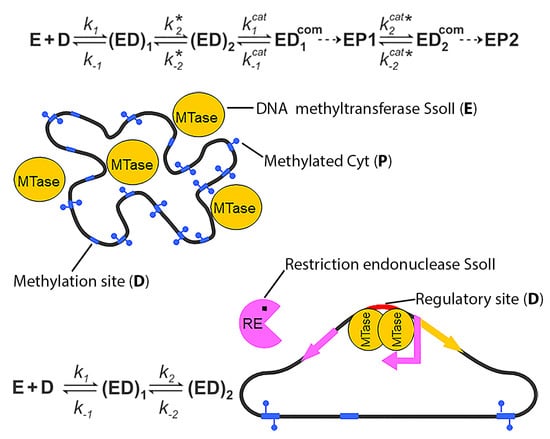
1. Introduction
2. Materials and Methods
2.1. DNA
2.2. Protein Purification
2.3. Steady-State Analyses of Interaction of M.SsoII with AdoMet and AdoHcy (Trp Fluorescence)
2.4. The Electrophoretic Mobility Shift Assay (EMSA)
2.5. The Methylation Assay
2.6. Inhibition of Substrate Methylation by Competitor DNA
2.7. Plasmid pBend2 Mutagenesis
2.8. The Circular Permutation Assay
2.9. Stopped-Flow Measurements
2.10. Kinetic Data Analysis
3. Results
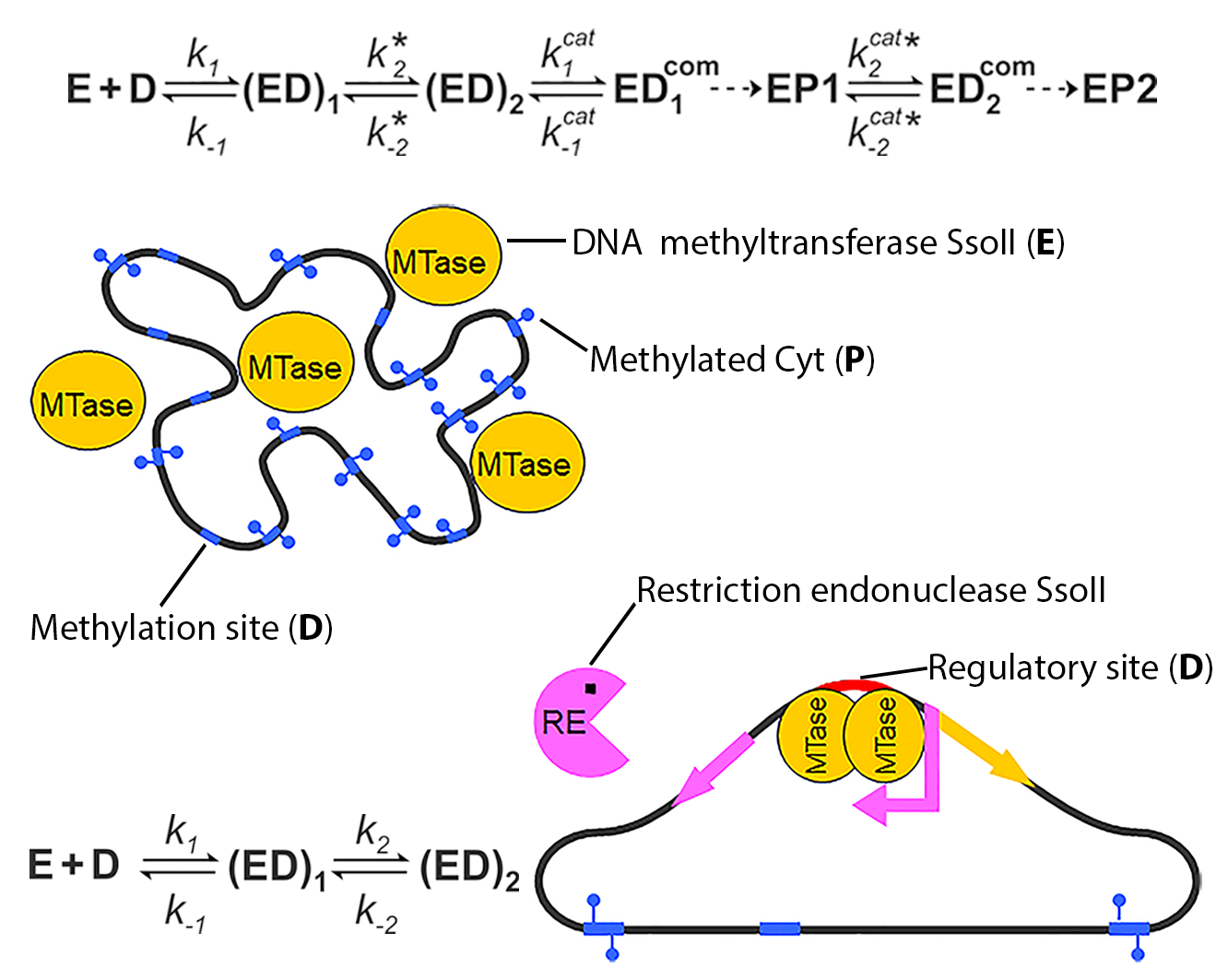
3.1. Cofactor Binding
3.2. Design of the DNA Duplexes
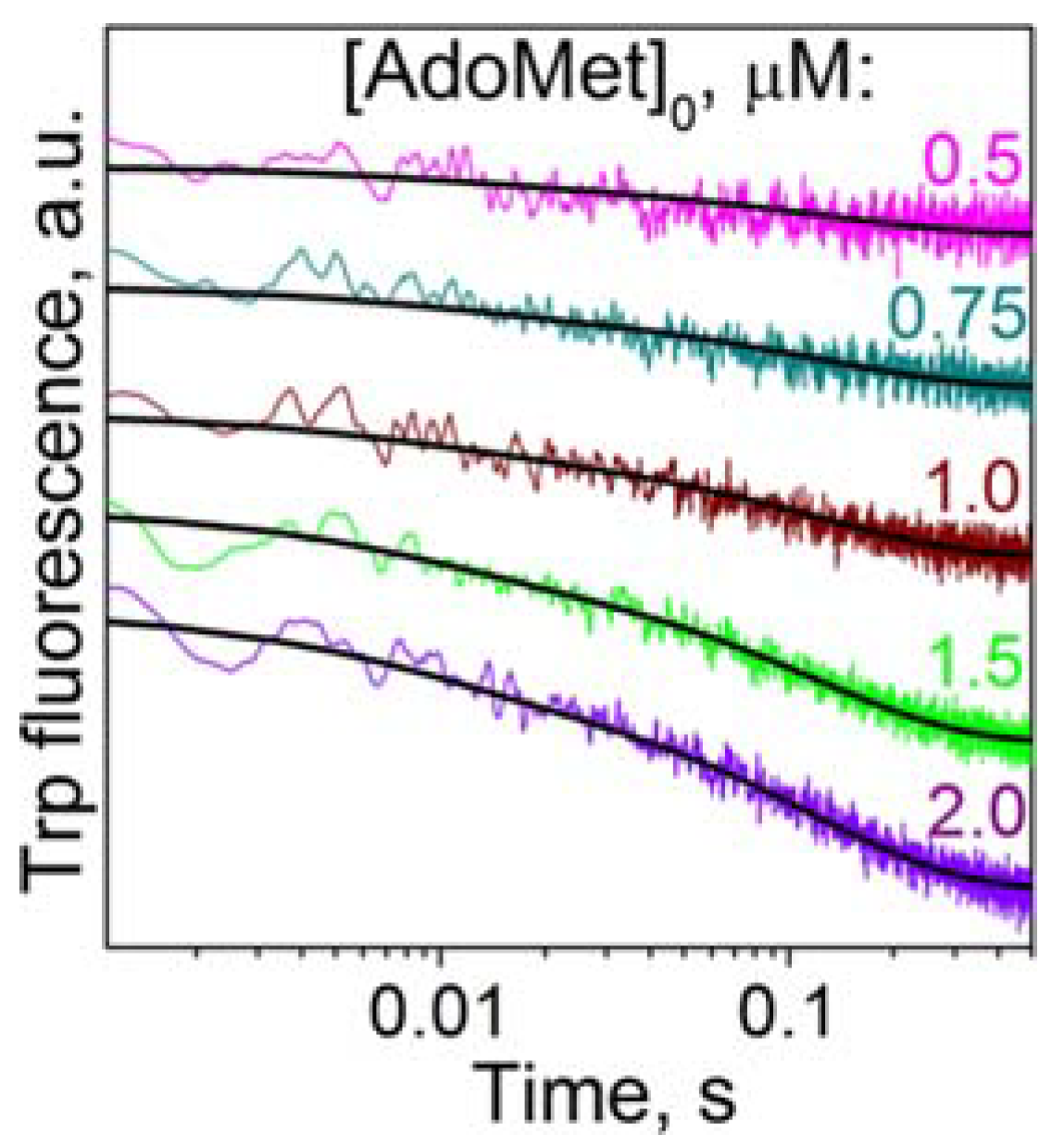
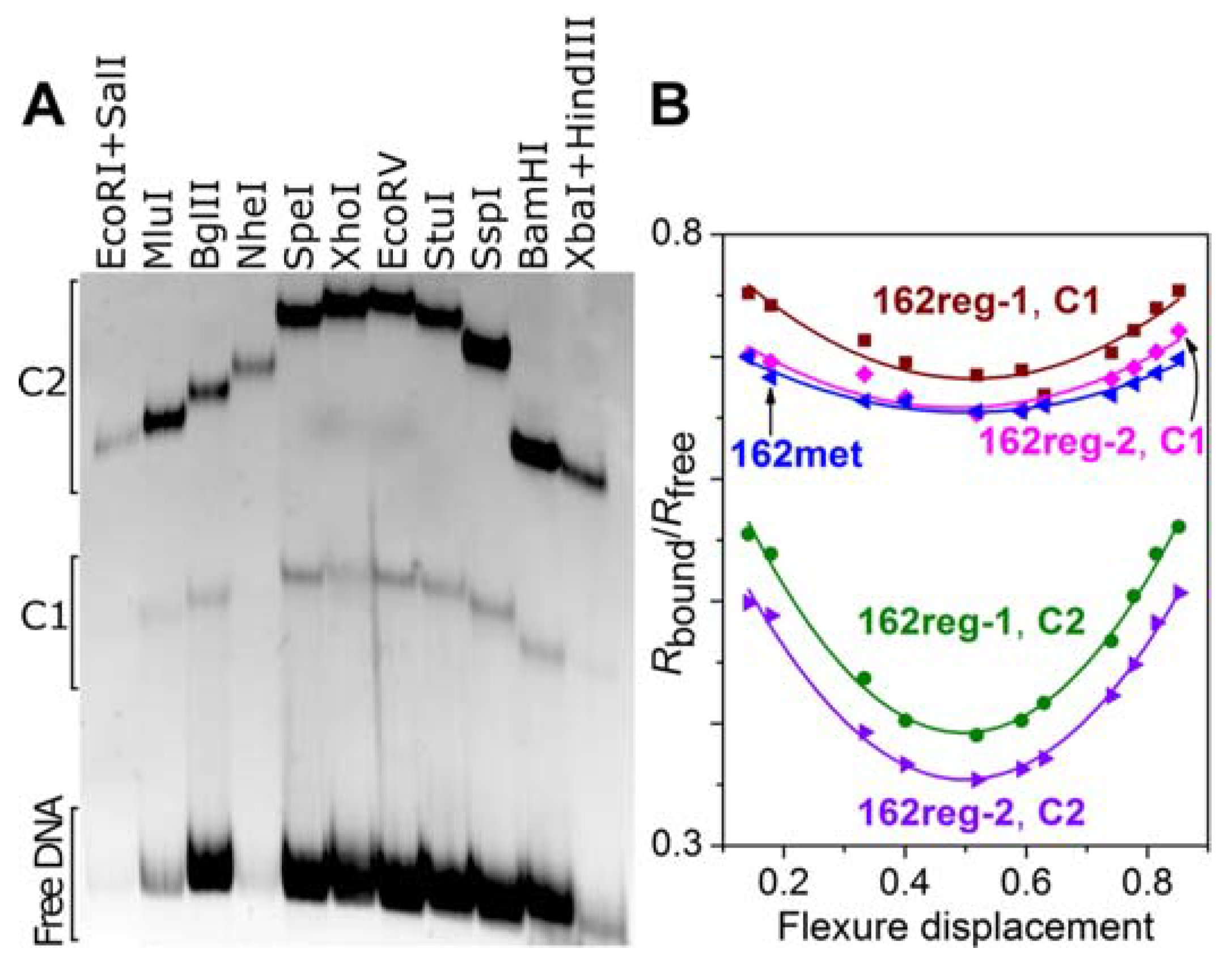
3.3. Formation of a Complex between M.SsoII and Various DNA Duplexes
3.4. DNA Bending by M.SsoII
3.5. DNA with the Regulatory Site Inhibits the MTase Activity
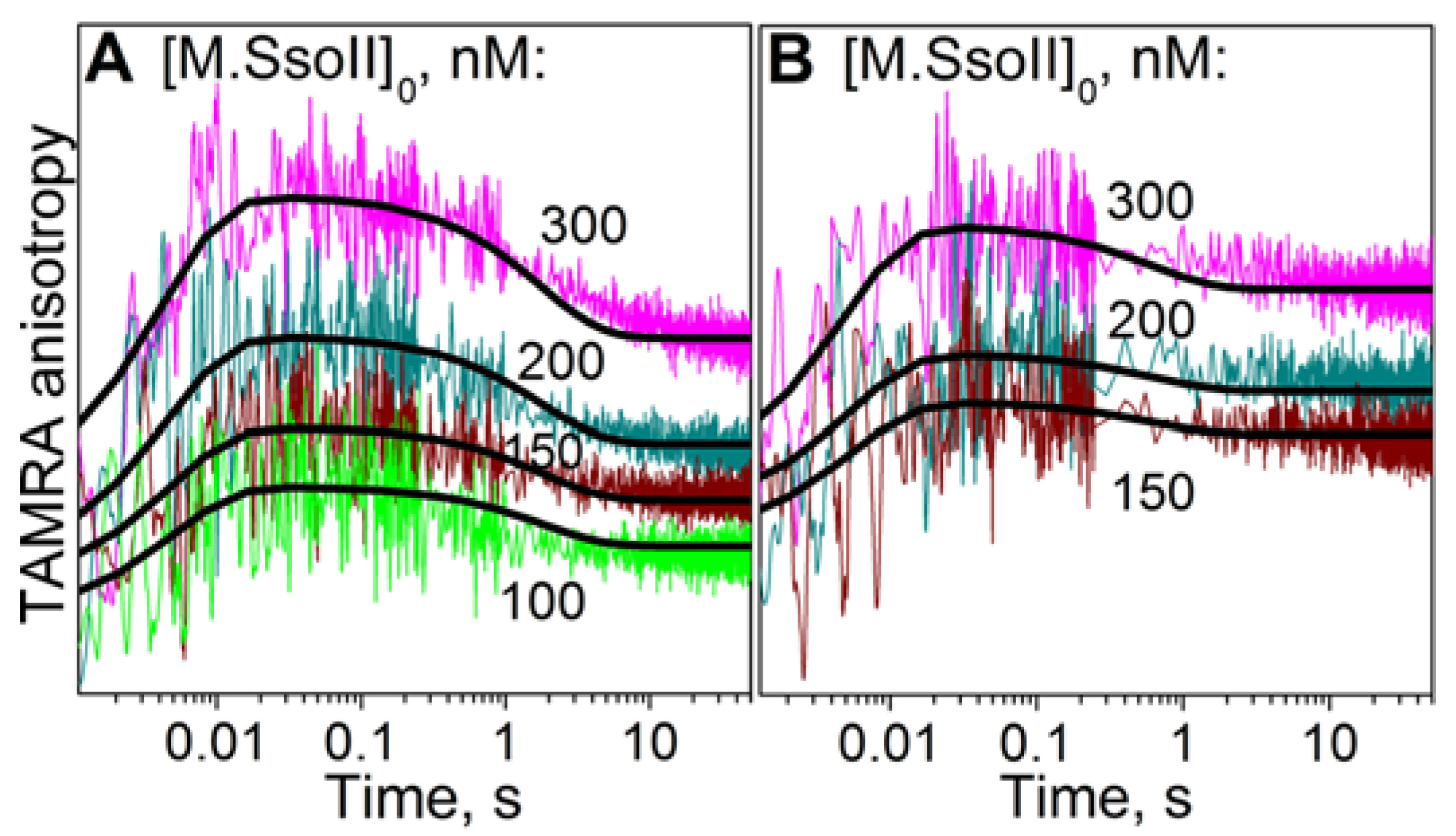
3.6. Kinetics of M.SsoII Binding to DNA with the Regulatory Site
3.7. Kinetics of M.SsoII Interaction with DNA Containing the Methylation Site
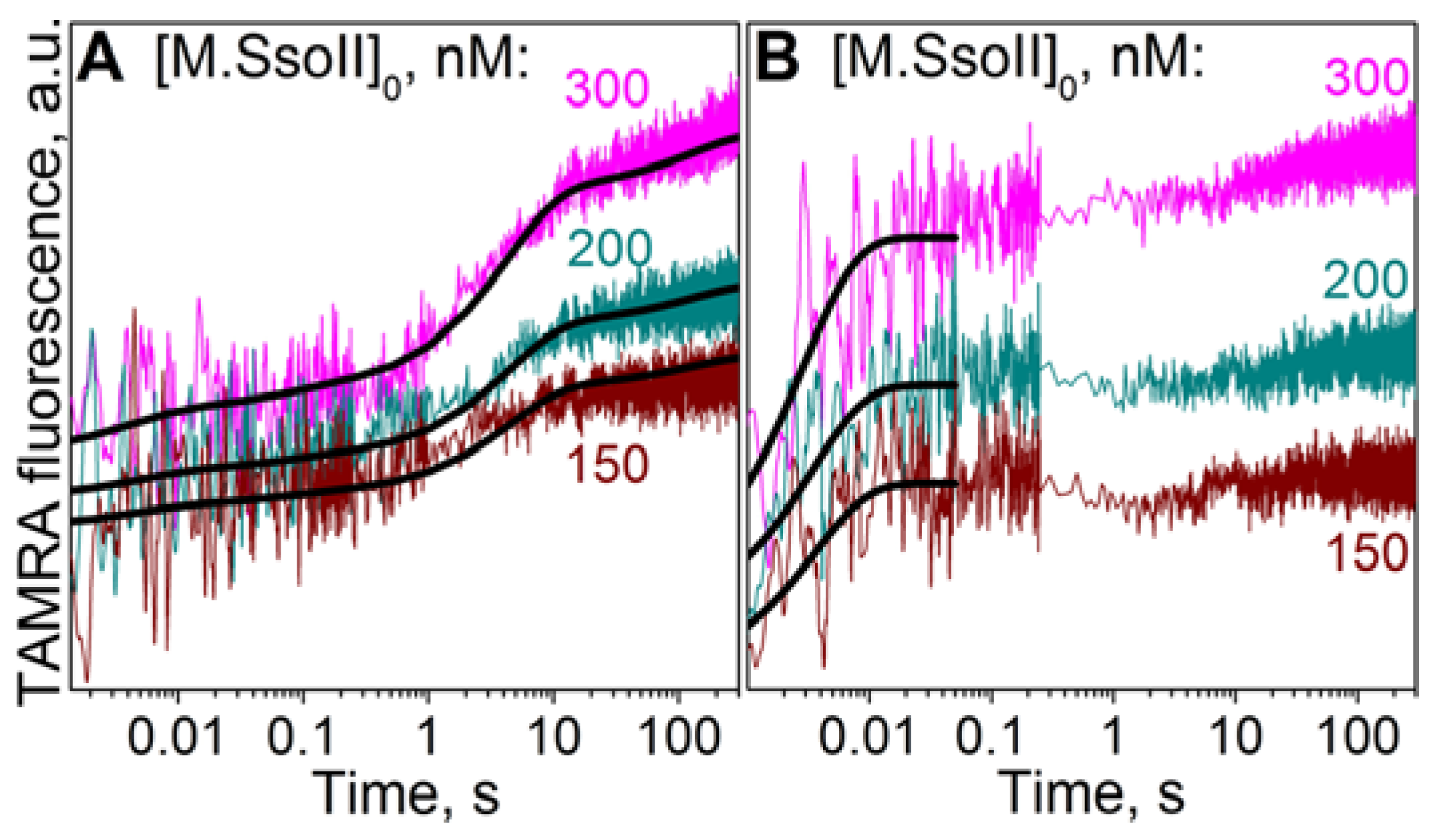
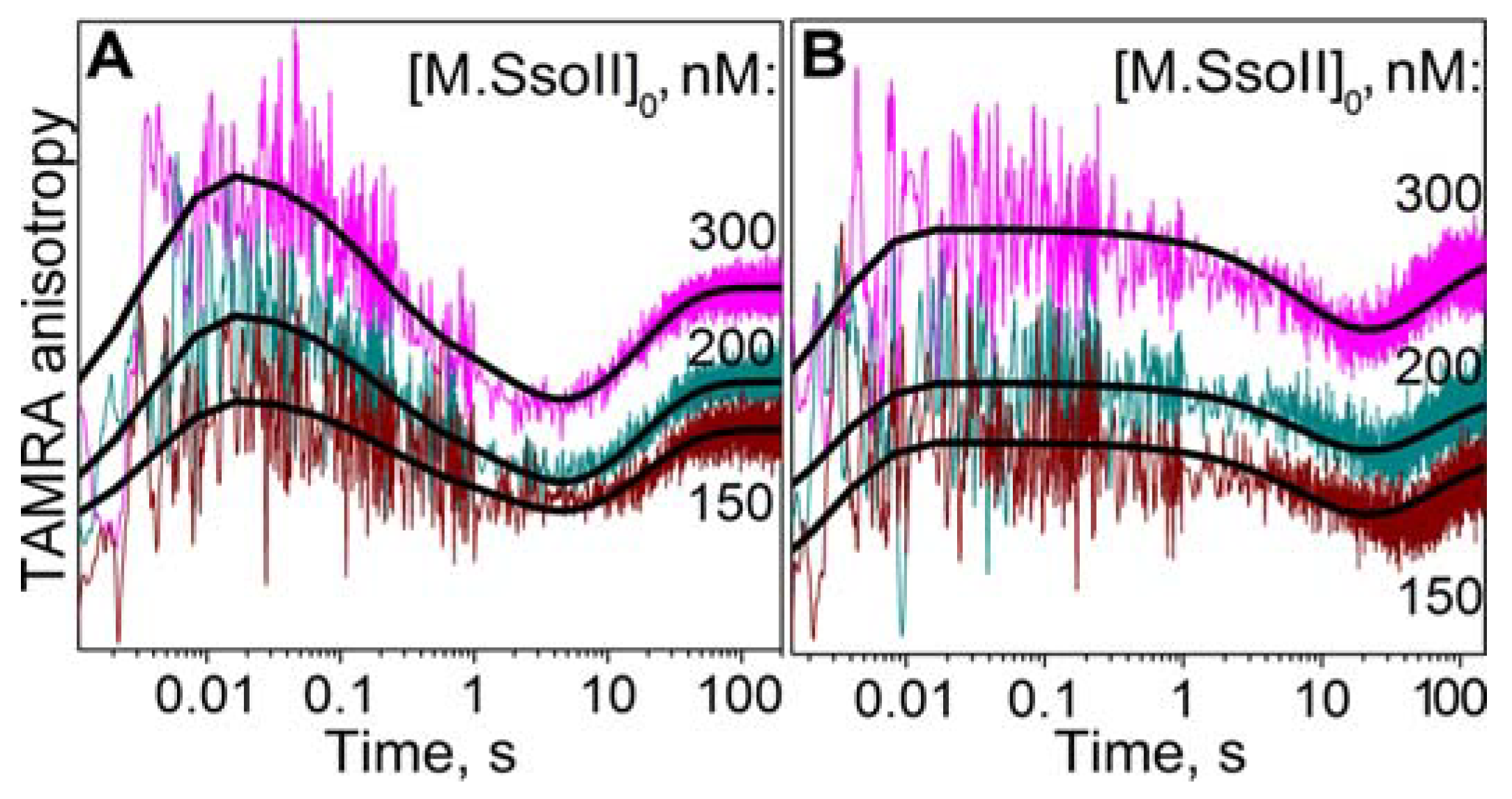
3.7.1. Interaction of M.SsoII with TAMRA-Labelled DNA Containing the Methylation Site in the Presence of AdoHcy
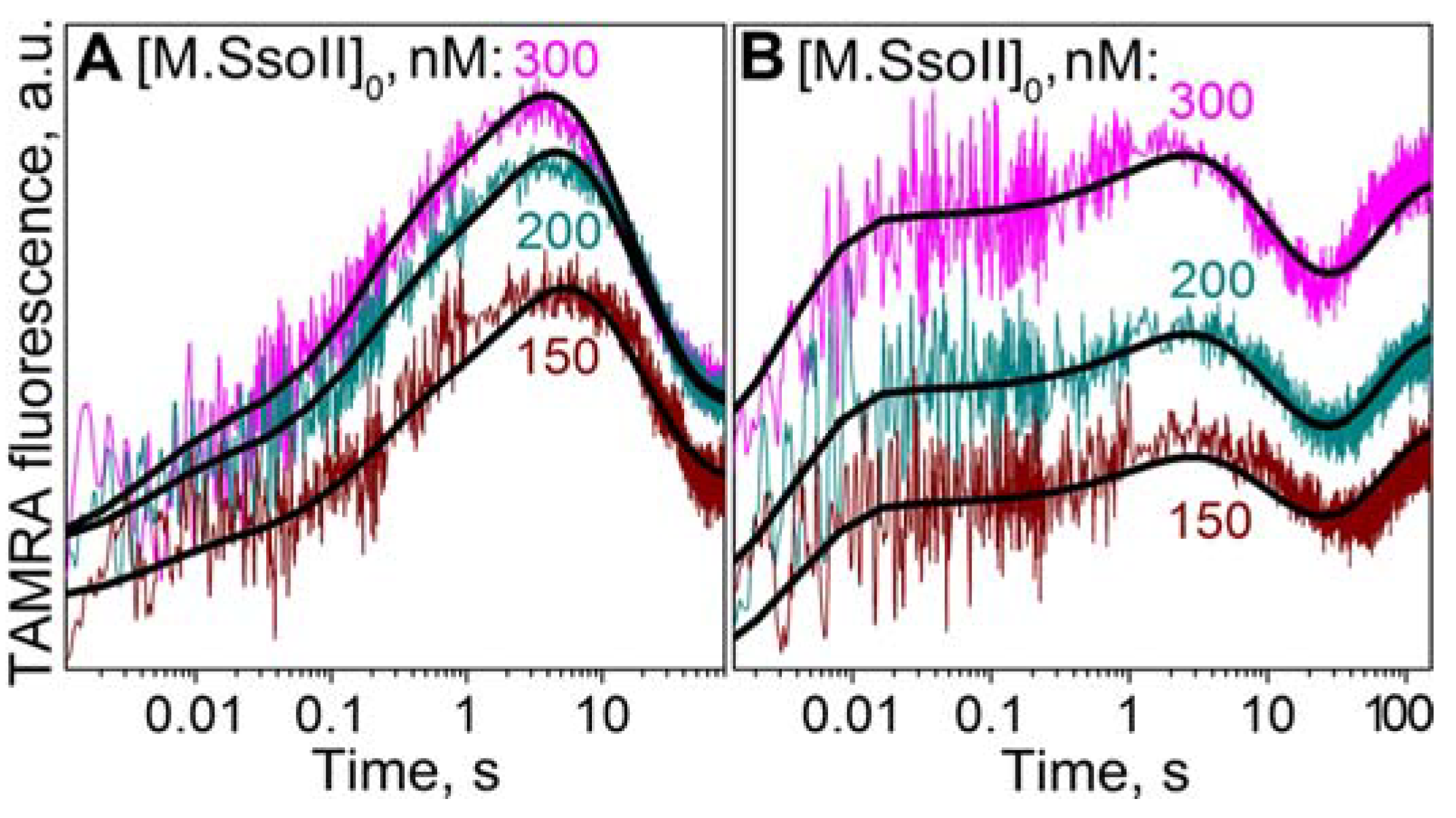
3.7.2. Interaction of M.SsoII with TAMRA-Labelled DNA Containing the Methylation Site in the Presence of AdoMet
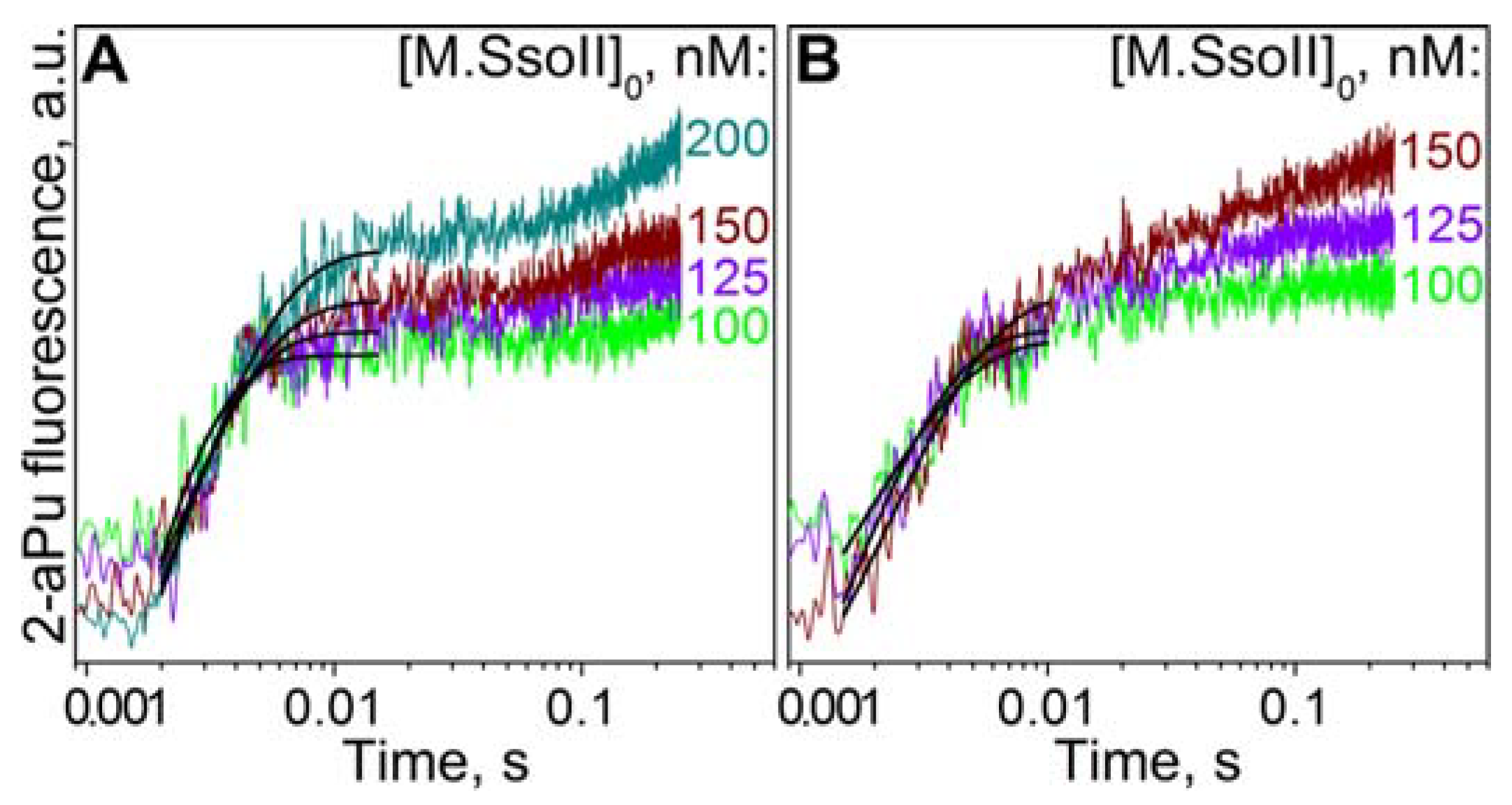
3.8. Kinetics of Flipping out of the Target Base
4. Discussion
4.1. Methyltransferase SsoII as an Enzyme that Modifies DNA
4.1.1. The Order of the DNA Substrate and AdoMet Binding
4.1.2. Kinetics of M.SsoII Interaction with DNA Substrates Containing the Unmethylated and Monomethylated CCNGG Site in the Presence of AdoMet or AdoHcy
4.2. Methyltransferase SsoII as a Transcription Factor
4.2.1. Kinetics of M.SsoII Interaction with DNA Duplexes Containing the Regulatory Site
4.2.2. Do (A/T)4 Tracts Flanking the Regulatory Site Influence the M.SsoII Binding
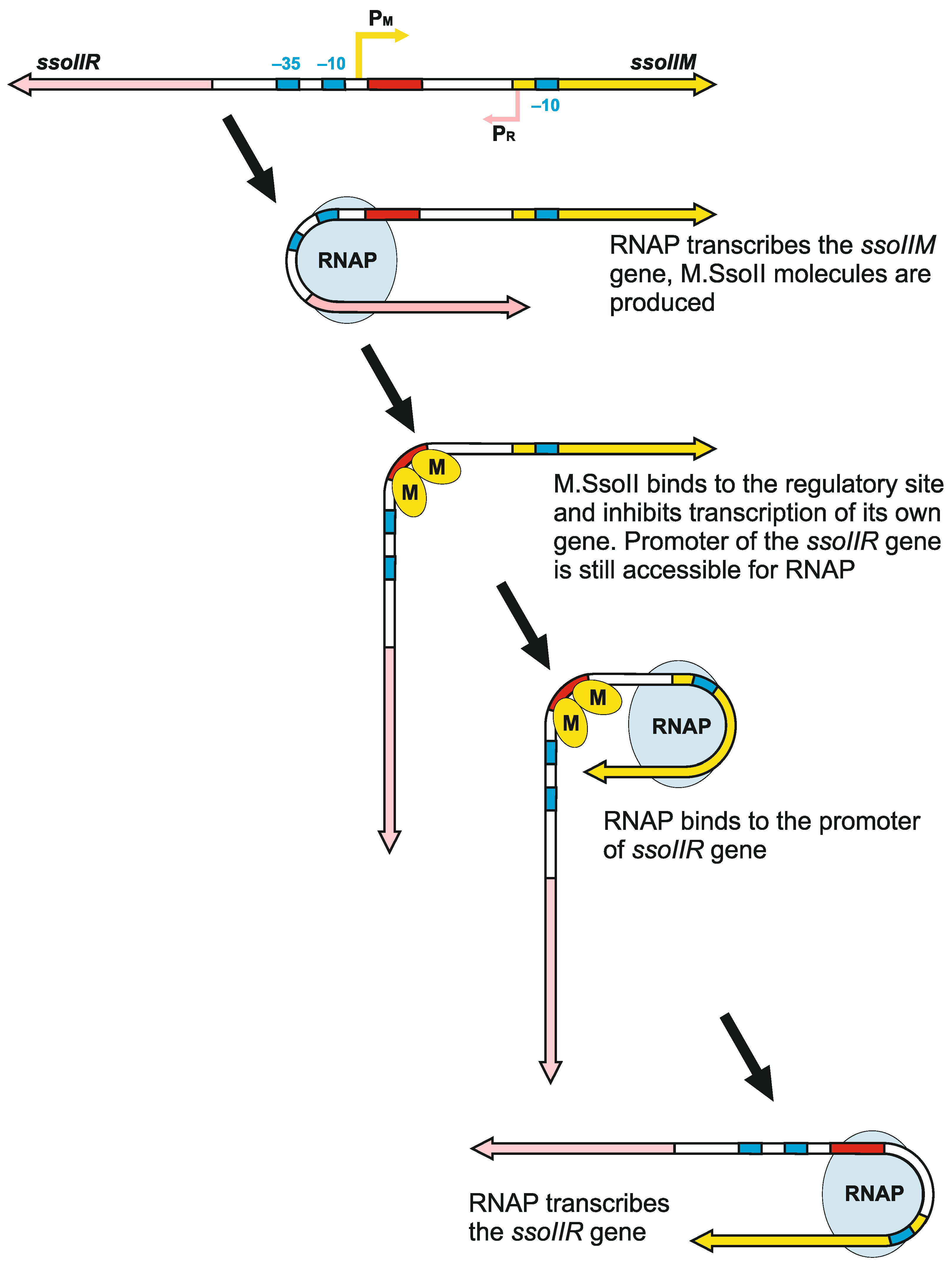
4.2.3. The Role of DNA Bending by M.SsoII in Transcriptional Regulation of the Genes in SsoII-Like RM Systems
4.3. How Does Methyltransferase SsoII Combine Its Two Mutually Exclusive Functions?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-aPu | 2-Aminopurine |
| AdoHcy | S-Adenosyl-l-homocysteine |
| AdoMet | S-Adenosyl-l-methionine |
| Cyt | Cytosine residue |
| EMSA | Electrophoretic mobility shift assay |
| MTase | DNA methyltransferase |
| M.HhaI | DNA methyltransferase HhaI |
| M.SsoII | DNA methyltransferase SsoII |
| PAGE | Polyacrylamide gel electrophoresis |
| R.SsoII | Restriction endonuclease SsoII |
| REase or R. | Restriction endonuclease |
| RM | Restriction–modification |
| RNAP | RNA polymerase |
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, E.A.; Brooks, J.E. Restriction modification systems: Where they are and what they do. In Bacterial Genomes; Springer: Boston, MA, USA, 1998; pp. 78–92. [Google Scholar]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Deng, Z. Twenty years hunting for sulfur in DNA. Protein Cell 2010, 1, 14–21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, T.G.; Yao, F.; Zhou, X.F.; Deng, Z.X.; You, D.L. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010, 38, 7133–7141. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Sinkunas, T.; Siksnys, V. Molecular mechanisms of CRISPR-mediated microbial immunity. Cell. Mol. Life Sci. 2014, 71, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 2013, 3, e26219. [Google Scholar] [CrossRef] [PubMed]
- Goeders, N.; Van Melderen, L. Toxin-Antitoxin Systems as Multilevel Interaction Systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; Jore, M.M.; Westra, E.R.; Zhu, Y.; Janssen, J.H.; Snijders, A.P.; Wang, Y.; Patel, D.J.; Berenguer, J.; Brouns, S.J.J.; et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 2014, 507, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, T.; Sberro, H.; Weinstock, E.; Cohen, O.; Doron, S.; Charpak-Amikam, Y.; Afik, S.; Ofir, G.; Sorek, R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015, 34, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Snir, S.; Koonin, E.V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 2011, 193, 6039–6056. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Abe, K.; Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010, 38, 2428–2443. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Ohashi, S.; Sadykov, M.R.; Mizutani-Ui, Y.; Kobayashi, I. IS-linked movement of a restriction-modification system. PLoS ONE 2011, 6, e16554. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, M.E.; Villion, M.; Magadan, A.H.; Moineau, S. CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat. Commun. 2013, 4, 2087. [Google Scholar] [CrossRef] [PubMed]
- Karyagina, A.S.; Lunin, V.G.; Degtyarenko, K.N.; Uvarov, V.Y.; Nikolskaya, II. Analysis of the nucleotide and derived amino acid sequences of the SsoII restriction endonuclease and methyltransferase. Gene 1993, 124, 13–19. [Google Scholar] [CrossRef]
- Davis, C.A.; Bingman, C.A.; Landick, R.; Record, M.T., Jr.; Saecker, R.M. Real-time footprinting of DNA in the first kinetically significant intermediate in open complex formation by Escherichia coli RNA polymerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7833–7838. [Google Scholar] [CrossRef] [PubMed]
- Denjmukhametov, M.M.; Brevnov, M.G.; Zakharova, M.V.; Repyk, A.V.; Solonin, A.S.; Petrauskene, O.V.; Gromova, E.S. The Ecl18kI restriction-modification system: Cloning, expression, properties of the purified enzymes. FEBS Lett. 1998, 433, 233–236. [Google Scholar] [CrossRef]
- Zakharova, M.V.; Beletskaya, I.V.; Denjmukhametov, M.M.; Yurkova, T.V.; Semenova, L.M.; Shlyapnikov, M.G.; Solonin, A.S. Characterization of pECL18 and pKPN2: A proposed pathway for the evolution of two plasmids that carry identical genes for a Type II restriction-modification system. Mol. Genet. Genom. 2002, 267, 171–178. [Google Scholar]
- Miyahara, M.; Ishiwata, N.; Yoshida, Y. StyD4I restriction-modification system of Salmonella typhi D4: Cloning and sequence analysis. Biol. Pharm. Bull. 1997, 20, 201–203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ibanez, M.; Alvarez, I.; RodriguezPena, J.M.; Rotger, R. A Co1E1-type plasmid from Salmonella enteritidis encodes a DNA cytosine methyltransferase. Gene 1997, 196, 145–158. [Google Scholar] [CrossRef]
- Konarev, P.V.; Kachalova, G.S.; Ryazanova, A.Y.; Kubareva, E.A.; Karyagina, A.S.; Bartunik, H.D.; Svergun, D.I. Flexibility of the linker between the domains of DNA methyltransferase SsoII revealed by small-angle X-ray scattering: Implications for transcription regulation in SsoII restriction-modification system. PLoS ONE 2014, 9, e93453. [Google Scholar] [CrossRef] [PubMed]
- Nikolskaya, I.I.; Lopatina, N.G.; Suchkov, S.V.; Kartashova, I.M.; Debov, S.S. Sequence specificity of isolated DNA-cytosine methylases from Shigella sonnei 47 cells. Biochem. Int. 1984, 9, 771–781. [Google Scholar]
- Kubareva, E.A.; Walter, J.; Karyagina, A.S.; Vorob’eva, O.V.; Lau, P.C.; Trautner, T. Determination of methylation site of DNA-methyltransferase NlaX by a hybrid method. Biotechniques 2002, 33, 526–531. [Google Scholar] [PubMed]
- Vorob’eva, O.V.; Kariagina, A.S.; Volkov, E.M.; Viriasov, M.B.; Oretskaia, T.S.; Kubareva, E.A. An analysis of methyltransferase SsoII-DNA contacts in the enzyme-substrate complex. Bioorganicheskaia Khimiia 2002, 28, 402–410. [Google Scholar] [PubMed]
- Vorob’eva, O.V.; Romanenkov, A.S.; Metelev, V.G.; Kariagina, A.S.; Lavrova, N.V.; Oretskaia, T.S.; Kubareva, E.A. Covalent binding of Cys142 from SsoII methyltransferase with DNA duplexes, containing a phosphoryldisulfide internucleotide group. Mol. Biol. 2003, 37, 906–915. [Google Scholar]
- Karyagina, A.; Shilov, I.; Tashlitskii, V.; Khodoun, M.; Vasil’ev, S.; Lau, P.C.; Nikolskaya, I. Specific binding of sso II DNA methyltransferase to its promoter region provides the regulation of sso II restriction-modification gene expression. Nucleic Acids Res. 1997, 25, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Shilov, I.; Tashlitsky, V.; Khodoun, M.; Vasil’ev, S.; Alekseev, Y.; Kuzubov, A.; Kubareva, E.; Karyagina, A. DNA-methyltransferase SsoII interaction with own promoter region binding site. Nucleic Acids Res. 1998, 26, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Vorob’eva, O.V.; Vasil’ev, S.A.; Kariagina, A.S.; Oretskaia, T.S.; Kubareva, E.A. Analysis of contacts between DNA and protein in a complex of SsoII mmethyltransferase-promoter region of the gene for the SsoII restriction-modification genes. Mol. Biol. 2000, 34, 1074–1080. [Google Scholar]
- Ryazanova, A.Y.; Molochkov, N.V.; Abrosimova, L.A.; Alexeevsky, A.V.; Karyagina, A.S.; Protsenko, A.S.; Friedhoff, P.; Oretskaya, T.S.; Kubareva, E.A. Secondary structure of SsoII-like (Cytosine-5)-DNA methyltransferases N-terminal region determined by Circular dichroism spectroscopy. Mol. Biol. 2010, 44, 807–816. [Google Scholar] [CrossRef]
- Ryazanova, A.Y.; Winkler, I.; Friedhoff, P.; Viryasov, M.B.; Oretskaya, T.S.; Kubareva, E.A. Crosslinking of (cytosine-5)-DNA methyltransferase SsoII and its complexes with specific DNA duplexes provides an insight into their structures. Nucleosides Nucleotides Nucleic Acids 2011, 30, 632–650. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, D.V.; Mokrishcheva, M.L.; Solonin, A.S. Binding of DNA methyltransferase M.Ecl18kI [corrected] to operator-promoter region decreases its methylating activity. Biochemistry 2012, 77, 307–311. [Google Scholar] [PubMed]
- Karyagina, A.S.; Lunin, V.G.; Levtchenko, I.; Labbe, D.; Brousseau, R.; Lau, P.C.; Nikolskaya, II. The SsoII and NlaX DNA methyltransferases: Overproduction and functional analysis. Gene 1995, 157, 93–96. [Google Scholar] [CrossRef]
- Yang, F.; Yang, J.; Zhang, X.; Chen, L.; Jiang, Y.; Yan, Y.; Tang, X.; Wang, J.; Xiong, Z.; Dong, J.; et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 2005, 33, 6445–6458. [Google Scholar] [CrossRef] [PubMed]
- Clewell, D.B.; Helinski, D.R. Effect of growth conditions on the formation of the relaxation complex of supercoiled ColE1 deoxyribonucleic acid and protein in Escherichia coli. J. Bacteriol. 1972, 110, 1135–1146. [Google Scholar] [PubMed]
- Hershfield, V.; Boyer, H.W.; Yanofsky, C.; Lovett, M.A.; Helinski, D.R. Plasmid ColEl as a molecular vehicle for cloning and amplification of DNA. Proc. Natl. Acad. Sci. USA 1974, 71, 3455–3459. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.; Johnson, D.H.; McDonald, H.; Brouillette, C.; Delucas, L.J. His-tag impact on structure. Acta Crystallogr. D Biol. Crystallogr. 2007, 63 Pt 3, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Piekarowicz, A.; Brzezinski, R. Cleavage and methylation of DNA by the restriction endonuclease HinfIII isolated from Haemophilus influenzae Rf. J. Mol. Biol. 1980, 144, 415–429. [Google Scholar] [CrossRef]
- Kumar, S.; Cheng, X.; Pflugrath, J.W.; Roberts, R.J. Purification, crystallization, and preliminary X-ray diffraction analysis of an M.HhaI-AdoMet complex. Biochemistry 1992, 31, 8648–8653. [Google Scholar] [CrossRef] [PubMed]
- Szczelkun, M.D.; Connolly, B.A. Sequence-specific binding of DNA by the EcoRV restriction and modification enzymes with nucleic acid and cofactor analogues. Biochemistry 1995, 34, 10724–10733. [Google Scholar] [CrossRef] [PubMed]
- Schluckebier, G.; Kozak, M.; Bleimling, N.; Weinhold, E.; Saenger, W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997, 265, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Weber, G. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 1973, 12, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Samworth, C.M.; Degli Esposti, M.; Lenaz, G. Quenching of the intrinsic tryptophan fluorescence of mitochondrial ubiquinol–cytochrome-c reductase by the binding of ubiquinone. Eur. J. Biochem. 1988, 171, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zwieb, C.; Wu, C.; Adhya, S. Bending of DNA by gene-regulatory proteins: Construction and use of a DNA bending vector. Gene 1989, 85, 15–23. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Ferrari, S.; Harley, V.R.; Pontiggia, A.; Goodfellow, P.N.; Lovell-Badge, R.; Bianchi, M.E. SRY, like HMG1, recognizes sharp angles in DNA. EMBO J. 1992, 11, 4497–4506. [Google Scholar] [PubMed]
- Fedorova, O.S.; Nevinsky, G.A.; Koval, V.V.; Ishchenko, A.A.; Vasilenko, N.L.; Douglas, K.T. Stopped-flow kinetic studies of the interaction between Escherichia coli Fpg protein and DNA substrates. Biochemistry 2002, 41, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Kuzmic, P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Timofeyeva, N.A.; Koval, V.V.; Knorre, D.G.; Zharkov, D.O.; Saparbaev, M.K.; Ishchenko, A.A.; Fedorova, O.S. Conformational dynamics of human AP endonuclease in base excision and nucleotide incision repair pathways. J. Biomol. Struct. Dyn. 2009, 26, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Toseland, C.P. Fluorescence to study the ATPase mechanism of motor proteins. EXS 2014, 105, 67–86. [Google Scholar] [PubMed]
- Vilkaitis, G.; Merkiene, E.; Serva, S.; Weinhold, E.; Klimasauskas, S. The mechanism of DNA cytosine-5 methylation. Kinetic and mutational dissection of Hhai methyltransferase. J. Biol. Chem. 2001, 276, 20924–20934. [Google Scholar] [CrossRef] [PubMed]
- Sankpal, U.T.; Rao, D.N. Mutational analysis of conserved residues in HhaI DNA methyltransferase. Nucleic Acids Res. 2002, 30, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, W.M., Jr.; Flynn, J.; Reich, N.O. Reconciling structure and function in HhaI DNA cytosine-C-5 methyltransferase. J. Biol. Chem. 2000, 275, 4912–4919. [Google Scholar] [CrossRef] [PubMed]
- Ryazanova, A.Y.; Kubareva, E.A.; Grman, I.; Lavrova, N.V.; Ryazanova, E.M.; Oretskaya, T.S.; Hianik, T. The study of the interaction of (cytosine-5)-DNA methyltransferase SsoII with DNA by acoustic method. Analyst 2011, 136, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Wilson, G.G.; Wende, W. Type II restriction endonucleases—A historical perspective and more. Nucleic Acids Res. 2014, 42, 7489–7527. [Google Scholar] [CrossRef] [PubMed]
- Haran, T.E.; Mohanty, U. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009, 42, 41–81. [Google Scholar] [CrossRef] [PubMed]
- Papapanagiotou, I.; Streeter, S.D.; Cary, P.D.; Kneale, G.G. DNA structural deformations in the interaction of the controller protein C.AhdI with its operator sequence. Nucleic Acids Res. 2007, 35, 2643–2650. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ball, N.J.; McGeehan, J.E.; Streeter, S.D.; Thresh, S.J.; Kneale, G.G. The structural basis of differential DNA sequence recognition by restriction-modification controller proteins. Nucleic Acids Res. 2012, 40, 10532–10542. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Bustamante, C.J.; Reich, N.O. Sequence-specific recognition of cytosine C5 and adenine N6 DNA methyltransferases requires different deformations of DNA. Proc. Natl. Acad. Sci. USA 1996, 93, 7618–7622. [Google Scholar] [CrossRef] [PubMed]
- Cal, S.; Connolly, B.A. The EcoRV modification methylase causes considerable bending of DNA upon binding to its recognition sequence GATATC. J. Biol. Chem. 1996, 271, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Dubey, A.K.; Bhattacharya, S.K. Angle and locus of the bend induced by the msp I DNA methyltransferase in a sequence-specific complex with DNA. Nucleic Acids Res. 1997, 25, 2025–2029. [Google Scholar] [CrossRef] [PubMed]
- Rasko, T.; Finta, C.; Kiss, A. DNA bending induced by DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 2000, 28, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Romanenkov, A.S.; Kisil, O.V.; Zatsepin, T.S.; Yamskova, O.V.; Karyagina, A.S.; Metelev, V.G.; Oretskaya, T.S.; Kubareva, E.A. DNA-methyltransferase SsoII as a bifunctional protein: Features of the interaction with the promoter region of SsoII restriction-modification genes. Biochemistry 2006, 71, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.F.; Landy, A. Empirical estimation of protein-induced DNA bending angles: Applications to lambda site-specific recombination complexes. Nucleic Acids Res. 1988, 16, 9687–9705. [Google Scholar] [CrossRef] [PubMed]
- Unruh, J.R.; Gokulrangan, G.; Lushington, G.H.; Johnson, C.K.; Wilson, G.S. Orientational dynamics and dye-DNA interactions in a dye-labeled DNA aptamer. Biophys. J. 2005, 88, 3455–3465. [Google Scholar] [CrossRef] [PubMed]
- Vamosi, G.; Gohlke, C.; Clegg, R.M. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: Multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 1996, 71, 972–994. [Google Scholar] [CrossRef]
- Gerasimaite, R.; Merkiene, E.; Klimasauskas, S. Direct observation of cytosine flipping and covalent catalysis in a DNA methyltransferase. Nucleic Acids Res. 2011, 39, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- Daujotyte, D.; Serva, S.; Vilkaitis, G.; Merkiene, E.; Venclovas, C.; Klimasauskas, S. HhaI DNA methyltransferase uses the protruding Gln237 for active flipping of its target cytosine. Structure 2004, 12, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Viollier, P. Two-in-one: Bifunctional regulators synchronizing developmental events in bacteria. Trends Cell Biol. 2012, 22, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Santi, D.V. Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 1987, 262, 4778–4786. [Google Scholar] [PubMed]
- Vilkaitis, G.; Dong, A.; Weinhold, E.; Cheng, X.; Klimasauskas, S. Functional roles of the conserved threonine 250 in the target recognition domain of HhaI DNA methyltransferase. J. Biol. Chem. 2000, 275, 38722–38730. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, C.P.; Sankpal, U.T.; Rao, D.N.; Surolia, A. Water-assisted dual mode cofactor recognition by HhaI DNA methyltransferase. J. Biol. Chem. 2002, 277, 4042–4049. [Google Scholar] [CrossRef] [PubMed]
- O’Gara, M.; Zhang, X.; Roberts, R.J.; Cheng, X. Structure of a binary complex of HhaI methyltransferase with S-adenosyl-l-methionine formed in the presence of a short non-specific DNA oligonucleotide. J. Mol. Biol. 1999, 287, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Javor, G.T. Depression of adenosylmethionine content of Escherichia coli by thioglycerol. Antimicrob. Agents Chemother. 1983, 24, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A. S-adenosylmethionine: Jack of all trades and master of everything? Biochem. Soc. Trans. 2006, 34 Pt 2, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Merkiene, E.; Klimasauskas, S. Probing a rate-limiting step by mutational perturbation of AdoMet binding in the HhaI methyltransferase. Nucleic Acids Res. 2005, 33, 307–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banavali, N.K.; MacKerell, A.D., Jr. Free energy and structural pathways of base flipping in a DNA GCGC containing sequence. J. Mol. Biol. 2002, 319, 141–160. [Google Scholar] [CrossRef]
- Parker, J.B.; Bianchet, M.A.; Krosky, D.J.; Friedman, J.I.; Amzel, L.M.; Stivers, J.T. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 2007, 449, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Barbic, A.; Zimmer, D.P.; Crothers, D.M. Structural origins of adenine-tract bending. Proc. Natl. Acad. Sci. USA 2003, 100, 2369–2373. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, A.; Zakharova, M.; Nagornykh, M.; Solonin, A.; Severinov, K. Transcription regulation of restriction-modification system Ecl18kI. Nucleic Acids Res. 2009, 37, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Burenina, O.Y.; Fedotova, E.A.; Ryazanova, A.Y.; Protsenko, A.S.; Zakharova, M.V.; Karyagina, A.S.; Solonin, A.S.; Oretskaya, T.S.; Kubareva, E.A. Peculiarities of the Regulation of Gene Expression in the Ecl18kI Restriction-Modification System. Acta Nat. 2013, 5, 70–80. [Google Scholar]
- Morozova, N.; Sabantsev, A.; Bogdanova, E.; Fedorova, Y.; Maikova, A.; Vedyaykin, A.; Rodic, A.; Djordjevic, M.; Khodorkovskii, M.; Severinov, K. Temporal dynamics of methyltransferase and restriction endonuclease accumulation in individual cells after introducing a restriction-modification system. Nucleic Acids Res. 2016, 44, 790–800. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
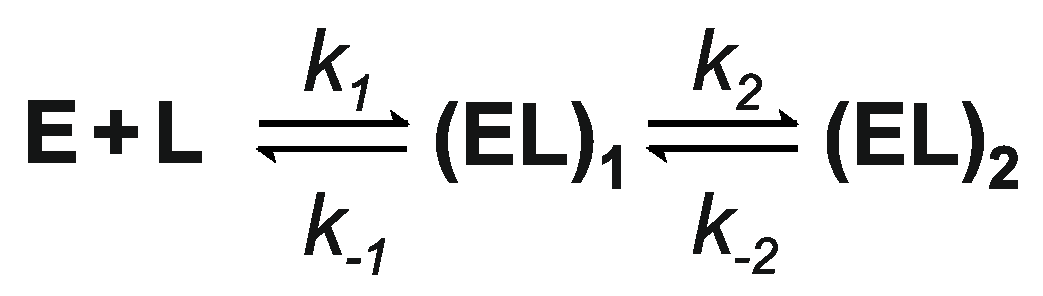{kind=link}
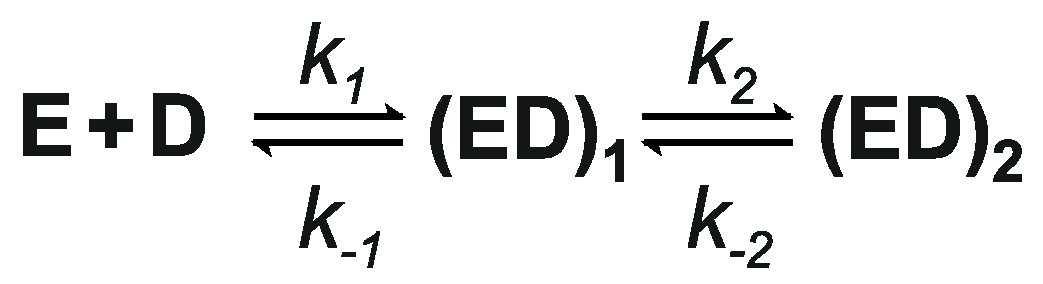{kind=link}
{kind=link}
{kind=link}
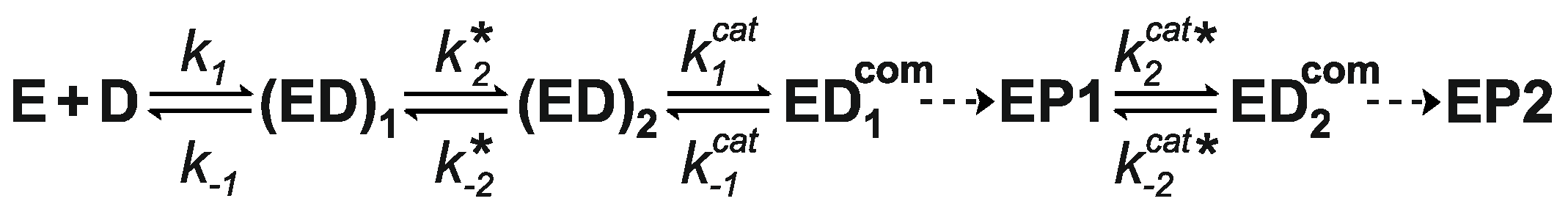{kind=link}
{kind=link}
| Ligand | k1, μM−1·s−1 | k−1, s−1 | k2, s−1 | k−2, s−1 |
|---|---|---|---|---|
| AdoMet | 6.9 ± 0.3 | 93 ± 4 | 7.0 ± 0.3 | 7.9 ± 0.3 |
| DNA–Protein Complex | DNA Bending Angle, Degrees | Number of Experiments | R2adjusted ** |
|---|---|---|---|
| 162reg-1 + M.SsoII | 46 ± 6 * | 5 | 0.739–0.974 |
| 162reg-1 + 2M.SsoII | 89 ± 5 | 6 | 0.946–0.996 |
| 162reg-2 + M.SsoII | 43 ± 6 | 6 | 0.714–0.909 |
| 162reg-2 + 2M.SsoII | 88 ± 4 | 6 | 0.966–0.998 |
| 162met + M.SsoII | 31 ± 9 | 5 | 0.595–0.948 |
| DNA Duplex | Cofactor | k1, μM−1·s−1 | k−1, s−1 | k2, s−1 | k−2, s−1 |
|---|---|---|---|---|---|
| 60reg-1 | AdoMet | 390 ± 8 | 93 ± 1 | 1.29 ± 0.04 | 11.0 ± 0.4 |
| AdoHcy | 352 ± 7 | 71 ± 1 | 2.30 ± 0.04 | 12.8 ± 0.3 | |
| — | 470 ± 10 | 127 ± 1 | 1.14 ± 0.05 | 10.0 ± 0.5 | |
| 60reg-2 | AdoMet | 390 ± 12 | 88 ± 1 | 1.13 ± 0.02 | 9.6 ± 0.2 |
| AdoHcy | 350 ± 11 | 52 ± 1 | 1.18 ± 0.02 | 7.6 ± 0.2 | |
| — | 430 ± 11 | 109 ± 1 | 1.06 ± 0.02 | 11.1 ± 0.2 | |
| 60reg-4 | AdoMet | 386 ± 7 | 154 ± 1 | 0.68 ± 0.02 | 6.5 ± 0.3 |
| AdoHcy | 342 ± 5 | 110 ± 1 | 1.15 ± 0.02 | 7.5 ± 0.2 | |
| — | 468 ± 9 | 195 ± 1 | 0.69 ± 0.03 | 5.4 ± 0.4 |
| AdoHcy | AdoMet | |||||||
|---|---|---|---|---|---|---|---|---|
| 60met-mC | 60met | 60met-mC | 60met | |||||
| Figure 6A, TAMRA Anisotropy Analysis | Figure 7A, TAMRA Fluorescence Analysis | Figure 6B, TAMRA Anisotropy Analysis | Figure 7B, TAMRA Fluorescence Analysis | Figure 9A, TAMRA Anisotropy Analysis | Figure 8A, TAMRA Fluorescence Analysis | Figure 9B, TAMRA Anisotropy Analysis | Figure 8B, TAMRA Fluorescence Analysis | |
| k1, μM−1·s−1 | 226 ± 3 | 226 a | 181 ± 3 | 324 ± 4 | 223 ± 4 | 230 ± 4 | 341 ± 7 | 338 ± 5 |
| k−1, s−1 | 158 ± 1 | 158 a | 143 ± 1 | 159 ± 1 | 183 ± 2 | 139 ± 1 | 254 ± 3 | 162 ± 1 |
| k2 *, s−1 | 0.22 ± 0.01 | 0.40 ± 0.02 | 0.18 ± 0.04 | |||||
| k−2 *, s−1 | 0.50 ± 0.02 | 1.5 ± 0.1 | 0.25 ± 0.16 | |||||
| k2 **, s−1 | 0.025 ± 0.003 | |||||||
| k−2 **,s−1 | 0.094 ± 0.007 | |||||||
| k2, s−1 | 3.0 ± 0.1 | 7.0 ± 0.2 | 7.6 ± 0.1 | |||||
| k−2, s−1 | 13.0 ± 0.1 | 3.7 ± 0.2 | 3.1 ± 0.1 | |||||
| k3, s−1 | 0.48 ± 0.004 | 0.46 ± 0.06 | 0.64 ± 0.05 | |||||
| k−3, s−1 | 0.21 ± 0.002 | 0.31 ± 0.03 | 0.19 ± 0.02 | |||||
| k4, s−1 | 0.0015 ± 0.0001 | |||||||
| k−4, s−1 | 0.0078 ± 0.0009 | |||||||
| , s−1 | 0.36 ± 0.02 | |||||||
| , s−1 | 0.053 ± 0.006 | |||||||
| , s−1 | 0.068 ± 0.001 | 0.057 ± 0.001 | ||||||
| , s−1 | 0.045 ± 0.001 | 0.030 ± 0.001 | ||||||
| *, s−1 | 0.013 ± 0.001 | 0.018 ± 0.001 | ||||||
| *, s−1 | 0.017 ± 0.001 | 0.021 ± 0.001 | ||||||
| 60met-mC-aPu | 60met-aPu | |||||
|---|---|---|---|---|---|---|
| AdoMet | AdoHcy | - | AdoMet | AdoHcy | - | |
| fl, s−1 | 483 ± 10 | 472 ± 8 | 494 ± 14 | 416 ± 12 | 640 ±33 | 408 ± 29 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timofeyeva, N.A.; Ryazanova, A.Y.; Norkin, M.V.; Oretskaya, T.S.; Fedorova, O.S.; Kubareva, E.A. Kinetic Basis of the Bifunctionality of SsoII DNA Methyltransferase. Molecules 2018, 23, 1192. https://doi.org/10.3390/molecules23051192
Timofeyeva NA, Ryazanova AY, Norkin MV, Oretskaya TS, Fedorova OS, Kubareva EA. Kinetic Basis of the Bifunctionality of SsoII DNA Methyltransferase. Molecules. 2018; 23(5):1192. https://doi.org/10.3390/molecules23051192
Chicago/Turabian StyleTimofeyeva, Nadezhda A., Alexandra Yu. Ryazanova, Maxim V. Norkin, Tatiana S. Oretskaya, Olga S. Fedorova, and Elena A. Kubareva. 2018. "Kinetic Basis of the Bifunctionality of SsoII DNA Methyltransferase" Molecules 23, no. 5: 1192. https://doi.org/10.3390/molecules23051192
APA StyleTimofeyeva, N. A., Ryazanova, A. Y., Norkin, M. V., Oretskaya, T. S., Fedorova, O. S., & Kubareva, E. A. (2018). Kinetic Basis of the Bifunctionality of SsoII DNA Methyltransferase. Molecules, 23(5), 1192. https://doi.org/10.3390/molecules23051192