
Novel-Substituted Heterocyclic GABA Analogues. Enzymatic Activity against the GABA-AT Enzyme from Pseudomonas fluorescens and In Silico Molecular Modeling

 , , and
, , and
Abstract
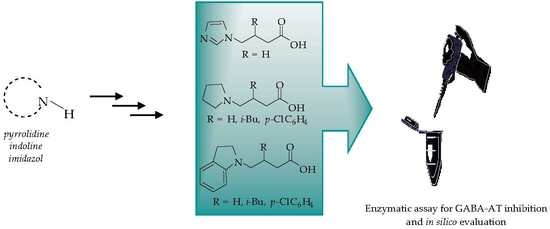
1. Introduction
2. Results and Discussion
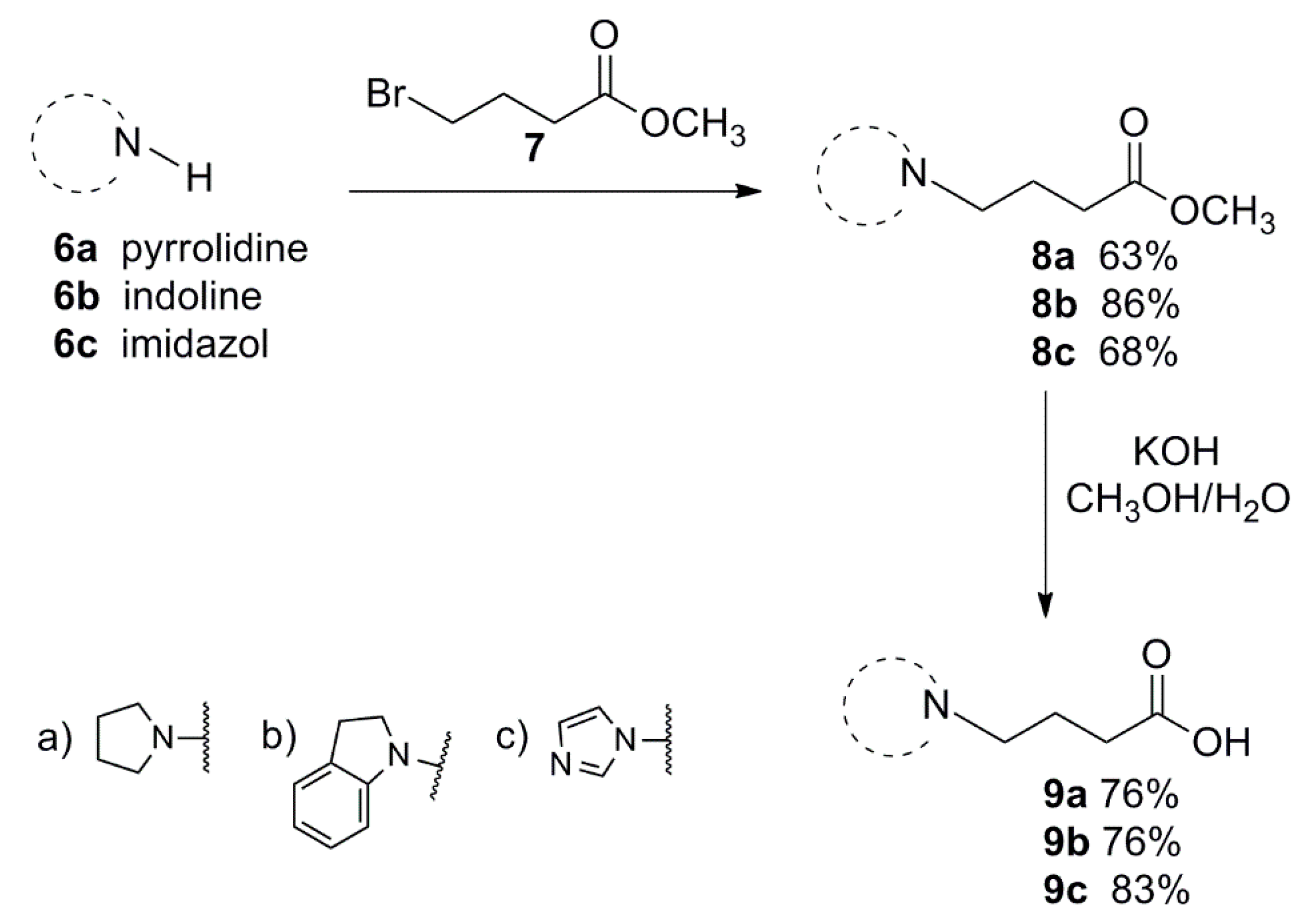
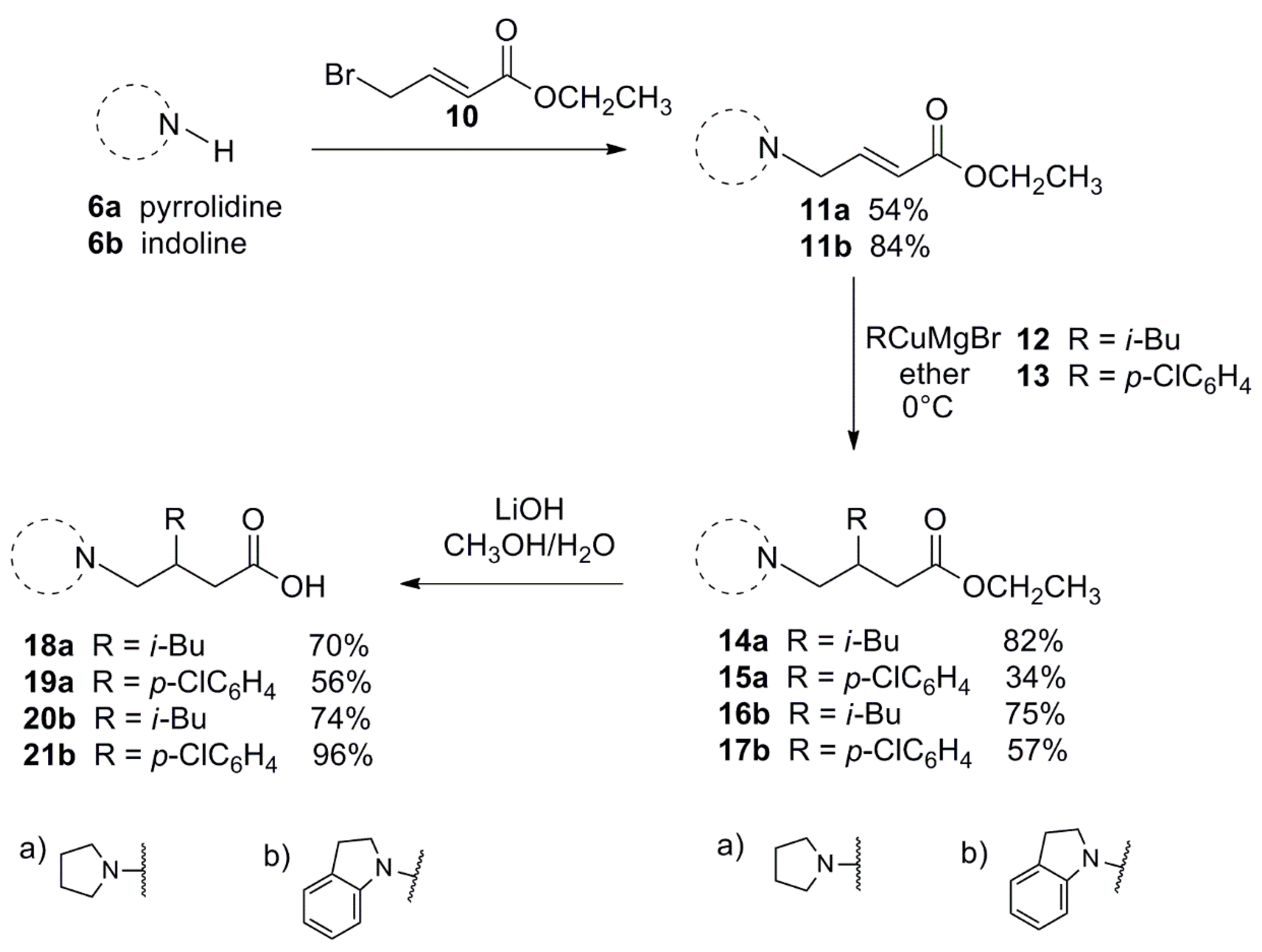
2.1. Chemistry
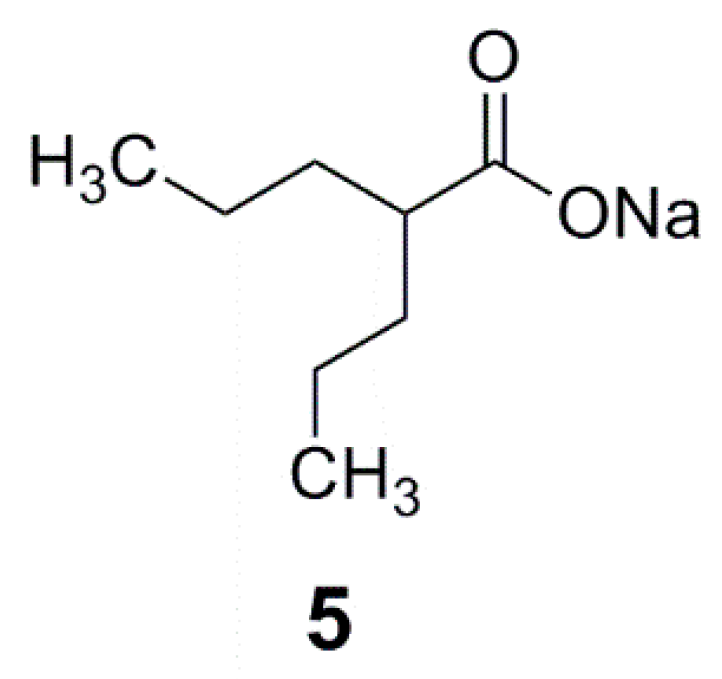
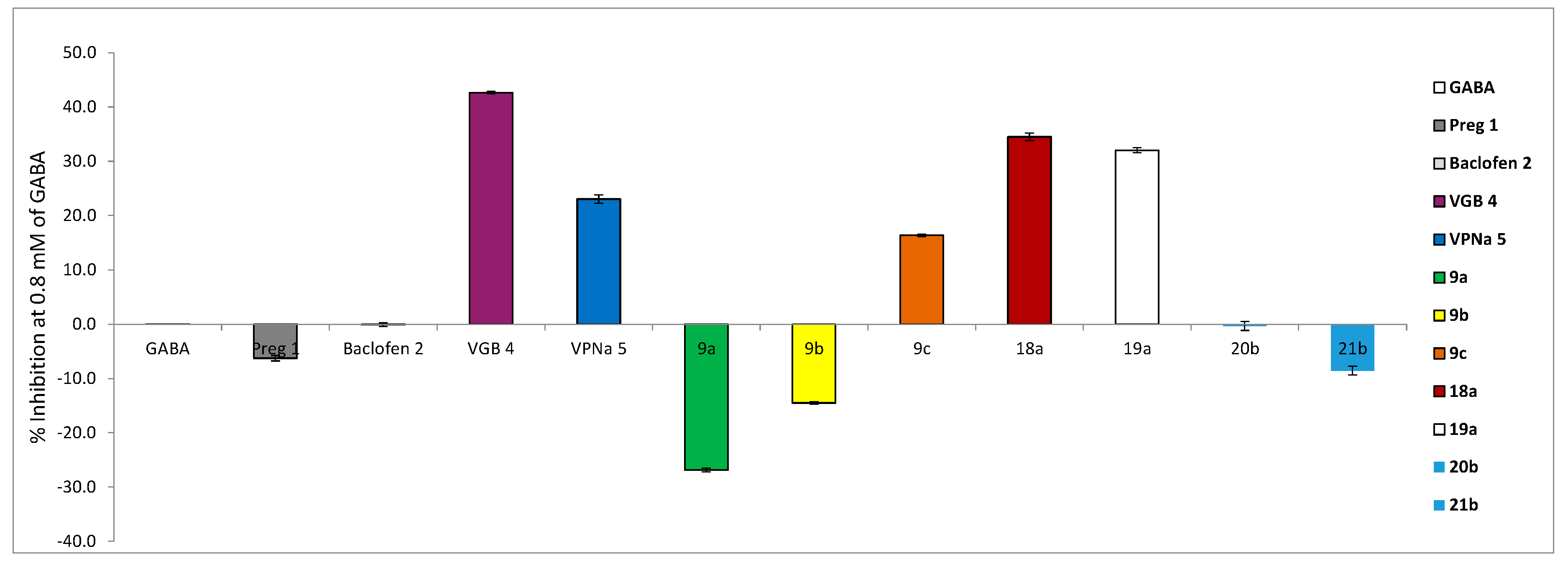
2.2. GABA-AT Inhibition
2.3. Computational Studies
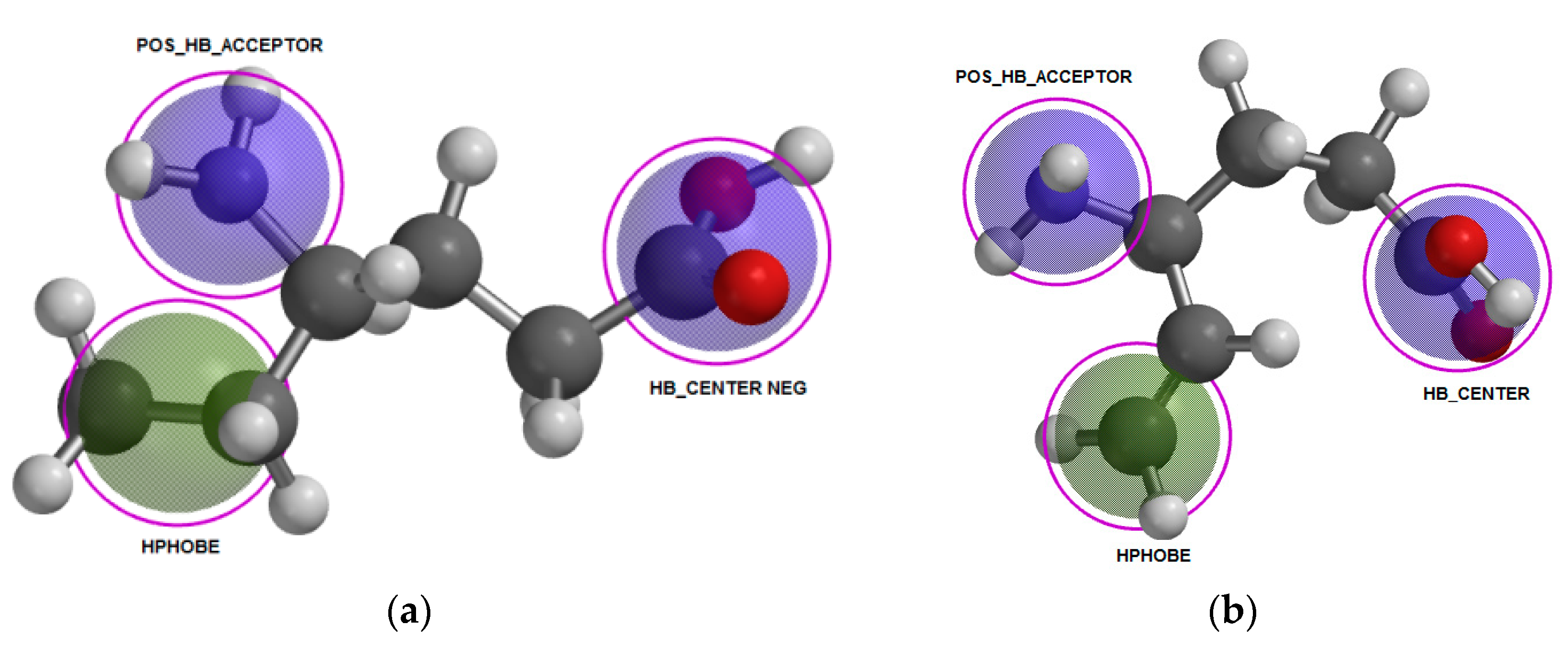
2.3.1. Conformational Analysis and Geometric Optimization
2.3.2. Natural Partial Charges
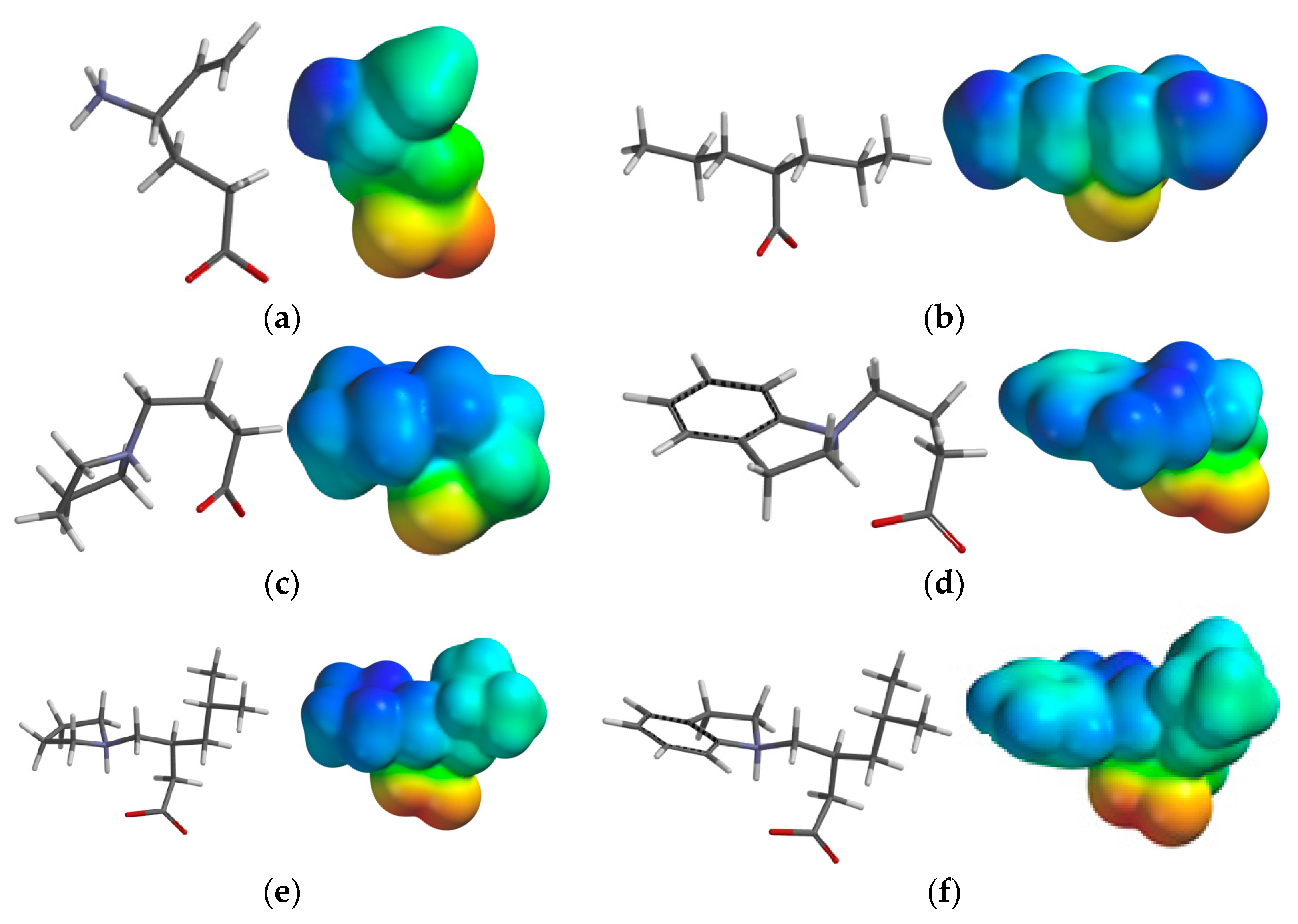
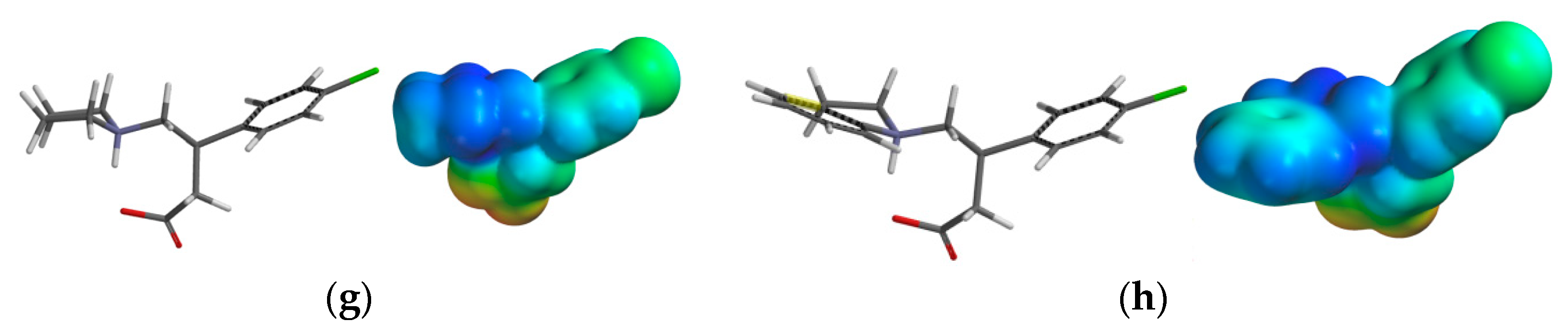
2.3.3. Electrostatic Potential Maps
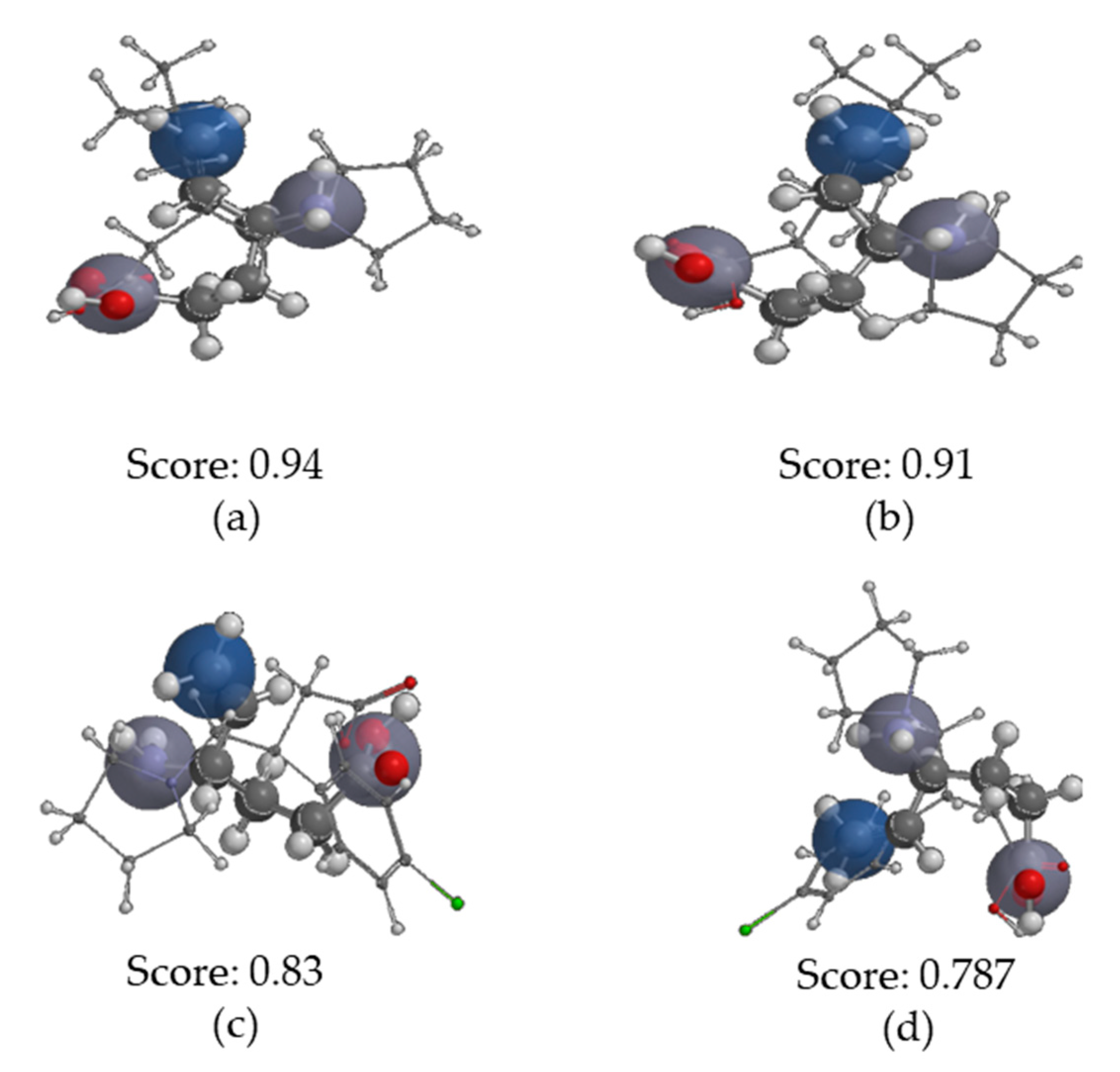
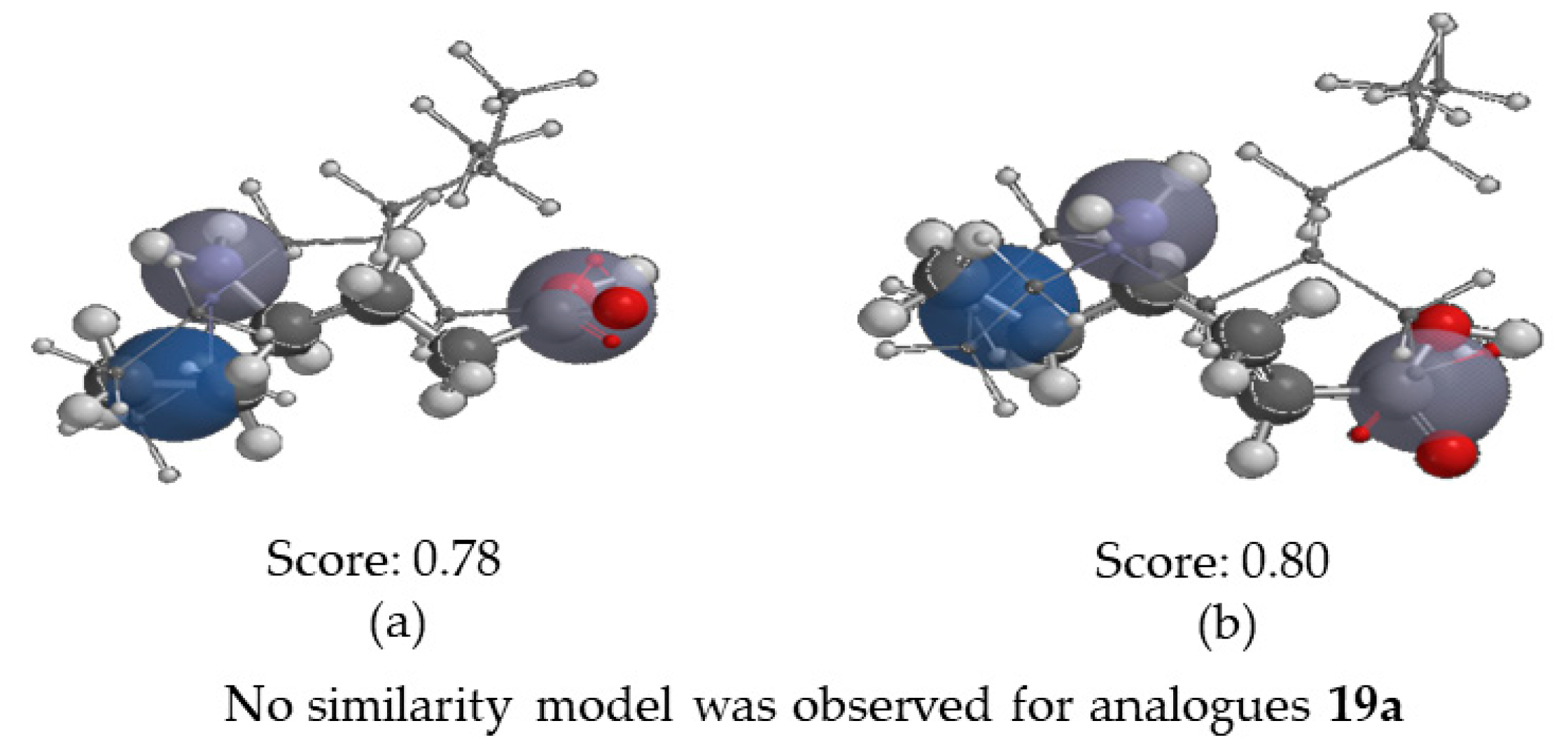
2.3.4. Molecular Similarity Analysis (MSA)
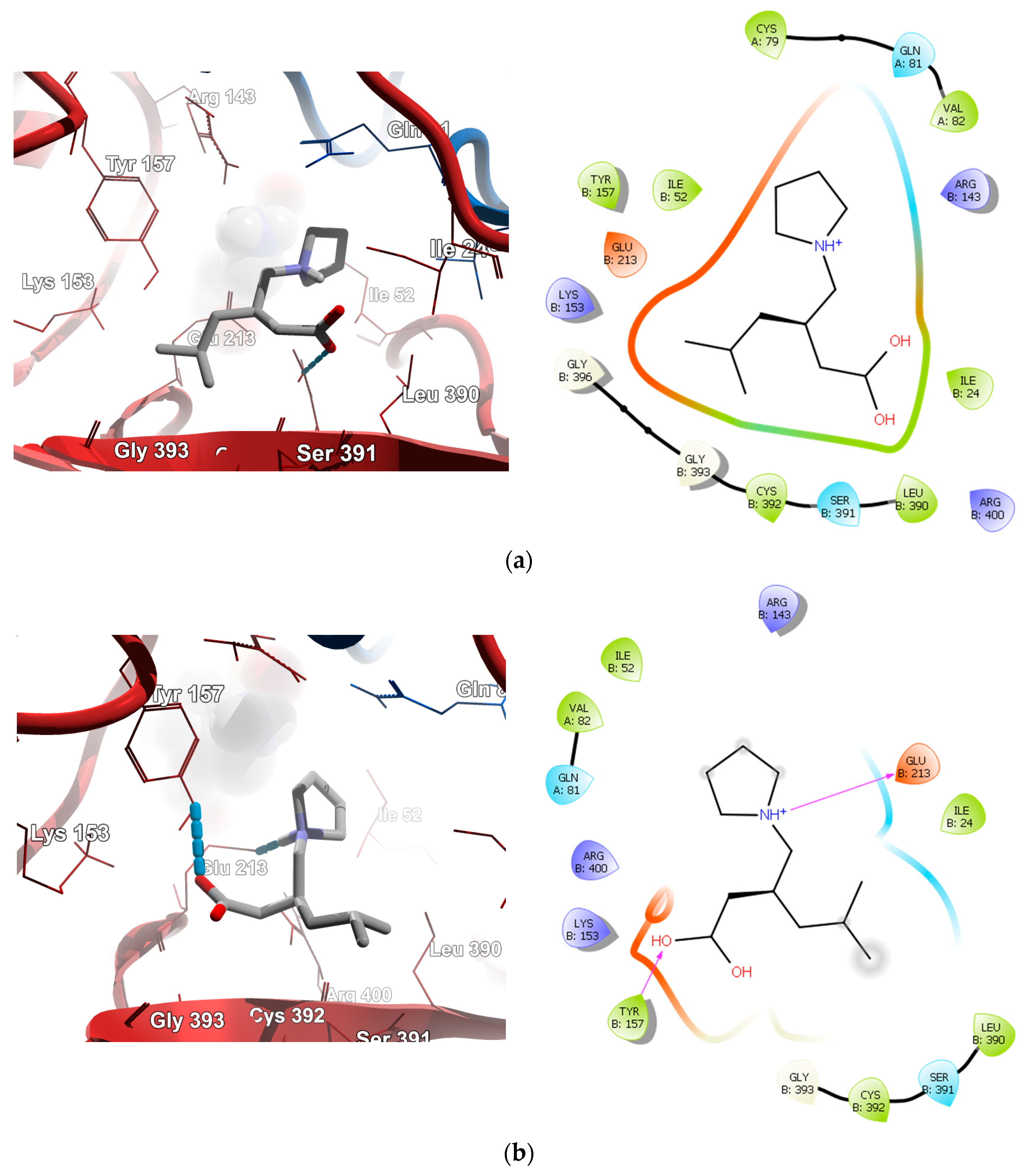
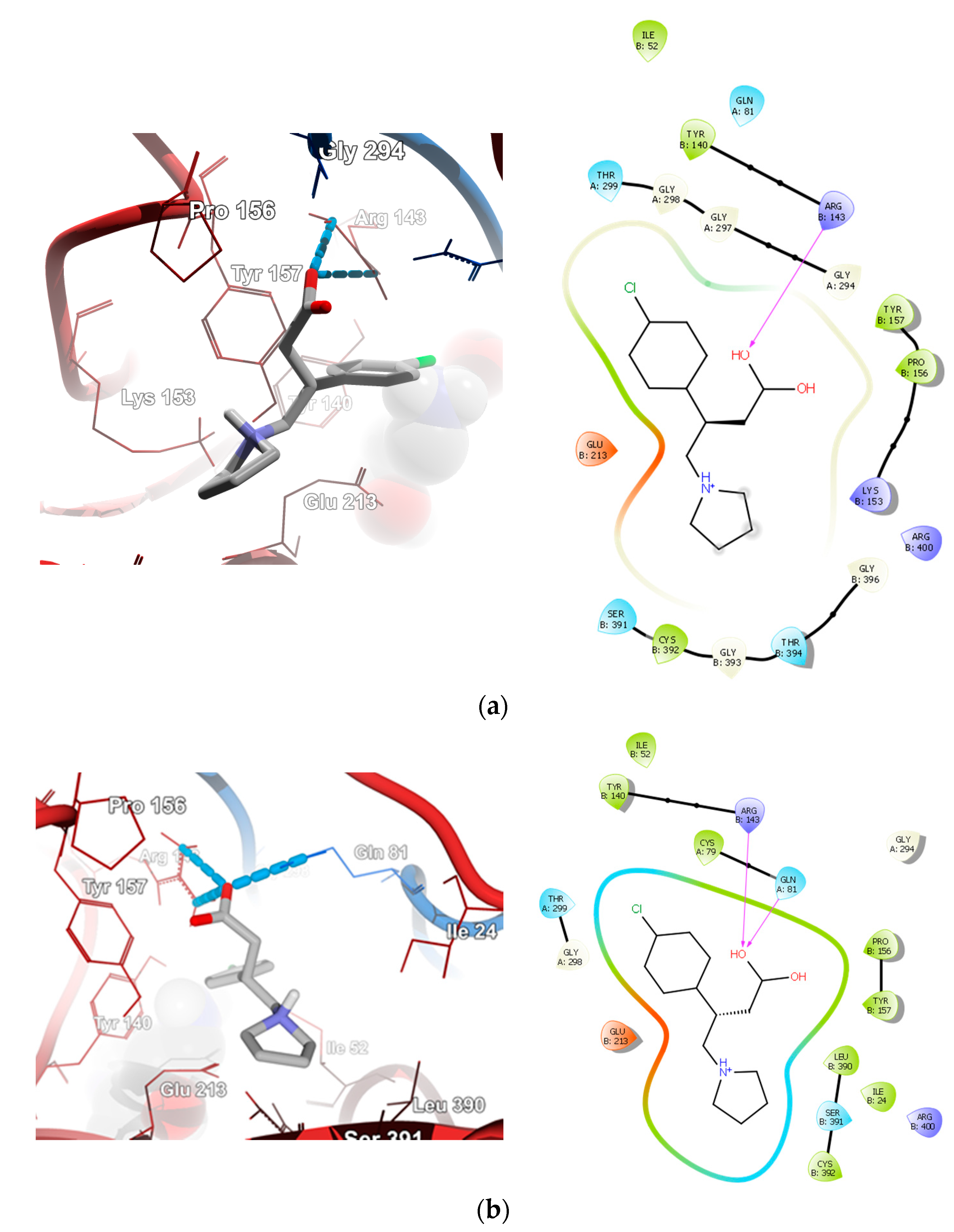
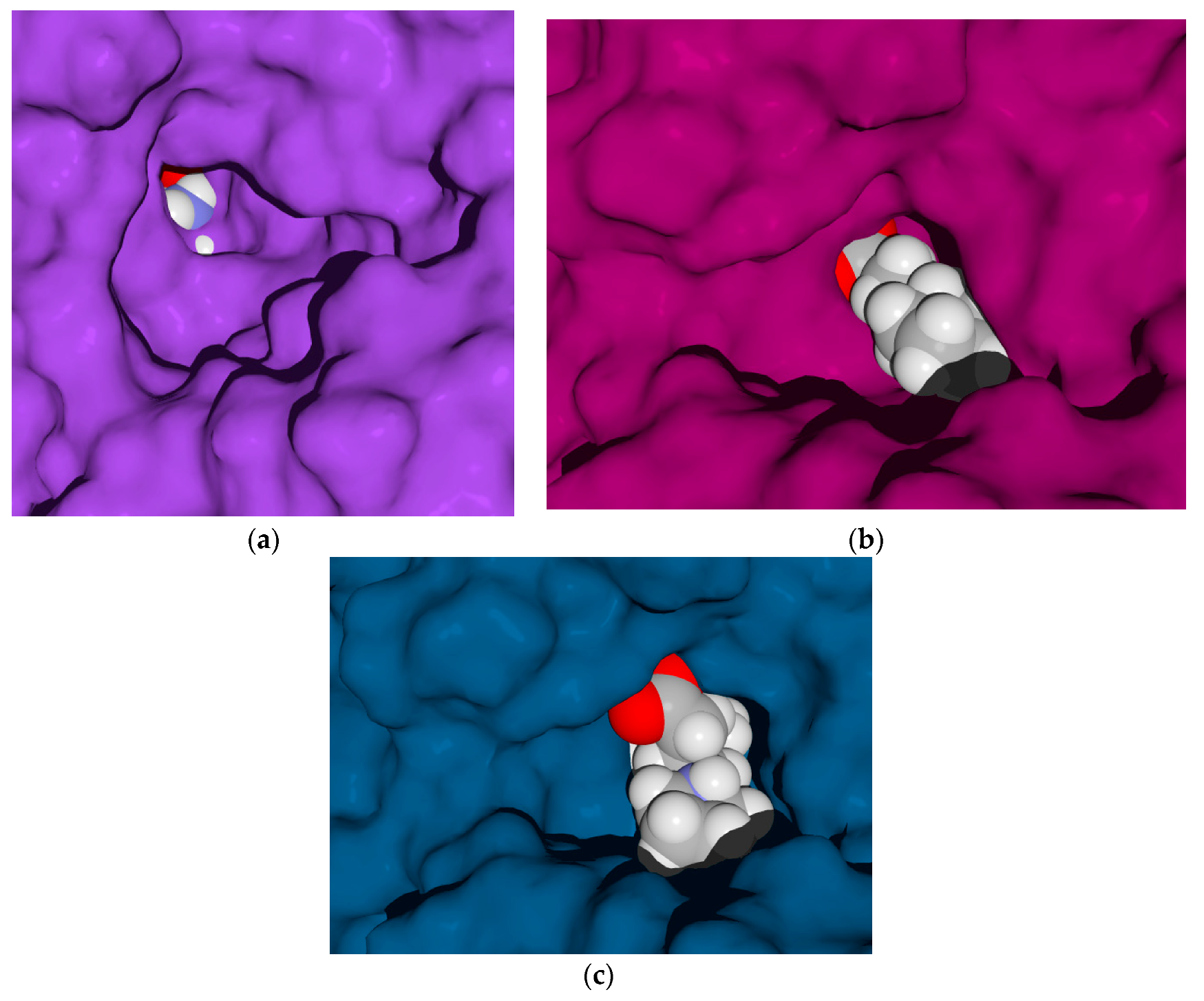
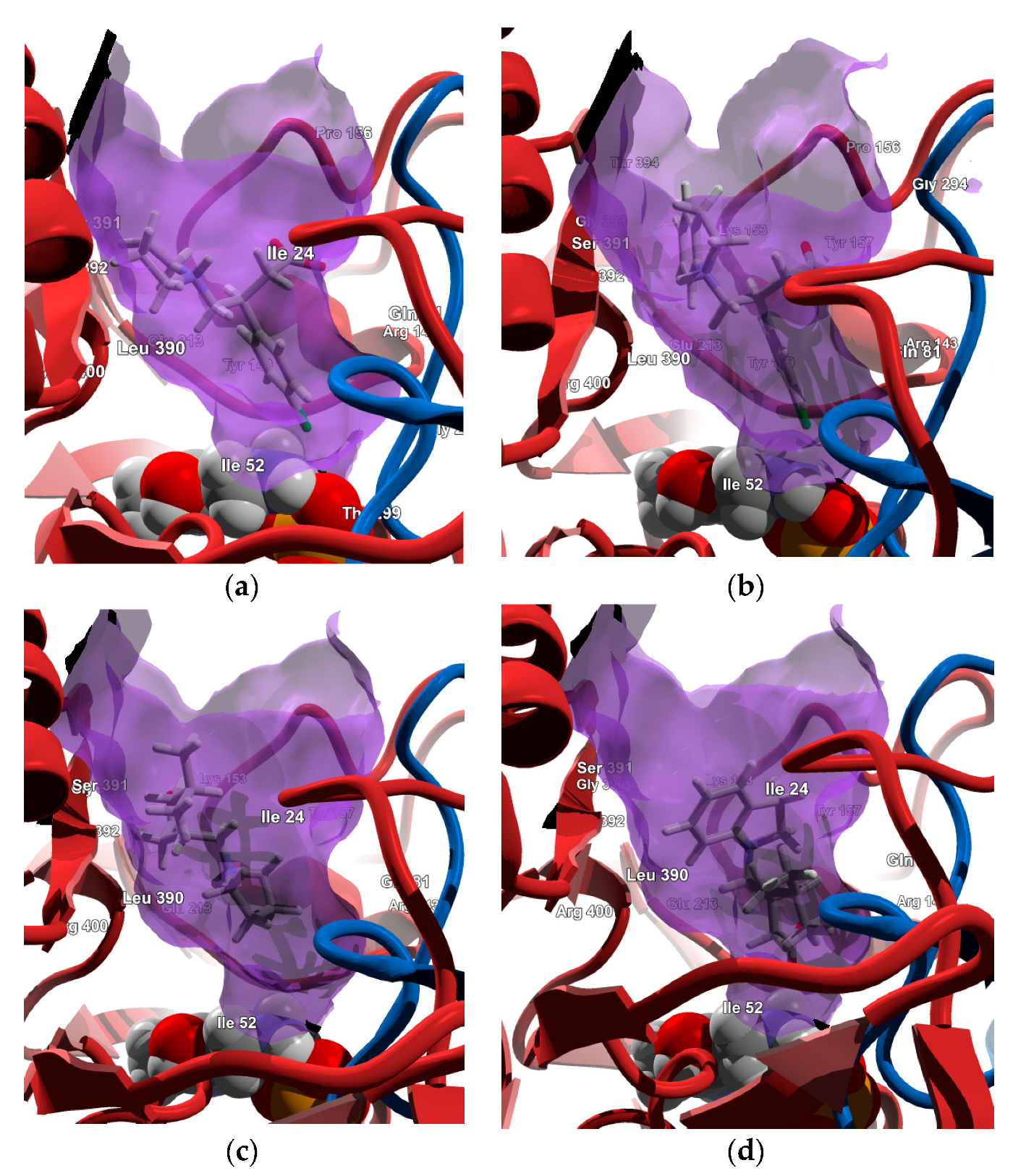
2.4. Molecular Docking Calculations
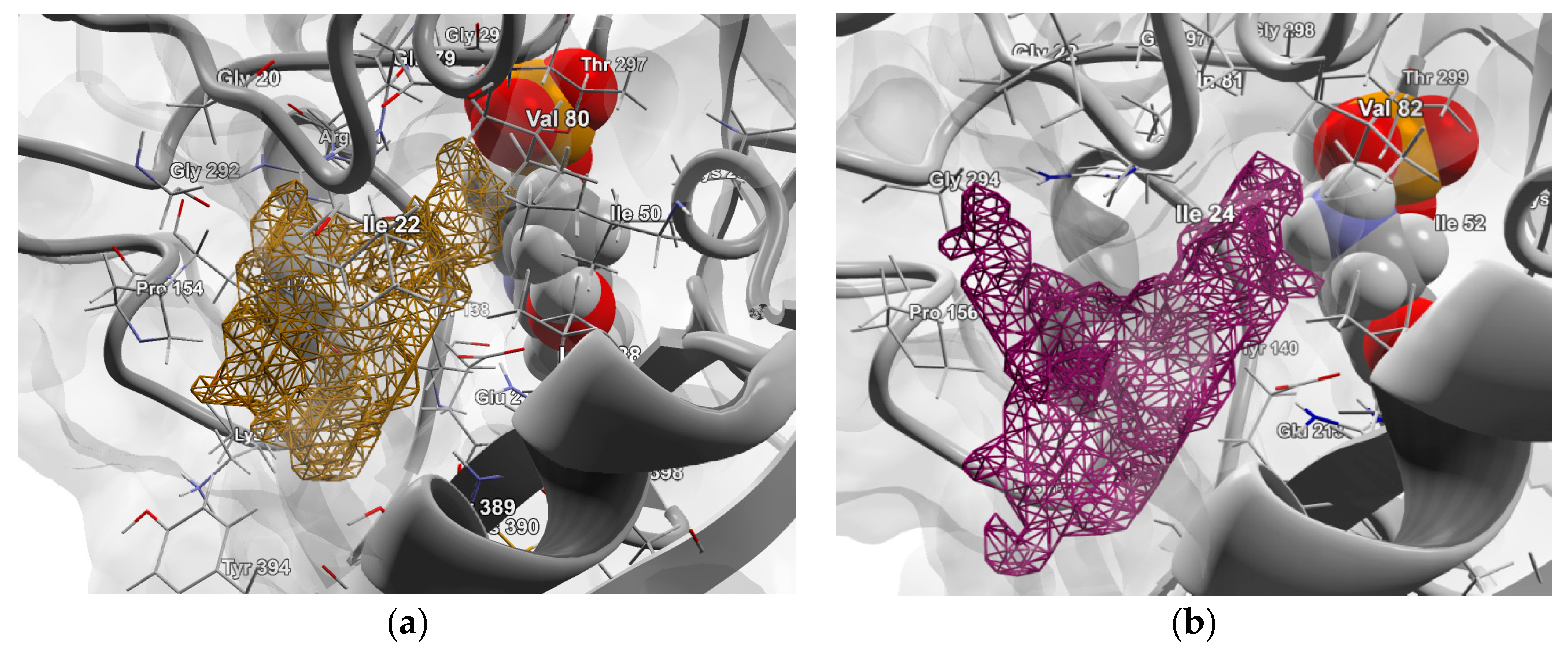
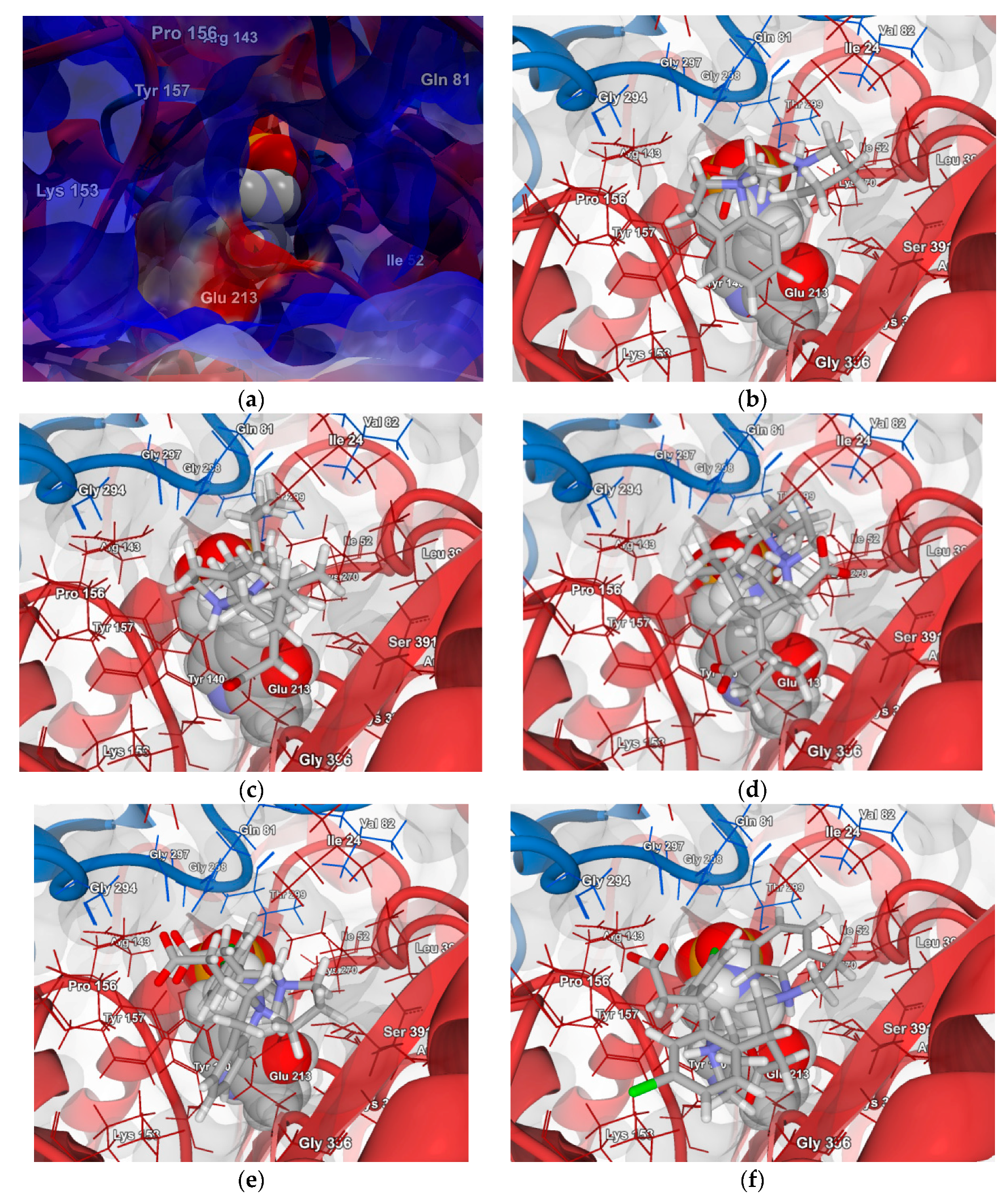
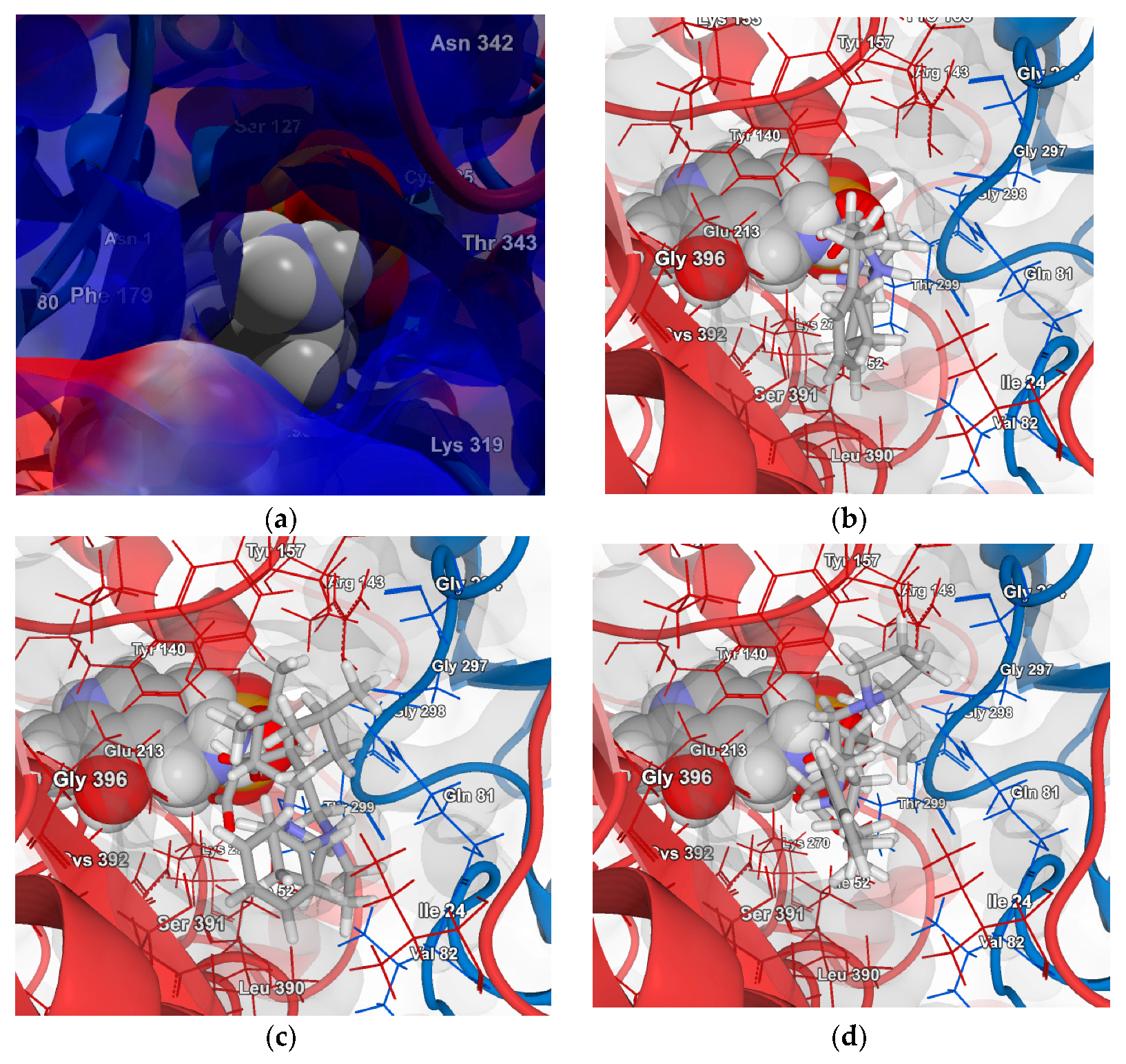
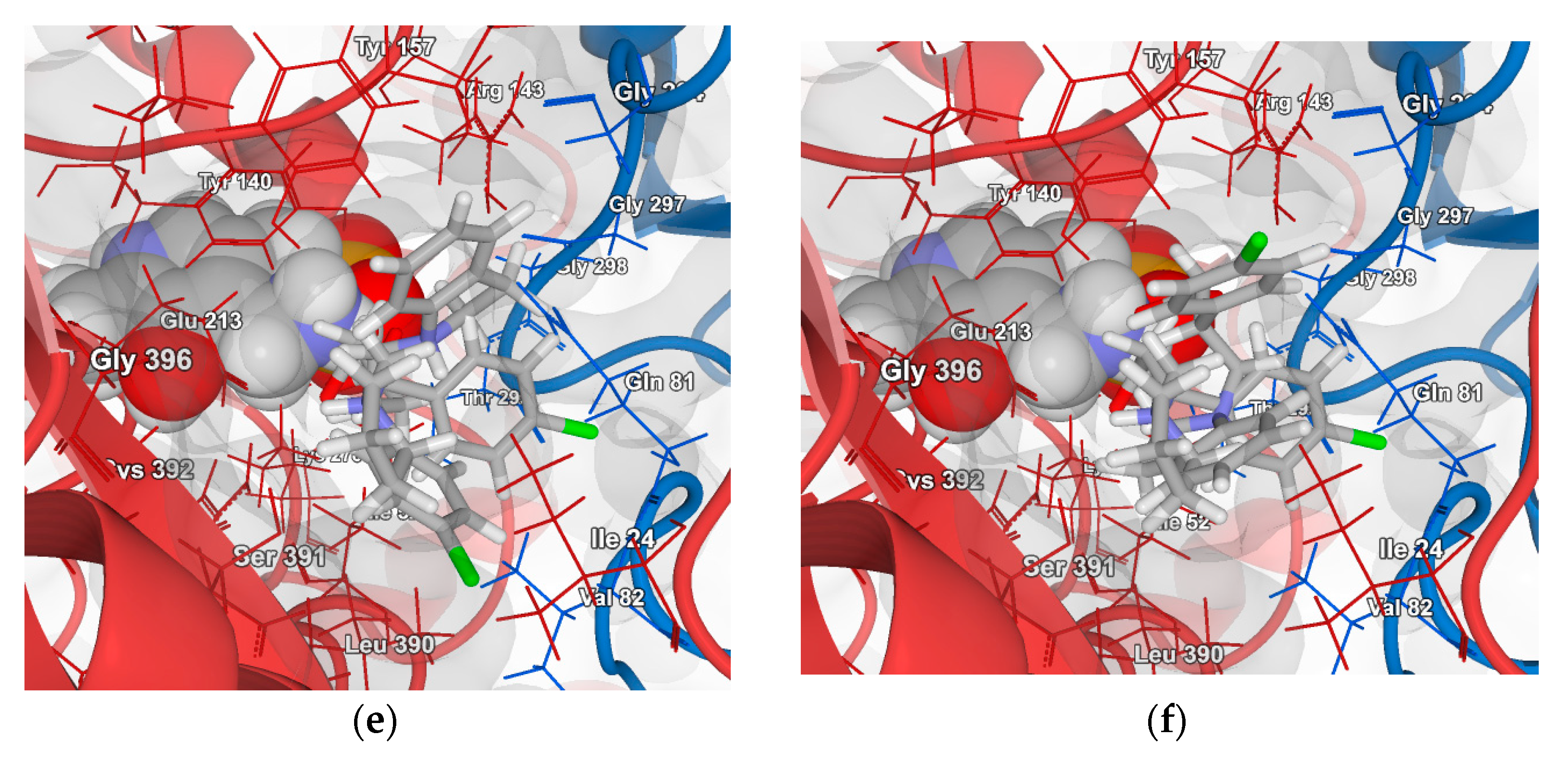
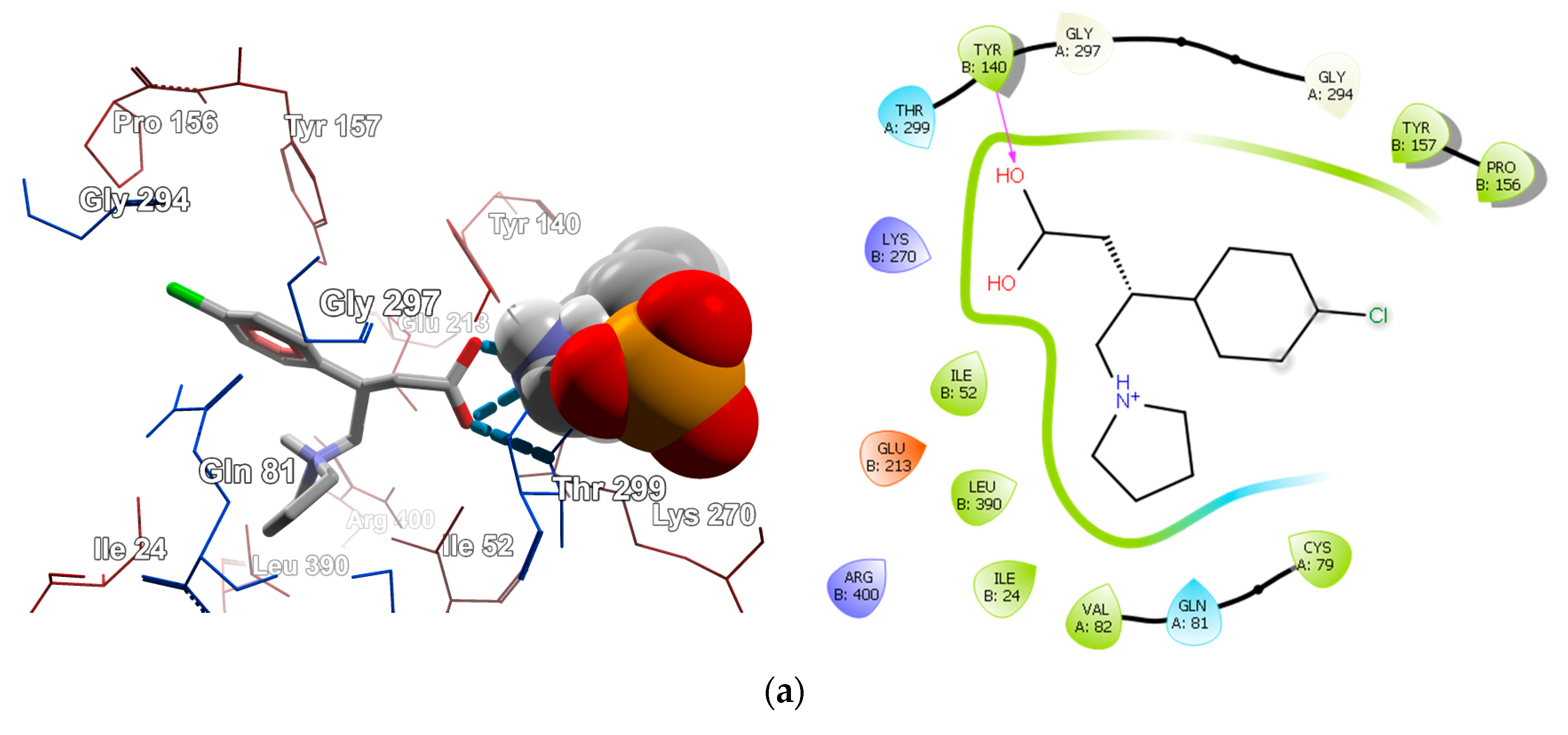
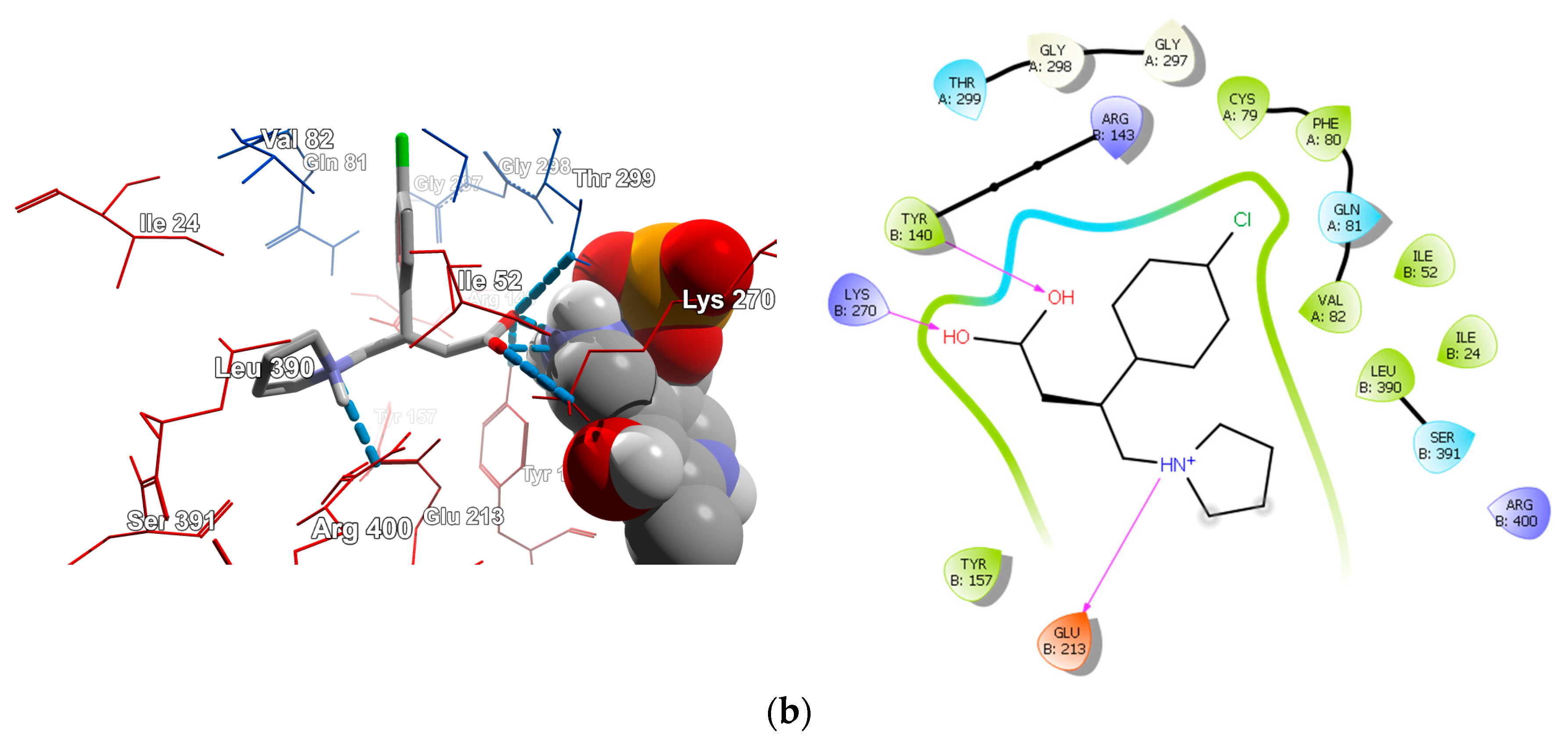
2.4.1. Molecular Docking over the Pseudomonas GABA-AT Model
2.4.2. Molecular Docking for Human GABA-AT Model
3. Materials and Methods
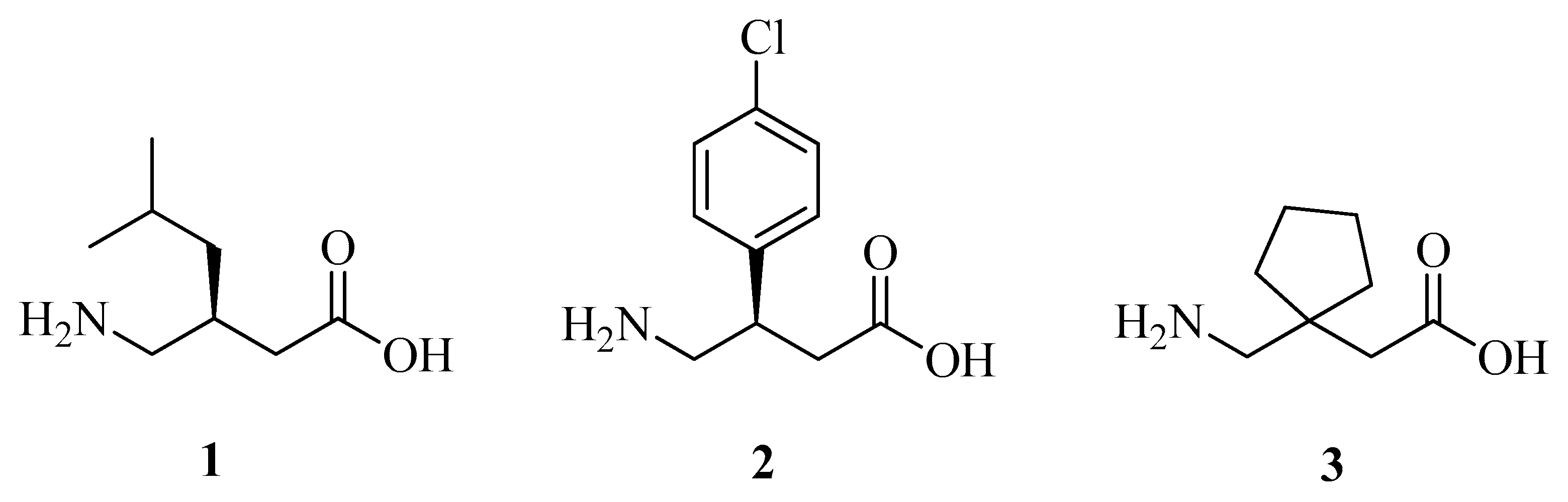
3.1. Chemistry
3.1.1. General Procedure for the Preparation of 8a–c
3.1.2. General Procedure for the Preparation of 9a–c
3.1.3. General Procedure for the Preparation of 11a, 11b
3.1.4. General Procedure for the Conjugate Additions to 11a and 11b
3.1.5. General Procedure for the Synthesis of 18–21
3.2. Enzymatic Assays
Enzyme Inhibitory Tests
3.3. Computational Studies
3.3.1. Conformational Analysis and Geometric Optimization
3.3.2. Determination of Natural Partial Charges and Electrostatic Potential Maps
3.3.3. Molecular Similarity Analysis
3.3.4. Homology Structural Modeling and Refinement
3.3.5. Molecular Docking Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kandel, E.R.; Schartz, J.H.; Jessell, T.M. Principles of Neural Science; McGraw-Hill Health Professions Division: New York, NY, USA, 2000. [Google Scholar]
- Niciu, M.J.; Kelmendi, B.; Sanacora, G. Overview of glutamatergic neurotransmission in the nrevous system. Pharm. Biochem. Behav. 2012, 100, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Amadasi, A.; Bertoldi, M.; Contestabile, R.; Bettati, S.; Cellini, B.; di Salvo, M.L.; Borri-Voltattorni, C.; Bossa, F.; Mozzarelli, A. Piridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324. [Google Scholar] [CrossRef] [PubMed]
- Baxter, C.F.; Roberts, E.J. The γ-Aminobutyric Acid-α-Ketoglutaric Acid Transaminase of Beef Brain. J. Biol. Chem. 1958, 233, 1135–1139. [Google Scholar] [PubMed]
- Bakay, R.A.; Harris, A.B. Neurotransmitter, receptor and biochemical changes in monkey cortical epileptic foci. Brain Res. 1981, 206, 387–404. [Google Scholar] [CrossRef]
- Aoyagi, T.; Wada, T.; Nagai, M.; Kojima, F.; Harada, S.; Takeuchi, T.; Takahashi, H.; Hirokawa, K.; Tsumita, T. Increased γ-aminobutyrate aminotransferase activity in brain of patients with Alzheimer’s disease. Chem. Pharm. Bull. 1990, 38, 1748–1749. [Google Scholar] [CrossRef] [PubMed]
- Nishino, N.; Fujiwara, H.; Noguchi-Kuno, S.A.; Tanaka, C. GABAA receptor but not muscarinic receptor density was decreased in the brain of patients with Parkinson’s disease. Jpn. J. Pharmacol. 1988, 48, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Ebato, H.; Seyfried, T.N.; Yu, R.K. Biochemical study of heterosis for brain myelin content in mice. J. Neurochem. 1983, 41, 440–446. [Google Scholar] [CrossRef]
- Gunne, L.-M.; Häggstrom, J.-E.; Sjöquist, B. Association with persistent neuroleptic-induced dyskinesia or regional changes in brain GABA synthesis. Nature 1984, 309, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.B.; Woodworth, G.F.; Kim, A.J. Evolving drug delivery strategies to overcome the blood brain barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Taylor, C.P.; Weber, M.; Piechan, J.; Prior, F.; Bian, F.; Cui, M.; Hoffman, D.; Donevan, S. Pregabalin is a potent and selective ligand for α2δ-1 andα2δ-2 calcium channels subunits. Eur. J. Pharm. 2011, 667, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.B. From basic science to blockbuster drug: The discovery of Lyrica. Angew. Chem. Int. Ed. 2008, 47, 3500–3504. [Google Scholar] [CrossRef] [PubMed]
- Reinares, M.; Rosa, A.R.; Franco, C.; Goikolea, J.M.; Fountoulakis, K.; Siamouli, M.; Gonda, X.; Frangou, S.; Vieta, E. A systematic review on the role of anticonvulsants in the treatment of acute bipolar depression. Int. J. Neuropsychopharmacol. 2013, 16, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Wensel, T.M.; Powe, K.W.; Cates, M.E. Pregabalin for the treatment of generalized anxiety disorder. Ann. Pharmacother. 2012, 46, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Satoh, J.; Arakawa, A.; Yoshiyama, T.; Suzuki, M. Pregabalin treatment for peripheral neuropathic pain. Drug Saf. 2012, 35, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Bowery, N.E. Baclofen: 10 years on. Trends Pharm. Sci. 1982, 31, 400–403. [Google Scholar] [CrossRef]
- Olpe, H.R.; Demie’ville, H.; Baltzer, V.; Bencze, E.L.; Koella, W.P.; Wolf, P.; Haas, H.L. The biological activity of d-Baclofen (Lipresal®). Eur. J. Pharmacol. 1978, 52, 133–136. [Google Scholar] [CrossRef]
- Mersfelder, T.L.; Nicholas, W.H. Gabapentin: Abuse, dependence and withdrawal. Ann. Pharmacother. 2016, 50, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kukkar, A.; Bali, A.; Sing, N.; Jaggi, A.S. Implications and mechanism of action of Gabapentin in neuropathic pain. Arch. Pharm. Res. 2013, 36, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Han, C.H.; Sweeny, M.; Cowles, V. Pharmacokinetics, efficacy, and tolerability of a once-daily gastroretentive dosage form of Gabapentin for the treatment of postherpetic neuralgia. J. Pharm. Sci. 2013, 102, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Bellioti, T.R.; Capiris, T.; Ekhato, V.; Kinsora, J.J.; Field, M.J.; Heffner, T.G.; Meltzer, L.T.; Schwarz, J.B.; Taylor, C.P.; Thorpe, A.J.; et al. Structure−activity relationships of Pregabalin and analogues that target the α2-δ protein. J. Med. Chem. 2005, 48, 2294–2307. [Google Scholar] [CrossRef] [PubMed]
- Schelkun, R.M.; Yuen, P.-W.; Wustrow, D.J.; Kinsora, J.; Su, T.-Z.; Vartanian, M.G. Heteroaromatic side-chain analogs of Pregabalin. Bioorg. Med. Chem. Lett. 2006, 16, 2329–2332. [Google Scholar] [CrossRef] [PubMed]
- Brow, K.M.; Roy, K.K.; Hockerman, G.H.; Doerksen, R.J.; Colby, D.A. Activation of the γ-Aminobutyric Acid Type B (GABAB) Receptor by Agonists and Positive Allosteric Modulators. J. Med. Chem. 2015, 58, 6336–6347. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.; Herdeis, C.; Osborne, H.B. GABAB-antagonistic activity of certain Baclofen homologues. Molecules 2013, 18, 10266–10284. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Peng, G.; Phan, T.; Dilip, U.; Chen, J.L.; Chernov-Rogan, T.; Zhang, X.; Grindstaff, K.; Amamalai, T.; Koller, K.; et al. Discovery of a novel potent GABAB receptor agonist. Bioorg. Med. Chem. Lett. 2011, 21, 6582–6585. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hafez, A.A.; Abdel-Wahab, B.A. 5-(4-chlorophenyl)-5,6-duhydro-1,3-oxazepin-7(4h)-one derivatives as lipophilic cyclic analogues of Baclofen: Design, synthesis and neuropharmacological evaluation. Bioorg. Med. Chem. 2008, 16, 7983–7991. [Google Scholar] [CrossRef] [PubMed]
- Constantino, G.; Macchiarulo, A.; Entrena Guadix, A.; Pelliciari, R. QSAR and molecular modeling studies of Baclofen analogues as GABAB agonists. Insights into the role of the aromatic moiety in GABAB binding in activation. J. Med. Chem. 2001, 44, 1827–1832. [Google Scholar] [CrossRef]
- Steffan, T.; Renukappa-Gutke, T.; Höfner, G.; Wanner, K.T. Design synthesis and SAR studies of GABA uptake inhibitors derived from 2-substituted pyrrolidine-2-yl acetic acids. Bioorg. Med. Chem. 2015, 23, 1284–1306. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.; Brehm, L.; Krogsgaard-Larsen, P. GABA agonists and uptake inhibitors. Synthesis, absolute stereochemistry, and enantioselectivity of (R)-(−)- and (S)-(+)-homo-.beta.-proline. J. Med. Chem. 1990, 33, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Mori, Y.; Nakamura, J.; Shibasaki, J. Synthesis and anticonvulsant activity of 1-acyl-2-pyrrolidinone derivatives. J. Med. Chem. 1991, 34, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Kolocouris, N.; Foscolos, G.; Papadopoulou-Daifoti, Z.; Vamvakides, A. Synthèse et étude pharmacologique de quelques nouveaux GABA-ergiques. Ann. Pharm. Françaises 1985, 43, 389–396. [Google Scholar]
- Matsuyama, K.; Yamashita, C.; Noda, A.; Goto, S.; Noda, H.; Ichimaru, Y.; Gomita, Y. Evaluation of isonicotinoyl-γ-aminobutyric acid (GABA) and nicotinoyl-GABA as pro-drugs of GABA. Chem. Pharm. Bull. 1984, 32, 4089–4095. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, P.; Hjeds, H.; Falch, E.; Jörgensen, F.S.; Nielsen, L. Advances in Drug Research; Testa, B., Ed.; Academic Press: New York, NY, USA, 1988; pp. 381–456. [Google Scholar]
- Shashoua, V.E.; Jacob, J.N.; Ridge, R.; Campbell, A.; Baldessarini, R.J. γ-Aminobutyric acid esters. 1. Synthesis, brain uptake and pharmacological studies of aliphatic and steroid esters of γ-aminobutyric acid. J. Med. Chem. 1984, 27, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Rekatas, G.V.; Demopoulos, V.J.; Kourounakis, P.N. Synthesis of N-acyl-2-pyrrolidinones from the corresponding N-acyl-GABA-derivatives. J. Heterocycl. Chem. 1996, 33, 989–990. [Google Scholar] [CrossRef]
- Mann, A.; Humblet, C.; Chambon, J.P.; Schlichter, R.; Desarmenien, M.; Feltz, P.; Wermuth, C.-G. Synthesis and activity of 5-(aminomethyl)-1,3-cyclohexanediones: Enolic analogs of γ-aminobutyric acid. J. Med. Chem. 1985, 28, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Silverman, R.B. Mechanism of inactivation of γ-aminobutyiric acid aminotransferase by 4-amino-5-hexynoic acid (γ-ethynylGABA). J. Am. Chem. Soc. 1991, 113, 9329–9340. [Google Scholar] [CrossRef]
- Hawker, D.D.; Silverman, R.B. Synthesis and evaluation of novel heteroaromatic substrates of GABA aminotransferase. Bioorg. Med. Chem. 2012, 20, 5763–5773. [Google Scholar] [CrossRef] [PubMed]
- Clift, M.D.; Silverman, R.B. Synthesis and evaluation of novel aromatic substrates and competitive inhibitors of GABA aminotransferase. Bioorg. Med. Chem. 2008, 18, 3122–3125. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Gerasimov, M.R.; Kvist, T.; Wellendorph, P.; Madsen, K.K.; Pera, E.; Lee, H.; Schousboe, A.; Chebib, M.; Brauner-Osborne, H.; et al. (1S, 3S)-3-Amino-4-difluoromethylenyl-1-cyclopentanoic Acid (CPP-115), a Potent γ-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Cocaine Addiction. J. Med. Chem. 2012, 55, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Silverman, R.B. Fluorinated Conformationally Restricted γ-Aminobutyric Acid Aminotransferase Inhibitors. J. Med. Chem. 2006, 49, 7404–7412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Silverman, R.B. Syntheses and evaluation of fluorinated conformationally restricted analogues of GABA as potential inhibitors of GABA aminotransferase. Bioorg. Med. Chem. 2005, 14, 2242–2252. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Silverman, R.B. New substrates and inhibitors of γ-aminobutyric acid aminotransferase containing bioisosteres of the carboxylic acid group: Design, synthesis, and biological activity. Bioorg. Med. Chem. 2006, 14, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Hawker, D.D.; Wu, R.; Doud, E.; Widom, J.; Sanishvili, R.; Liu, D.; Lelleher, N.L.; Silverman, R.B. Design and mechanism of tetrahydrothiophene-based-γ-aminobutyiric acid aminotransferase inactivators. J. Am. Chem. Soc. 2015, 137, 4525–4533. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Le, H.V.; Wu, R.; Doud, E.; Sanishvili, R.; Kellie, J.F.; Compton, P.D.; Pachaiyappan, B.; Liu, D.; Kelleher, N.L.; et al. Mechanism of inactivation of GABA aminotransferase by (E)- and (Z)-(1S,3S)-3-amino-4-fluoromethylenyl-1-cyclopentanoic acid. ACS Chem. Biol. 2015, 10, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.K.; Sinha, B.N.; Khosa, R.L. γ-Aminobutyiric acids as novel GABA-AT inhibitors. Med. Chem. Res. 2013, 22, 134–146. [Google Scholar] [CrossRef]
- Bansal, S.K.; Sinha, B.N.; Khosa, R.L. Schiff bases of GABA. Med. Chem. Res. 2012, 21, 3063–3072. [Google Scholar] [CrossRef]
- Pinto, A.; Tamborini, L.; Pennacchietti, E.; Caluccia, A.; Silvestri, A.; Cullia, G.; De Micheli, C.; Conti, A.; De Biase, D. Bicyclic γ-amino acids as inhibitors ot γ-aminobutyrate aminotransferase. J. Enzyme Inh. Med. Chem. 2016, 31, 295–301. [Google Scholar]
- Rall, T.W.; Schleifer, L.S. Goodman & Gilman. The Pharmacological Basis of Therapeutics; Rall, T.W., Nies, A.S., Taylor, P., Eds.; McGraw-Hill: New York, NY, USA, 1991; pp. 436–462. [Google Scholar]
- Rekatas, G.V.; Tani, E.; Demopoulos, V.J.; Kourounakis, P.N. Synthesis of GABA-valproic acid derivatives and evaluation of their anticonvulsant and antioxidant activity. Arch. Pharm. Med. Chem. 1996, 329, 393–398. [Google Scholar] [CrossRef]
- Tunnicliff, G.; Crites, G. Chemical Inactivation of Bacterial GABA Aminotransferase. Biochem. Mol. Biol. Int. 1998, 46, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman-Saba, A.J.; Suliman, F.E.O.; Barghouthi, S. Kinetic Studies on the Inhibition of GABA-T by γ-Vinyl GABA and Taurine. J. Enzym. Inhib. Med. Chem. 2003, 18, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswari, P.; Sriram, D.; Thirumurugan, R.; Jit, L.R.J.S.; Ravagendran, J.V.; Kavya, R.; Rakhra, K.; Saraswat, V. Synthesis of N4-(2,4-dimethylphenyl) semicarbazones as 4-aminobutyrate aminotransferase inhibitors. Acta Pharm. 2006, 56, 259–272. [Google Scholar] [PubMed]
- Wustrow, D.J.; Bryans, J.S. 3-Substituted GABA Analogs with Central Nervous System Activity: A Review. Med. Res. Rev. 1999, 19, 149–177. [Google Scholar]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do Structurally Similar Molecules Have Similar Biological Activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Peterson, P.E.; Carter, R.J.; Zhou, X.; Langston, J.A.; Fisher, A.J.; Toney, M.D. Crystal Structures of Unbound and Aminooxyacetate-Bound Escherichia coli γ-Aminobutyrate Aminotransferase. Biochemistry 2004, 43, 10896–10905. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J. Spartan Software; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N.V. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Petersson, G.A.; Tensfeldt, T.G.; Montgomery, J.A., Jr. A complete basis set model chemistry. III. The complete basis set-quadratic configuration interaction family of methods. J. Chem. Phys. 1991, 94, 6091–6101. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 9a, 9b, 9c, 18a, 19a, 20b, 21b are available from the authors. |






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
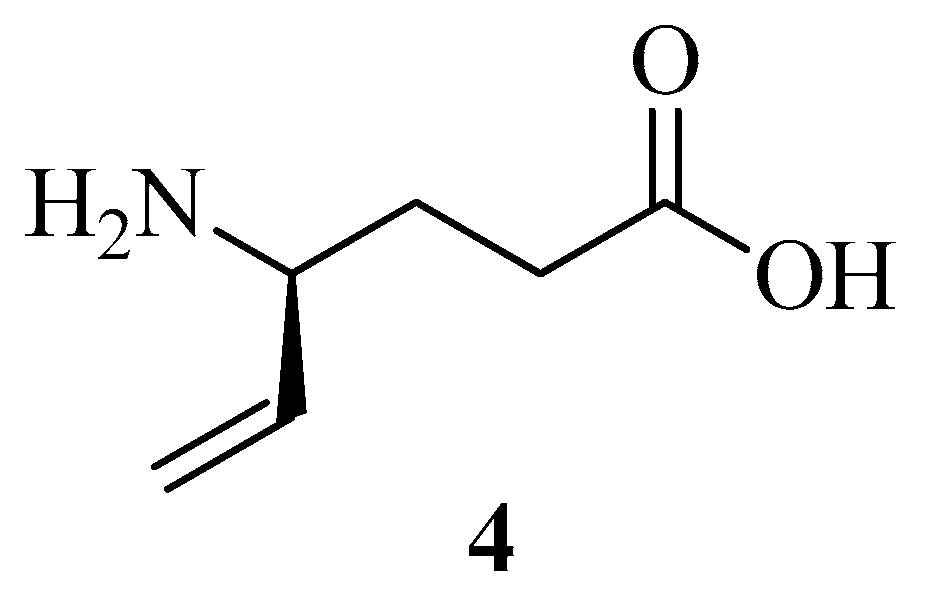
| Natural Charges | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| VGB 4 | 1 | 2 | 9b | 21b | 20b | 9a | 19a | 18a | |
| Neutral N | −0.878 | −0.923 | −0.914 | −0.523 | −0.52 | −0.521 | −0.534 | −0.548 | −0.549 |
| Neutral O | −0.731 | −0.709 | −0.741 | −0.737 | −0.736 | −0.731 | −0.716 | −0.74 | −0.735 |
| Molecule | RBF | AMR () |
|---|---|---|
| 9a | 0.148 | 40.969 |
| 9b | 0.125 | 56.516 |
| 18a | 0.154 | 59.191 |
| 19a | 0.132 | 70.341 |
| 20b | 0.136 | 74.738 |
| 21b | 0.116 | 85.887 |
| PDB_ID | Resolution (Å) | Organism | % Identity Respect to Human | % Identity Respec to P. fluorencens |
|---|---|---|---|---|
| 4zsw | 1.70 | Wild boar | 95.46 | 27.52 |
| 4y0h | 1.63 | Wild boar | 95.46 | 27.52 |
| 4y0i | 1.66 | Wild boar | 95.46 | 27.52 |
| 1ohv | 2.30 | Wild boar | 95.46 | 27.52 |
| 1ohw | 2.30 | Wild boar | 95.46 | 27.52 |
| 1sf2 | 2.40 | E. coli | 26.35 | 73.82 |
| 4ffc | 1.80 | M. abscessus | 27.99 | 42.79 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tovar-Gudiño, E.; Guevara-Salazar, J.A.; Bahena-Herrera, J.R.; Trujillo-Ferrara, J.G.; Martínez-Campos, Z.; Razo-Hernández, R.S.; Santiago, Á.; Pastor, N.; Fernández-Zertuche, M. Novel-Substituted Heterocyclic GABA Analogues. Enzymatic Activity against the GABA-AT Enzyme from Pseudomonas fluorescens and In Silico Molecular Modeling. Molecules 2018, 23, 1128. https://doi.org/10.3390/molecules23051128
Tovar-Gudiño E, Guevara-Salazar JA, Bahena-Herrera JR, Trujillo-Ferrara JG, Martínez-Campos Z, Razo-Hernández RS, Santiago Á, Pastor N, Fernández-Zertuche M. Novel-Substituted Heterocyclic GABA Analogues. Enzymatic Activity against the GABA-AT Enzyme from Pseudomonas fluorescens and In Silico Molecular Modeling. Molecules. 2018; 23(5):1128. https://doi.org/10.3390/molecules23051128
Chicago/Turabian StyleTovar-Gudiño, Erika, Juan Alberto Guevara-Salazar, José Raúl Bahena-Herrera, José Guadalupe Trujillo-Ferrara, Zuleyma Martínez-Campos, Rodrigo Said Razo-Hernández, Ángel Santiago, Nina Pastor, and Mario Fernández-Zertuche. 2018. "Novel-Substituted Heterocyclic GABA Analogues. Enzymatic Activity against the GABA-AT Enzyme from Pseudomonas fluorescens and In Silico Molecular Modeling" Molecules 23, no. 5: 1128. https://doi.org/10.3390/molecules23051128
APA StyleTovar-Gudiño, E., Guevara-Salazar, J. A., Bahena-Herrera, J. R., Trujillo-Ferrara, J. G., Martínez-Campos, Z., Razo-Hernández, R. S., Santiago, Á., Pastor, N., & Fernández-Zertuche, M. (2018). Novel-Substituted Heterocyclic GABA Analogues. Enzymatic Activity against the GABA-AT Enzyme from Pseudomonas fluorescens and In Silico Molecular Modeling. Molecules, 23(5), 1128. https://doi.org/10.3390/molecules23051128