
HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
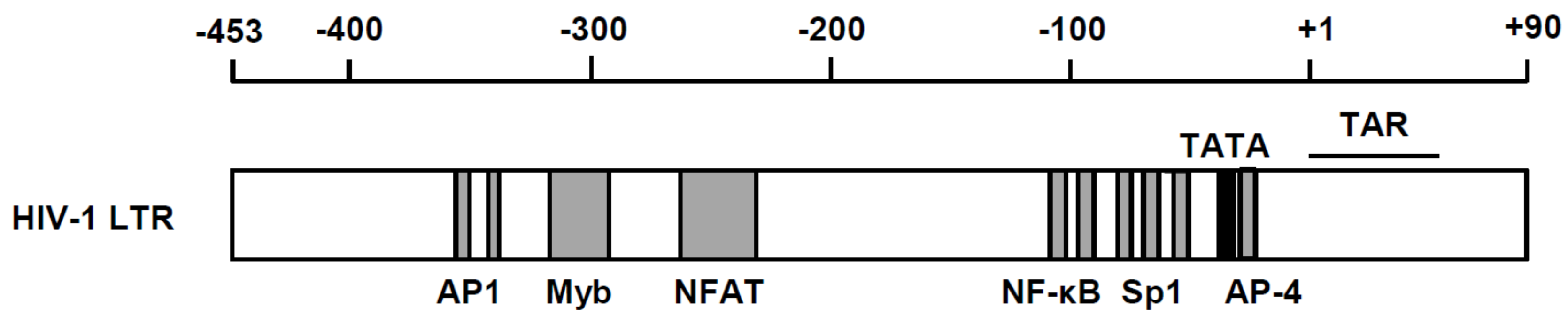
2. Significance of Transcriptional Regulation of HIV Gene Expression in Viral Life Cycle
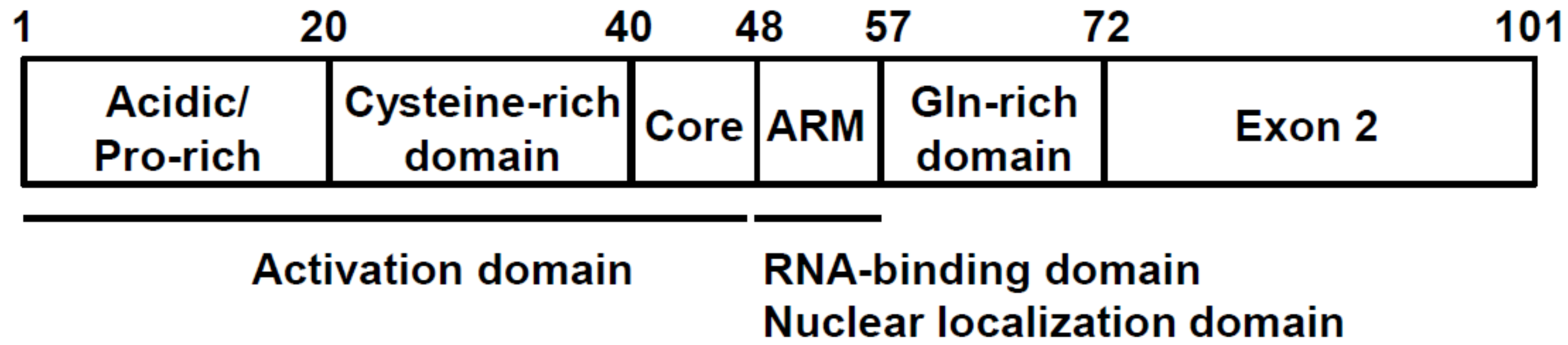
3. Tat Functional Domains and Its Binging Partners
4. P-TEFb as a Crucial Cofactor of Tat for Its Action
5. P-TEFb as a Positive Regulator of Transcription Elongation and the Involvement of Brd4
6. Negative Regulation of P-TEFb by HEXIM1 and 7SK Small Nuclear Ribonucleoprotein (7SK snRNP)
7. 3D Structure of Tat-P-TEFb Complex
8. Identification of Critical Regions for Tat Transcriptional Activity within the Tat/P-TEFb Complex by In Vitro Study
9. Identification of Critical Regions for Tat Transcriptional Activity within the Tat/P-TEFb Complex by Molecular Dynamics (MD) Simulation
10. Experimental Approaches for Studying PPI between Tat and P-TEFb in Cells
11. Development of Specific Therapy
11.1. “Shock and Kill” or “Block and Lock”?
11.2. “Shock and Kill” Approach
11.3. Use of Natural Products for “Shock and Kill” Therapy
11.4. “Block and Lock” Approach and Tat/TAR/P-TEFb Interaction
12. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CycT1 | cyclin T1 |
| HIV | Human immunodeficiency virus type 1 |
| LRA | latency reversing agents |
| P-TEFb | positive transcriptional elongation factor b |
| RNAPII | RNA polymerase II |
| TAR | transactivation response |
References
- Joint United Nations Programme on HIV/AIDS (UNAIDS). Global Report: UNAIDS Report on the Global AIDS Epidemic 2013. Available online: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf (accessed on 1 March 2018).
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Di Mascio, M.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Utz, P.J.; Durand, D.B.; Toole, J.J.; Emmel, E.A.; Crabtree, G.R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.; Kunkel, S.; Nabel, G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 1989, 86, 2336–2340. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, D.M.; Hannibal, M.C.; Smith, M.J.; Cossman, R.; Nabel, G.J. Activation of the human immunodeficiency virus type 1 enhancer is not dependent on NFAT-1. J. Virol. 1992, 66, 3961–3965. [Google Scholar] [PubMed]
- Margolis, D.M.; Somasundaran, M.; Green, M.R. Human transcription factor YY1 represses human immunodeficiency virus type 1 transcription and virion production. J. Virol. 1994, 68, 905–910. [Google Scholar] [PubMed]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef] [PubMed]
- Karn, J.; Stoltzfus, C.M. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Perspect. Med. 2012, 2, a006916. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Victoriano, A.F.; Okamoto, T. Transcriptional control of HIV replication by multiple modulators and their implication for a novel antiviral therapy. AIDS Res. Hum. Retroviruses 2012, 28, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Ochiai, K.; Okamoto, T. Reactivation of latent HIV-1 infection by the periodontopathic bacterium Porphyromonas gingivalis involves histone modification. J. Immunol. 2009, 182, 3688–3695. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Wong-Staal, F. Demonstration of virus-specific transcriptional activator(s) in cells infected with HTLV-III by an in vitro cell-free system. Cell 1986, 47, 29–35. [Google Scholar] [CrossRef]
- Okamoto, T.; Benter, T.; Josephs, S.F.; Sadaie, M.R.; Wong-Staal, F. Transcriptional activation from the long-terminal repeat of human immunodeficiency virus in vitro. Virology 1990, 177, 606–614. [Google Scholar] [CrossRef]
- Arya, S.K.; Gallo, R.C.; Hahn, B.H.; Shaw, G.M.; Popovic, M.; Salahuddin, S.Z.; Wong-Staal, F. Homology of genome of AIDS-associated virus with genomes of human T-cell leukemia viruses. Science 1984, 225, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.A.; Sodroski, J.G.; Haseltine, W.A. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 1985, 41, 813–823. [Google Scholar] [CrossRef]
- Jeang, K.T.; Xiao, H.; Rich, E.A. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J. Biol. Chem. 1999, 274, 28837–28840. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.G.; Collalti, E.; Ratner, L.; Gallo, R.C.; Wong-Staal, F. A molecular clone of HTLV-III with biological activity. Nature 1985, 316, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Shojania, S.; O’Neil, J.D. HIV-1 Tat is a natively unfolded protein: The solution conformation and dynamics of reduced HIV-1 Tat-(1-72) by NMR spectroscopy. J. Biol. Chem. 2006, 281, 8347–8356. [Google Scholar] [CrossRef] [PubMed]
- Jean, M.J.; Power, D.; Kong, W.; Huang, H.; Santoso, N.; Zhu, J. Identification of HIV-1 Tat-Associated Proteins Contributing to HIV-1 Transcription and Latency. Viruses 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Gautier, V.W.; Gu, L.; O’Donoghue, N.; Pennington, S.; Sheehy, N.; Hall, W.W. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology 2009, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Ruben, S.; Perkins, A.; Purcell, R.; Joung, K.; Sia, R.; Burghoff, R.; Haseltine, W.A.; Rosen, C.A. Structural and functional characterization of human immunodeficiency virus Tat protein. J. Virol. 1989, 63, 1–8. [Google Scholar] [PubMed]
- Jeang, K.T.; Chun, R.; Lin, N.H.; Gatignol, A.; Glabe, C.G.; Fan, H. In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J. Virol. 1993, 67, 6224–6233. [Google Scholar] [PubMed]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [PubMed]
- Truant, R.; Cullen, B.R. The arginine-rich domains present in human immunodeficiency virus type 1 Tat and Rev function as direct importin beta-dependent nuclear localization signals. Mol. Cell. Biol. 1999, 19, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Endo-Munoz, L.; Warby, T.; Harrich, D.; McMillan, N.A. Phosphorylation of HIV Tat by PKR increases interaction with TAR RNA and enhances transcription. Virol. J. 2005, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Himiari, Z.; Tsimbalyuk, S.; Forwood, J.K. Structural Basis for Importin-alpha Binding of the Human Immunodeficiency Virus Tat. Sci. Rep. 2017, 7, 1650. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sharp, P.A. Tat-SF1: Cofactor for stimulation of transcriptional elongation by HIV-1 Tat. Science 1996, 274, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Green, M.R. The HIV-1 Tat cellular coactivator Tat-SF1 is a general transcription elongation factor. Genes Dev. 1998, 12, 2992–2996. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, H.B.; Robinson, T.J.; Gordan, R.; Hartemink, A.J.; Garcia-Blanco, M.A. Identification of Tat-SF1 cellular targets by exon array analysis reveals dual roles in transcription and splicing. RNA 2011, 17, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.A.; Roeder, R.G. A novel RNA polymerase II-containing complex potentiates Tat-enhanced HIV-1 transcription. EMBO J. 1999, 18, 3688–3701. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar] [CrossRef] [PubMed]
- Grana, X.; De Luca, A.; Sang, N.; Fu, Y.; Claudio, P.P.; Rosenblatt, J.; Morgan, D.O.; Giordano, A. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 3834–3838. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.F.; Price, D.H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Cujec, T.P.; Peterlin, B.M. Effects of human chromosome 12 on interactions between Tat and TAR of human immunodeficiency virus type 1. J. Virol. 1994, 68, 6505–6513. [Google Scholar] [PubMed]
- Newstein, M.; Stanbridge, E.J.; Casey, G.; Shank, P.R. Human chromosome 12 encodes a species-specific factor which increases human immunodeficiency virus type 1 tat-mediated trans activation in rodent cells. J. Virol. 1990, 64, 4565–4567. [Google Scholar] [PubMed]
- Herrmann, C.H.; Rice, A.P. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J. Virol. 1995, 69, 1612–1620. [Google Scholar] [PubMed]
- Mancebo, H.S.; Lee, G.; Flygare, J.; Tomassini, J.; Luu, P.; Zhu, Y.; Peng, J.; Blau, C.; Hazuda, D.; Price, D.; et al. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 1997, 11, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.T.; Ivanov, D.; Guo, J.; Nee, E.; Gaynor, R.B. Role of the human and murine cyclin T proteins in regulating HIV-1 tat-activation. J. Mol. Biol. 1999, 288, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Gahmen, U.; Lu, H.; Zhou, Q.; Alber, T. AFF4 binding to Tat-P-TEFb indirectly stimulates TAR recognition of super elongation complexes at the HIV promoter. Elife 2014, 3, e02375. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, A.; Krasnopolsky, S.; Taube, R. Super elongation complex promotes early HIV transcription and its function is modulated by P-TEFb. Transcription 2017, 8, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Li, T.; Rahl, P.B.; Adamson, T.E.; Loudas, N.B.; Guo, J.; Varzavand, K.; Cooper, J.J.; Hu, X.; Gnatt, A.; et al. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol. Cell 2012, 45, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Rasmussen, E.B.; Lis, J.T. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc. Natl. Acad. Sci. USA 1993, 90, 7923–7927. [Google Scholar] [CrossRef] [PubMed]
- Rougvie, A.E.; Lis, J.T. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 1988, 54, 795–804. [Google Scholar] [CrossRef]
- Krumm, A.; Meulia, T.; Brunvand, M.; Groudine, M. The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 1992, 6, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Plet, A.; Eick, D.; Blanchard, J.M. Elongation and premature termination of transcripts initiated from c-fos and c-myc promoters show dissimilar patterns. Oncogene 1995, 10, 319–328. [Google Scholar] [PubMed]
- Strobl, L.J.; Eick, D. Hold back of RNA polymerase II at the transcription start site mediates down-regulation of c-myc in vivo. EMBO J. 1992, 11, 3307–3314. [Google Scholar] [PubMed]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Klatt, A.; Gilmour, D.S.; Henderson, A.J. Negative elongation factor NELF represses human immunodeficiency virus transcription by pausing the RNA polymerase II complex. J. Biol. Chem. 2007, 282, 16981–16988. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.E.; Okamoto, T.; Reuter, P.; Ugarkovic, D.; Schroder, H.C. Functional characterization of Tat protein from human immunodeficiency virus. Evidence that Tat links viral RNAs to nuclear matrix. J. Biol. Chem. 1990, 265, 3803–3808. [Google Scholar] [PubMed]
- Biglione, S.; Byers, S.A.; Price, J.P.; Nguyen, V.T.; Bensaude, O.; Price, D.H.; Maury, W. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology 2007, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar] [PubMed]
- Lu, X.; Zhu, X.; Li, Y.; Liu, M.; Yu, B.; Wang, Y.; Rao, M.; Yang, H.; Zhou, K.; Wang, Y.; et al. Multiple P-TEFbs cooperatively regulate the release of promoter-proximally paused RNA polymerase II. Nucleic Acids Res. 2016, 44, 6853–6867. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res 2013, 41, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.P. Cyclin-dependent kinases as therapeutic targets for HIV-1 infection. Expert Opin. Ther. Targets 2016, 20, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.P.; Kimata, J.T. Subversion of Cell Cycle Regulatory Mechanisms by HIV. Cell Host Microbe 2015, 17, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Bres, V.; Yoh, S.M.; Jones, K.A. The multi-tasking P-TEFb complex. Curr. Opin. Cell Biol. 2008, 20, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Lenasi, T.; Barboric, M. P-TEFb stimulates transcription elongation and pre-mRNA splicing through multilateral mechanisms. RNA Biol. 2010, 7, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Schroeder, S.; Ott, M. CYCLINg through transcription: Posttranslational modifications of P-TEFb regulate transcription elongation. Cell Cycle 2010, 9, 1697–1705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yik, J.H. The Yin and Yang of P-TEFb regulation: Implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 2006, 70, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.; Peterlin, B.M. Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. J. Biol. Chem. 2013, 288, 14400–14407. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Euphorbia Kansui Reactivates Latent HIV. PLoS ONE 2016, 11, e0168027. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Barboric, M.; Yik, J.H.; Czudnochowski, N.; Yang, Z.; Chen, R.; Contreras, X.; Geyer, M.; Matija Peterlin, B.; Zhou, Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007, 35, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Sedore, S.C.; Byers, S.A.; Biglione, S.; Price, J.P.; Maury, W.J.; Price, D.H. Manipulation of P-TEFb control machinery by HIV: Recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 2007, 35, 4347–4358. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Luo, Z.; Peterlin, B.M. Genetic analysis of the structure and function of 7SK small nuclear ribonucleoprotein (snRNP) in cells. J. Biol. Chem. 2014, 289, 21181–21190. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, K.; Hirokawa, T.; Hibi, Y.; Okamoto, T. Molecular dynamics simulation and experimental verification of the interaction between cyclin T1 and HIV-1 Tat proteins. PLoS ONE 2015, 10, e0119451. [Google Scholar] [CrossRef] [PubMed]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Babayeva, N.D.; Suwa, Y.; Baranovskiy, A.G.; Price, D.H.; Tahirov, T.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb and AFF4. Cell Cycle 2014, 13, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.E.; Wei, P.; KewalRamani, V.N.; Mayall, T.P.; Herrmann, C.H.; Rice, A.P.; Littman, D.R.; Jones, K.A. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes Dev. 1998, 12, 3512–3527. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D.; Grdina, T.A.; Bogerd, H.P.; Cullen, B.R. Recruitment of a protein complex containing Tat and cyclin T1 to TAR governs the species specificity of HIV-1 Tat. EMBO J. 1998, 17, 7056–7065. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Taube, R.; Wimmer, J.; Cujec, T.P.; Peterlin, B.M. Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc. Natl. Acad. Sci. USA 1999, 96, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, D.; Kwak, Y.T.; Nee, E.; Guo, J.; Garcia-Martinez, L.F.; Gaynor, R.B. Cyclin T1 domains involved in complex formation with Tat and TAR RNA are critical for tat-activation. J. Mol. Biol. 1999, 288, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, K.; Hibi, Y.; Imai, K.; Victoriano, A.F.; Kurimoto, E.; Kato, K.; Okamoto, T. Functional characterization of human cyclin T1 N-terminal region for human immunodeficiency virus-1 Tat transcriptional activation. J. Mol. Biol. 2011, 410, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Irwin, D.; Geyer, M.; Peterlin, B.M. Optimized chimeras between kinase-inactive mutant Cdk9 and truncated cyclin T1 proteins efficiently inhibit Tat transactivation and human immunodeficiency virus gene expression. J. Virol. 2002, 76, 10873–10881. [Google Scholar] [CrossRef] [PubMed]
- Jadlowsky, J.K.; Nojima, M.; Okamoto, T.; Fujinaga, K. Dominant negative mutant cyclin T1 proteins that inhibit HIV transcription by forming a kinase inactive complex with Tat. J. Gen. Virol. 2008, 89, 2783–2787. [Google Scholar] [CrossRef] [PubMed]
- Jadlowsky, J.K.; Nojima, M.; Schulte, A.; Geyer, M.; Okamoto, T.; Fujinaga, K. Dominant negative mutant cyclin T1 proteins inhibit HIV transcription by specifically degrading Tat. Retrovirology 2008, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, A.; Verstraete, N.; Galker, S.; Maatook, M.; Bensaude, O.; Taube, R. A single point mutation in cyclin T1 eliminates binding to Hexim1, Cdk9 and RNA but not to AFF4 and enforces repression of HIV transcription. Retrovirology 2014, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Verstraete, N.; Kuzmina, A.; Diribarne, G.; Nguyen, V.T.; Kobbi, L.; Ludanyi, M.; Taube, R.; Bensaude, O. A Cyclin T1 point mutation that abolishes positive transcription elongation factor (P-TEFb) binding to Hexim1 and HIV tat. Retrovirology 2014, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Toth, G.; Borics, A. Closing of the flaps of HIV-1 protease induced by substrate binding: A model of a flap closing mechanism in retroviral aspartic proteases. Biochemistry 2006, 45, 6606–6614. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Okur, A.; Rizzo, R.C.; Simmerling, C. HIV-1 protease flaps spontaneously close to the correct structure in simulations following manual placement of an inhibitor into the open state. J. Am. Chem. Soc. 2006, 128, 2812–2813. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, F.; Kentsis, A.; Osman, R.; Pan, Z.Q. Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL.hypoxia-inducible transcription factor-1alpha complex. J. Biol. Chem. 2005, 280, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, K.; Hirokawa, T.; Okamoto, T. MD simulation of the Tat/Cyclin T1/CDK9 complex revealing the hidden catalytic cavity within the CDK9 molecule upon Tat binding. PLoS ONE 2017, 12, e0171727. [Google Scholar] [CrossRef] [PubMed]
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 2000, 7, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Marcello, A.; Cinelli, R.A.; Ferrari, A.; Signorelli, A.; Tyagi, M.; Pellegrini, V.; Beltram, F.; Giacca, M. Visualization of in vivo direct interaction between HIV-1 TAT and human cyclin T1 in specific subcellular compartments by fluorescence resonance energy transfer. J. Biol. Chem. 2001, 276, 39220–39225. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu. Rev. Biophys. 2008, 37, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Luo, Z.; Schaufele, F.; Peterlin, B.M. Visualization of positive transcription elongation factor b (P-TEFb) activation in living cells. J. Biol. Chem. 2015, 290, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, K.; Omagari, K.; Okuda, T.; Hibi, Y.; Okamoto, T. Quantification of the HIV transcriptional activator complex in live cells by image-based protein-protein interaction analysis. Genes Cells 2016, 21, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Schutt, A.D.; Holly, M.; Slice, L.W.; Sherman, M.I.; Richman, D.D.; Potash, M.J.; Volsky, D.J. Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science 1991, 254, 1799–1802. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, H.; Rossi, J.J. RNAi in combination with a ribozyme and TAR decoy for treatment of HIV infection in hematopoietic cell gene therapy. Ann. N. Y. Acad. Sci. 2006, 1082, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. MBio 2017, 8, e00133–17. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Van Lint, C. Preclinical shock strategies to reactivate latent HIV-1: An update. Curr. Opin. HIV AIDS 2016, 11, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. HIV-1 eradication strategies: Design and assessment. Curr. Opin. HIV AIDS 2013, 8, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the search for a cure for HIV-1 infection: Targeting the latent reservoir for HIV-1. J. Allergy Clin. Immunol. 2014, 134, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the effort to cure HIV infection: Going beyond N = 1. J. Clin. Investig. 2016, 126, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Delagreverie, H.M.; Delaugerre, C.; Lewin, S.R.; Deeks, S.G.; Li, J.Z. Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents. Open Forum Infect. Dis. 2016, 3, ofw189. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2017, 15, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Poveda, E. Ingenol derivates promising for HIV eradication. AIDS Rev. 2014, 16, 246. [Google Scholar] [PubMed]
- Spivak, A.M.; Bosque, A.; Balch, A.H.; Smyth, D.; Martins, L.; Planelles, V. Ex Vivo Bioactivity and HIV-1 Latency Reversal by Ingenol Dibenzoate and Panobinostat in Resting CD4(+) T Cells from Aviremic Patients. Antimicrob. Agents Chemother. 2015, 59, 5984–5991. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lu, P.; Qu, X.; Shen, Y.; Zeng, H.; Zhu, X.; Zhu, Y.; Li, X.; Wu, H.; Xu, J.; et al. Reactivation of HIV-1 from Latency by an Ingenol Derivative from Euphorbia Kansui. Sci. Rep. 2017, 7, 9451. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Valente, S.T. Didehydro-Cortistatin A: A new player in HIV-therapy? Expert Rev. Anti Infect. Ther. 2016, 14, 145–148. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asamitsu, K.; Fujinaga, K.; Okamoto, T. HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies. Molecules 2018, 23, 933. https://doi.org/10.3390/molecules23040933
Asamitsu K, Fujinaga K, Okamoto T. HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies. Molecules. 2018; 23(4):933. https://doi.org/10.3390/molecules23040933
Chicago/Turabian StyleAsamitsu, Kaori, Koh Fujinaga, and Takashi Okamoto. 2018. "HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies" Molecules 23, no. 4: 933. https://doi.org/10.3390/molecules23040933
APA StyleAsamitsu, K., Fujinaga, K., & Okamoto, T. (2018). HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies. Molecules, 23(4), 933. https://doi.org/10.3390/molecules23040933