2.1. Olefin Metathesis
Olefin metathesis, a carbon-carbon double bond breaking and reforming sequence, became an important method to the synthetic organic and polymer chemist. Olefin metathesis can broadly be classified (See
Scheme 1) into ring-closing metathesis (RCM), ring-opening metathesis (ROM), ring-opening cross metathesis (ROCM), ring-opening metathesis polymerization (ROMP), acyclic diene metathesis polymerization (ADMET), and cross-metathesis (CM) [
17]. In this section, we shall present the preparation of transition metal-pyridinyl alcohol complexes and their catalytic activity in the homogeneous metathesis reactions.
The ROMP of norbornenes and 2,3-disubstituted norbornadienes has been catalysed by tungsten(VI) alkylidene complexes [
18]. The reactivity of these complexes is highly associated with the anciliary ligands present [
19]. Complex
1, a five-coordinate tungsten(VI) alkylidene complex, was synthesized by Van der Schaaf et al. (
Figure 1) [
20] The complex catalyses the ROMP of norbornene, but it is inert for simple linear alkenes.
Van der Schaaf and co-workers [
21] were interested in improving the catalytic activity of
1 for the metathesis of olefins by manipulating the ligands. They synthesized complex
2, which is an intermediate in the synthetic procedure of complex
3, using the pyridinyl alcoholato ligand
4 (see
Scheme 2).
The electronic advantage obtained from attaching two alkyl groups on the alkylidene ligand (in complex
2) is that the electron deficiency of W decreases, and therefore, the donative bond of the imido nitrogen to the W centre weakens [
21].
For the purpose of catalysing the RCM of diolefins, synthesis of polymers, isomerisation of olefins and CM of olefins, six Grubbs 1 (
Gr1) type complexes
5a–
f that contained different types of pyridinyl alcoholato ligands were synthesized by Van der Schaaf et al. [
22] The complexes were synthesized in such a way that the formation of the lithium salts of the corresponding pyridinyl alcohols was followed by a reaction with the ruthenium compound [RuCl
2(=CHC
6H
5)(P(
i-Pr
3))
2]. With the same procedure, the sulphur analogues of the first two alcoholato-ligands (
6a,b) were also synthesized in good yields [
22].
The ruthenium-complexes 5a and 5b were used in the cyclisation of diethyldiallyl malonate into 3-cyclopentene-1,1-dicarboxylic acid diethyl ester (100%, at 60 °C, in Cl3CCH3) and cyclisation of N,N′-di-2-propenylcarbamic acid 1,1-dimethylethyl ester into 2,5-dihydro-1H-pyrrole-1-carboxylic acid 1,1-dimethylethyl ester (100%, at 60 °C, in CHCl3). Complex 5a was also used in the cyclisation of the 5-hexenyl ester of 10-undecenoic acid into oxacyclohexadec-11-en-2-one (50% at 60 °C in toluene) and polymerization of dichloropentadiene.
The tungsten(VI) phenylimido alkylidene complexes
7a,b,
8a,b and
9a–
d (
Figure 2) containing a monoanionic
O,
N-chelating ligand (pyridinyl alcoholato) were also synthesized by Van der Schaaf et al. [
23] Complexes
7a and
8a were synthesized by refluxing the corresponding pyridinyl alcohol lithium salt via transmetallation to the trialkyl tungsten phenylimido chloride [W(CH
2SiMe
3)
3Cl(=NPh)] in THF for one hour. Complex
7a was produced as a mixture of
anti:
syn (1:10) rotamers.
The geometry of 7a (syn isomer) is a distorted square pyramidal in which the alkylidene function occupies the apical position. The metal atom is slightly above the basal plane defined by the N-bonded phenyl imido group, the CH2SiMe3 group and the alkoxy oxygen with the 2-pyridinyl nitrogen of the chelating ligand.
The six coordinated complex 8a is slightly distorted octahedron in which the phenylimido N and the alkoxy groups occupy trans positions. The three CH2SiMe3 groups and the pyridinyl nitrogen are bonded in the equatorial plane. The five coordinated complex 8b, on the other hand, was synthesized by reacting the lithium salt of the pyridinyl alcohol with the trialkyl tungsten phenylimido chloride at room temperature. Refluxing complex 8b for four hours resulted in complex 7b (anti:syn, 4:6). The six coordinated complex 9 was also synthesized at room temperature via reacting the lithium salt of the corresponding pyridinyl alcohol with dialkyl-tert-butoxytungsten phenylimido chloride [W(CH2SiMe3)2Cl(=NPh)(OCMe3)]. The substituents in complex 9 play a less important role in W-complexes.
Complexes 7a,b catalysed the ROMP of norbornene to give polymeric cyclopentenes at 70 °C (≥90% cis-vinylene bonds). The alkyl substituents (R1 and R2) on the α-C of the pyridinyl alcoholato ligand can be easily varied, making these ligands readily tuneable. This tuning has enabled the isolation and characterisation of alkylidenes 7a,b and their precursor complexes.
The decreasing order of the relative basicity of the methoxide ligands in the complexes 7a,b is assumed to be OCH(CMe3)(2-py) > OCPh2-(2-py). The strong basicity of the ligand in 8b decreased the acidity of W and therefore the pyridine nitrogen does not coordinate to W. In 8a, the pyridine nitrogen coordinates due to less basicity of the ligand, which makes the W more acidic. Complex 9 is thermally more stable than 8a,b. None of complexes 9a–d gave an alkylidene complex, even after refluxing for 24 h. Generally, metal atoms in a high valent early-transition-metal complex decrease in electron deficiency when an alkylidene functionality is formed from two alkyl groups via an Hα-abstraction reaction. These electronic advantages for a d0-metal complex are illustrated by the two molecular structures of 7a and 8a. The syn rotamers result in a five coordinate complex and the potentially bidentate ligands in the anti rotamers monodentate bond due to steric reasons. Complexes 7a,b showed low activity in the ROMP of norbornene at low temperature, but significantly increased the rate of ROMP when increasing the temperature due to the high Lewis acidity of W to interact readily with the olefinic double bond of norbornene. The complexes can polymerise the ROM reaction of strained cyclic olefins other than the ROMP of norbornene; however, they are inert towards linear olefins.
The dendritic ruthenium complexes
10a,
b (
Figure 3) were synthesized from the dendritic pyridinyl alcohols
11a,
b (
Figure 3) via treatment with
n-BuLi and PhC(H)=RuCl
2(PR
3)
2 [
24]. Complexes
10a,b catalysed the RCM reaction (at 80 °C) of diethyl diallyl malonate with 100% conversion after 30 min. The result obtained is comparable to the unimolecular catalyst. The conversion to diethyl-3-cyclopentene dicarboxylate stopped after about 20% conversion in an experiment that was carried out in a solution where the catalyst is separated from the substrate and product by a nanofiltration membrane MPS-60. The use of a dendritic pyridinyl alcoholato ligand is very important to easily remove the catalyst from the product.
Ruthenium alkylidene complexes bearing one N-heterocyclic carbene (NHC) and one pyridinyl alcoholato ligand,
12a–
d (
Figure 4) were synthesized by two different procedures by Denk et al. [
25] The first procedure involves treatment of the
Gr1 complex with the lithium salt of the pyridinyl alcoholato ligand
13 followed by the addition of the NHC ligand. In the second procedure, the
Gr1 complex was treated with the NHC ligand, followed by a reaction with the lithium salt of the pyridinyl alcoholato ligand
13. Complexes
12a,b were synthesized via the first procedure with 73%
12a (the yield of
12b was not reported), while complexes
12c,d were synthesized through the second procedure and resulted in 73% and 46% yield, respectively [
25].
The complexes were tested for the ROMP of norbornene and cyclooctene at room temperature and at 60 °C. Complexes
12a and
12d resulted in oligomers at room temperature, while
12b and
12c yielded 57% and 5% (norbornene) and 18% and 14% (cyclooctene), respectively. At 60 °C, norbornene underwent ROMP with
12a (100%),
12b (98%),
12c (99%) and
12d (100%) and cyclooctene
12a (75%),
12b (78%),
12c (72%) and
12d (80%) [
17,
25]. The W and Ru complexes of pyridinyl alcoholato ligands in complexes
7 and
12 share similarities in yielding less at low temperatures and high at high temperature in the ROMP of norbornene, which was comparable to
Gr2.
Jordaan and Vosloo [
26,
27] synthesized four different types of pyridinyl alcoholato ligands using Herrmann et al.’s [
28] procedure for the purpose of synthesising the Grubbs-type catalysts for the metathesis of 1-octene. They prepared the lithium salts of the four pyridinyl alcoholato ligands in a similar procedure to Van der Schaaf et al. [
22], which were then reacted with the
Gr1,
Gr1-
py and
Gr2 to produce the ruthenium alkylidene complexes
14–
16 (
Figure 4) bearing the stable NHC and chelating pyridinyl alcoholato ligands. The incorporation of pyridinyl alcoholato ligands with the Grubbs-type catalysts has shown an increase in the thermal stability, activity (in the metathesis of 1-octene) and lifetime of the catalyst [
26,
27,
29].
Vosloo and co-workers [
30,
31] further investigated the activity, stability and selectivity of
Gr2-type catalysts for the metathesis of 1-octene. With this aim, five different types of pyridinyl alcoholato ligands and the corresponding
Gr2-type complexes
17a–
c (
Figure 5) were synthesized. The same procedure as Jordaan [
26,
27] was followed, however, the synthesis of complexes using pyridinyl alcohols made up of adamantanone and camphor was not successful [
30]. The complexes
17a–
c showed low activities compared to
16d for the metathesis reaction of 1-octene. They showed, however, high stability and turnover numbers (TONs) compared to
Gr2. At high temperature, the activity of the complexes increased with decreasing selectivity. The combination of the NHC ligand and the chelating ability of the pyridinyl alcoholato ligands are responsible for the stability and activity of the catalyst at high temperatures. Incorporating other electron-donating (OMe) and electron-withdrawing (Cl) at the 2- or 4-position on one of the α-phenyl groups of
16d showed improved catalytic performance for 1-octene metathesis at temperatures ranging from 80–110 °C [
32]. The 4-methoxy-substituted complex outperformed the rest at 110 °C (96% conversion with 95% selectivity towards the major products 7-tetradecene and ethene). A modelling study suggested that the improved catalytic performance can be attributed to steric repulsion between the substituted phenyl group and the NHC ligand resulting in strengthening the Ru-N bond [
32]. Slugovc and Wattel [
33] patented a number of 8-quinolinolate
Gr2 derivatives for use in ROMP reactions. The latter was found to be quite inactive (<1% conversion) for 1-octene metathesis at 60 °C [
31].
The removal of trace amounts of residual ruthenium catalyst and/or catalyst degradation products in the area of pharmaceutical application is one of the challenging aspects in olefin metathesis reactions. One important method of ruthenium removal is adsorption onto surfaces [
34]. The minimum possible amount of ruthenium catalyst residue for end products in the pharmaceutical industries should be less than 10 ppm. However, the adsorption techniques used could not do so. To fill this gap, Schachner et al. [
35] synthesized six new ruthenium complexes that contained pyridinyl alcoholato ligands from Grubbs’ 1st and 2nd generation ruthenium carbenes, as shown in
Scheme 3 and
Table 1.
The catalytic activities of the precatalysts were evaluated with the ROMP of cyclooctene, CM of 5-decene with 5-hexenylacetate and the RCM of 5-hexen-1-yl-10-undecenoate and most of them showed superior activity compared to the commercially available precatalysts in CM and RCM reactions. Even though they showed moderate activity in ROMP, it could be considered as an important aspect from an industrial application point of view. All complexes showed a very high affinity to untreated, unmodified and commercially available chromatography-grade silica [
35]. This led the residual ruthenium concentration in the unprocessed reaction mixture to below 10 ppm, which is a prerequisite for fine-chemical application [
13].
With the aim of developing heterogeneous catalysts for metathesis, Cabera et al. [
36,
37] made a systematic evaluation of the activity and stability of five Grubbs-type precatalysts
16b,
20a,
21 and
22a,b that contained pyridinyl alcoholato ligands for a given metathesis (RO-RCM and CM) within the context of a biphasic solid/liquid system. The reactions were done by adsorbing the substrate and catalyst on a thin layer silica plate and developing the TLC in EtOAc/hexane (1:7
v/
v) for CM and hexane for RO-RCM. The substrate for self-CM was methyl 9-dodecene and for RO-RCM
cis-cyclooctene. None of them gave a product spot at room temperature for the CM. Only
22a showed activity for the RO-RCM of
cis-cyclooctene forming 1,8-cyclohexadecadiene among many oligomeric multiples of cyclooctene.
Manipulation of the ligands around the transition metal (W and Ru) complexes showed increment both in the activity and stability on the metathesis reactions of olefins. The chelation ability of the pyridinyl alcohol nitrogen combined with the stable NHC ligand is responsible for the increment in the activity and stability of the catalysts. The investigations of Vosloo and co-workers [
26,
27,
29,
30,
31,
32] were focused on the steric and electronic effects of the groups on the carbon atom to which the alcohol function was attached in the pyridinyl alcohol. Even though the electronic effect of the substituents did not show significant influence on the catalyst activity, the steric effect is clearly seen on the
16d precatalyst stability.
We did not find any literature on the investigation of the electronic and steric effects of substituents (on the pyridine ring) on the chelation efficiency of pyridinyl alcoholato ligands. Therefore, the chelation ability of the pyridinyl alcohols needs further investigation by synthesising catalysts with electron-withdrawing and/or electron-donating groups on the pyridine ring. The hemilabile nature of the pyridinyl alcoholato ligands could possibly be manipulated by putting either electron-donating or electron-withdrawing groups on the pyridine ring.
2.2. Olefin Polymerization
The pyridinyl alcohols
23a–
g (
Figure 6) were used by Kim et al. [
38] in order to synthesize group four metal (pyCAr
2O)
2M(NR
2)
2 complexes containing bidentate pyridine. They reacted M(NMe
2)
4 (where M = Ti, Zr, and/or Hf) with the pyridinyl alcohols
23a–
g to obtain complexes
24a–
g in very good yields.
All ligands 23a–g reacted with Ti(NMe2)4 to yield the complexes 24a–g with 98%, 88%, quantitative, 82%, 95%, 55% and 59% respectively. With Zr ligands 23a, 23b and 23e resulted complex 24a–c with 92%, 67% and 62% yields using the same procedure, respectively. Ligands 23c,d resulted in the complex (pyCAr2O)xZr(NMe2)4-x (x = 1–3) with 65% yield. Using three- and four-equivalents of 23c and 23e can result in the tris and tetrakis ligand species of Zr.
The complexes (pyCAr2O)2Hf(NMe2)2 of 23a (92%) and 23e (58%) were synthesized and have similar structures as that of Ti and Zr complexes, while 23b and 23c yielded mixtures of products. Three equivalents of 23b,c resulted in the tris(ligand)species (py(Ar2O)3Hf(NMe2) in 54% and 82%, respectively. The formation of the product mixtures is due to comparable rates of reaction of pyridinyl alcohol with (pyCAr2O)M(NMe2)2, M(NMe2)4 and/or (pyCAr2O)M(NMe2)3. More acidic pyridinyl alcohols are less selective for the desired (pyCAr2O)M(NMe2)2 species.
X-ray crystallography revealed that these complexes have
C2-symmetric structures in the solid state and in solution, but undergo facile inversion of configuration at the metal, with racemisation of configuration of barriers in the range of 12–14 kcal/mol. The Ti and Zr complexes of the type (pyCAr
2O)
2M(NMe
2)
2 of pyridinyl ligands
23a,
23b,
23d, and
23e (
23d,e only for Ti) were tested in the polymerization reaction of ethylene in the presence of the Lewis acid Al(
i-Bu)
3 and methylalumoxane (MAO) as cocatalysts and resulted in a 0.17–1.5 g yield in toluene at 1 atm ethylene and 43 °C with Zr catalysts more highly active than Ti complexes [
38]. The polymerization of ethylene has been investigated using the in situ alkylation/activation protocol developed for Cp
2Zr(NMe
2)
2 metallocene amide compounds [
39]. The AlR
3 reagent and activator would generate (pyCAr
2O)
2M(R)
+ or (pyCAr
2O)
2M(H)
+ species in situ. The Ti and Zr complexes are activated in situ for the polymerization of ethylene.
A Zr complex
25 (
Figure 6) having distorted octahedral structure with
trans-O,
cis-N,
cis-C ligand arrangement, but that undergoes inversion of configuration at the metal centre at elevated temperature resulted in a reaction with pyridinyl alcoholato ligands
26a,
b and
27 with Zr(CH
2Ph)
4 in toluene or benzene [
40]. Alkyl abstraction from
25 by treatment with the benzene solution of B(C
6F
5)
3 or Zr-R protonolysis reactions resulted in cationic five-coordinate species (pyCR
2O)
2Zr(CH
2)Ph
+. These species adopted distorted square pyramidal structures in which the ligand arrangement is strongly influenced by the π-donor properties of the alkoxide ligand. The cationic complexes obtained via the aforementioned reaction of
25 (of ligands
26a and
27) with the Lewis acid B(C
6F
5)
3 catalyse ethylene polymerization with very low activity resulting in much lower molecular weight polymers, while the cationic complex resulted from
25 with ligand
26b did not polymerise ethylene at all. This clearly showed that the electron withdrawing CF
3 is beneficial and also the presence of relatively electron-donating pyCMe
2O
- ligand in the cationic complex made of
25 (with ligand
26b). The detection of the benzyl end groups by
1H NMR suggested that the polymerization is initiated via insertion into the Zr-CH
2Ph bond. The cationic complex of
25 that is prepared with ligand
26a undergoes polymerization of 1-hexene to result in polyhexene in moderate activity under mild conditions (CD
2Cl
2, 23 °C). This trend is opposite to that reported for metallocene catalysts, in which incorporation of electron-donating substituents on the cyclopentadiene (Cp) ligands generally increases activity in the absence of overriding steric factors [
41].
The Zr complex
28 [Zr(BMP)(CH
2Ph)
2] (
Figure 7) was synthesized from 2,6-
bis[(1
S,2
S,5
R)-(−)-menthoxo]pyridinyl diol, (BMP)H
2, via treatment with Zr(CH
2Ph)
4 [
42]. Treatment of
28 by one equivalent of B(C
6F
5)
3 in C
6D
6 or C
6D
5Br resulted in complex
29. The reaction of
29 with Et
2O in C
6D
5Br yielded complex
30a. Complex
30b was prepared by abstraction of one of the benzyl ligands with [CPh
3]
+[B(C
6F
5)
4]
- from
28. Complex
29 undergoes single insertion of ethylene into its Zr-carbon bond to yield [Zr(BMP)(CH
2CH
2CH
2Ph)][(η
6-PhCH
2)B(C
6F
5)
3]. α-Olefins and cyclooctadiene, on the other hand, afforded diastereomeric mixtures of the general formula [Zr(BMP)(η
1:η
6-CHRCHR’CH
2Ph)]
+ [B(CH
2Ph)(C
6F
5)
3]
-. It was reported that the complexes might lead to higher insertion stereoselectivities of olefins and actual polymerization [
42].
Vinyl-type norbornene polymers (PNBs) with high molecular weight and relatively narrow molecular weight distributions resulted from the polymerization of norbornene with Ni and Pd complexes bearing (imino)pyridinyl alcoholato tridentate ligands [
43]. The five- and six-coordinated Ni complexes, and cationic Pd complexes formed with the [PdCl
4]
2− counter ion were activated with excess methylaluminoxane (MAO). The polymerization reaction was performed at an Al/catalyst ratio of 1000:1 at 30 °C in toluene. The complexes are synthesized by mixing the corresponding pyridinyl alcohol with one equivalent of NiCl
2·6H
2O, NiBr
2 or Ni(OAc)
2·4H
2O (for
31c) in ethanol at room temperature (
Scheme 4). The reaction of pyridinyl alcohols and PdCl
2 in ethanol at room temperature proceeded relatively slowly; therefore the preparation of palladium complexes
31d and
32d was requisite to be carried out at 60 °C for 3 h.
The
N and
O atoms of the (imino)pyridinyl alcoholato ligand are coordinated to the metal centre with a distorted trigonal bipyramid for the nickel chloride complex
31a, a distorted octahedron for the nickel acetate complex
31c and a square planar arrangement for the cation moiety of palladium complex
32d (see
Figure 8).
A series of cobalt(II) and nickel(II) complexes
35–
40 supported by (imino)- and (amino)pyridinyl alcoholato ligands were synthesized by Ai et al. [
44] as it is shown in
Scheme 5 and
Scheme 6 and tested for 1,3-butadiene polymerization reaction. The 2-arylimino-6-(alcohol) pyridines (
L1–
L4) or 2-arylamino-6-(alcohol) pyridines (
L5 and
L6) reacted with one equivalent of CoCl
2·6H
2O or NiCl
2·6H
2O in absolute ethanol at room temperature.
All the complexes adopted a distorted trigonal bipyramidal configuration with the equatorial plane formed by the pyridinyl nitrogen atom and two chlorine atoms. On activation with ethylaluminum sesquichloride (EASC), the cobalt complexes displayed high catalytic activity and selectivity (>96%) under Al/Co molar ratio of 40:1 at 25 °C. In comparison with the Co-complexes, the Ni-complexes resulted in relatively lower catalytic activity, cis-1,4 content and lower molecular weight under similar reaction conditions. The conversion of butadiene, microstructure and molecular weight of the resulting polymers were affected by the reaction parameters and size of substituents on the aromatic ring.
Ai et al. [
45] synthesized a series of ion-pair cobalt complexes
41–
46 supported by benzimidazolyl-pyridinyl alcoholato ligands and tested them in the polymerization of 1,3-butadiene (BD). The polymerization of BD by a pyridinyl alcoholato ligand supported cobalt catalyst resulted in
cis-1,4-polybutadiene with high selectivity (>95%) under Al/catalyst molar ration of 50:1 at 30 °C upon activation with the cocatalyst EASC. In 1 h, 91–100% conversion of BD/catalyst ratio of 2500:1 and toluene as solvent is reported. Increasing bulkiness of substituents in the ligand structure led to lower catalytic activity. The ion-pair cobalt complexes were synthesized by reacting the corresponding pyridinyl alcohols with one equivalent of CoCl
2·6H
2O at room temperature in anhydrous ethanol or methanol (
Scheme 7). All complexes possessed cationic octahedral geometry bearing two pyridinyl alcoholato ligands with [CoCl
4]
2− as the counter ion.
The only pyridinyl alcohols involved in the preparation of Zr, Ti, Hf, Co, Ni and Pd complexes for olefin polymerization catalysis are the ones we have presented. Compared to the large number and varieties of pyridinyl alcohols, a very limited investigation has been done so far. It is therefore worthwhile to consider more pyridinyl alcohols with different electronic and steric effects and further investigate this area.
Enantioselective reactions have been given a great deal of attention in the past few decades. Biologically important organic compounds have chiral centres that became a challenge for the synthetic organic chemist. The synthesis of chiral organic compounds therefore needs chiral catalysts at large. Many research groups invested their time in looking for the best type of chiral catalyst that can result in excellent chemical yield (c.y.) and enantiomeric excess (ee).
Chiral pyridinyl alcohols have a great deal of attention in the construction of chiral transition metal catalysts. Significant progress has been made in utilising pyridinyl alcohols as chiral ligands in the various asymmetric reactions. Hereunder, we present the use of pyridinyl alcohols as chiral ligands in the various homogeneous asymmetric transition metal catalysed reactions.
2.3. Olefin Epoxidation
There are many methods for the preparation of epoxides. The most extensive industrial method is the oxidation of alkenes in liquid phase using organic hydroperoxides in the presence of a catalyst [
46]. Indeed, hydrocarbon-soluble organometallic compounds of the transition metals can be employed as homogeneous catalysts.
Hawkins and Sharpless [
47] synthesized the pyridinyl diol
48 (
Figure 9) for the purpose of testing the titanium(IV)-pyridine diol complex
47 as a model for titanium-tartrate asymmetric epoxidation catalyst. The model complex was synthesized by reacting
48 with Ti(O
i-Pr)
4 in CH
2Cl
2. The asymmetric epoxidation of (
E)-
α-phenylcinnamyl alcohol was catalysed by adding 3 moles of tertiary butylhydroperoxide (TBHP) to a mixture of X (X = 1.25, 1.50, 2.00) equivalents of
48 and Ti(O
i-Pr)
4, in CH
2Cl
2 at −5 °C. The epoxide
49 was obtained in 41% ee, 40% ee, and 41% ee, respectively (increasing the concentration of
48). With the same procedure, 3 moles of tritylhydroperoxide (Ph
3COOH) were added keeping the concentration of
48, 1.5 equivalents and varying the concentration of Ti(O
i-Pr)
4, 3 mM, 13 mM, and 52 mM. The epoxide
50 was obtained in 64% ee, 64% ee and 52% ee, respectively.
The yield obtained with Ph
3COOH, 64% ee agrees with the model complex
47 based on the titanium-tartrate system, while the 41% ee obtained with TBHP disagrees. This switch in face selectivity suggested an alkyl hydroperoxide dependent change in mechanism [
47].
Tungsten(VI) compounds are well known for their catalytic activity in olefin oxidation reactions since Milas reported the metal-catalysed dihydroxylation of olefins with H
2O
2 and WO
3 in the 1930s [
48]. Herrmann et al. [
28] came up with a result that revealed the nature of the ligand has an important influence on the product selectivity in addition to the metal oxidation state of the catalyst in terminal olefin epoxidation. During the investigation that was made to resolve the great challenges in the catalytic oxidation of terminal olefins to epoxides, by means of molecular oxygen, they synthesized pyridinyl alcoholato ligands
4 and
51a,
b. The dioxomolybdenum(VI) complex
52 was synthesized by reacting MoO
2(acac)
2 with two moles of the pyridinyl alcohol in MeOH, within 30 min at room temperature [
28]. The geometry of complex
52 is distorted octahedral coordination geometry with the oxo-ligands forming a
cis-dioxo unit that is common to all [MoO
2]
2+ complexes. While the nitrogen atoms of the pyridine are positioned
trans to the oxo-ligand each, the alkoxy functions of the ligands are placed perpendicular to the plane defined by the dioxomolybdenum core. The solubility in non-polar aprotic solvents such as alkenes or alkanes and its stability during the course of the reaction make the complex valuable in catalytic applications. In the catalytic autoxidation reaction of 1-octene, they [
28] obtained significant selectivity improvements (38 to 55%) with elemental oxygen. They also synthesized WOCl
3L-type complexes via the treatment of WOCl
4 with the corresponding pyridinyl alcohol in CH
3Cl under mild conditions.
Two years later, Herrmann et al. [
49] prepared dioxotungsten(VI) complexes of the type WO
2L
2 (
52) with pyridinyl alcoholate ligands from WO
2(acac)
2, by ligand exchange, with two equivalents of pyridinyl alcohol in methanol. They synthesized the complexes dioxo
bis[2-(2′-pyridinyl)propan-2-olato-
N,
O]tungsten(VI), dioxo
bis{di[4″,4″-di(methoxy)phenyl]-(2′-pyridinyl) methanolato-
N,O}tungsten(VI), dioxo
bis[9-(2′-pyridinyl)fluoren-9-olato-
N,
O]tungsten(VI) and dioxo
bis[5-(2′-pyridinyl)-10,11-dihydrodibenzo[a,d]cyclopentan-5-olato-
N,O]tungsten(VI). The complexes were tested as catalysts (1 mol%) in olefin epoxidation with
tert-butyl hydroperoxide (
t-BOOH) (oxidant) and
cis-cyclooctene (substrate) at 70 °C without solvent being in good (50–65%) conversion with excellent (100%) selectivity towards the epoxide was achieved.
Oxovanadium(IV) compounds as oxidation catalysts were first discovered by Katsuki and Sharpless [
50] where they applied it in the regioselective epoxidation of allylic alcohols. The 2-[(−)-menthyl]-pyridine oxo-complexes of vanadium(IV),
53 and molybdenum(VI),
54 (
Figure 10) were synthesized from VO(acac)
2 and/or MoO
2(acac)
2 and 2-[(−)-menthyl]pyridine in 50% and 73% yield, respectively [
51]. The vanadium(IV) complex is a square pyramidal and the Mo(VI) has a distorted octahedral arrangement. Both complexes were found to be active in the epoxidation of terminal olefins (e.g., 1-hexene 20% conversion and 25% ee) with
t-BOOH as an oxidant. They also oxidised the sulphides (PhSMe) similarly to the corresponding sulphoxide (90% selectivity) with an ee of up to 18% using H
2O
2 as the oxidant.
Complexes
55 and
56 were prepared from the 2,6-
bis[(1
S,2
S,5
R)-(−)-menthyl]pyridine by treat ment with VO(O
i-Pr)
3 and/or MoO
2(acac)
2 respectively by Bellemin-Laponnaz et al. [
52]. The complexes demonstrated the ability to catalyse the asymmetric oxidation of prochiral olefins with
t-BOOH as the oxidant.
Chiral dioxomolybdenum(VI) and chiral dioxotungsten(VI) complexes of type
52 were also synthesized by Herrmann et al. [
53] to investigate their catalytic potential in the asymmetric epoxidation of unfunctionalised olefins. The pyridinyl alcoholato ligands
57a,
b and
58 were used as ligands for the complex formation. Herrmann et al. [
53] tested three different metal precursors bearing the
cis-dioxo metal fragment for the formation of the complexes; (i) MoO
2Cl
2, 2-equivalents of lithium salt of ligand, THF, and refluxing for 30 min, (ii) MO
2(acac)
2, 2-equivalents of ligand, MeOH, 30 min, at room temperature, and (iii) Na
2(MO
4), 0.07-equivalent of ligand, H
2O, pH 4, 12 h at room temperature. Although MoO
2(acac)
2 and WO
2(acac)
2 are much easier to handle than the dioxodichlorides (because of their high resistance to air and moisture), the metal salts Na
2[MoO
4] and Na
2[WO
4] are more advantageous as they are economical, easy to be handled, and ecologically friendly. In both complexes, a distorted octahedral coordination geometry was formed around the metal centres by the two anionic
N,O-chelating ligands and two oxo-ligands [
53].
Upon epoxidising
trans-methyl styrene with the complexes
54, the catalytic activity for molybdenum complexes resulted in good yields (71–81%) of the (
S,
S)-epoxide. Though the yields of tungsten complexes were only 31 to 36%, it is worthwhile to know the fact that tungsten systems were used for the first time in the asymmetric epoxidation of olefins. The bulkier the ligand, the higher the ee and complexes bearing the same type of ligand resulted in similar ee. Therefore, it is worthwhile to conclude that the enantioselectivity depends more strongly on the ligand than on the metal [
53].
Kuhn et al. [
54] prepared chiral bis(oxazoline) and pyridinyl alcoholate dioxo-molybdenum(VI) complexes
59 (MoO
2Cl(THF)L type) and
60 (MoO
2L
2 type) via treatment of MoO
2Cl
2(THF)
2 with the corresponding 2-pyridinyl alcohol ligands (
57a,
b,
61,
62, and
63a,
b) in CH
2Cl
2 and/or with 2-equivalents of chiral pyridinyl ligands in TlOEt (method 1) or CH
2Cl
2 (method 2) respectively, in good yield (
Figure 11). The catalytic potential of the complexes was evaluated in the asymmetric epoxidation of
trans-
β-methyl styrene using
t-BOOH as oxidant. Conversions between 23–81% and ee 1–11% of the (
S,
S) configuration were observed in all cases except the complexes of the type MoO
2Cl (THF)L, where L is pyridinyl alcohol made up of (+)-8-phenylisomenthone (
62) and (−)-thujone (
63b), which resulted in the (
R,
R)-configuration. Low substrate conversion (23–47%) and ee (1–5%) were observed for complexes of the type MoO
2L
2,
60 where the ligand is
57a,
61 and
62. Where L =
57b has shown a substrate conversion of 51% and 23% ee (
S,
S)-configuration, which is the highest ee value.
The MoO
2Cl
2L (where L = 2-pyridinyl alcohol ligand) type complex
64 was synthesized via the reaction of MoO
2Cl
2(THF)
2 with 2-pyridinyl alcohol in CH
2Cl
2. Using 2 moles of 2-pyridinyl alcohol in CH
2Cl
2 and refluxing resulted in
65 (MoO
2L
2 type) [
55]. The (1
R,2
S,5
S)-8-trimethyl silyloxy-1-(2-pyridinyl)menthol was prepared according to Herrmann et al. {28,53] and Kuhn et al. [
54]. Complex
65 was tested for epoxidation of a variety of olefins using
t-BOOH as oxidant at 55 °C under air atmosphere (1 atm) without solvent. The epoxidation results obtained were: cyclooctene (100% conversion and 89.8% ee), styrene (49.8% conversion and 54.0% ee),
α-pinene (63.0% conversion and 11.7% ee) and (
R)-(+)-limonene (89.5% conversion and 82.6% ee). The catalytic activity of
65 was slightly lower than
64. Ring-opening activity was also observed for
α-pinene oxide-producing campholenic aldehyde and epoxy campholenic aldehyde.
Fridgen et al. [
56] synthesized molybdenum(VI) complexes,
66–
68 (
Figure 12) with chiral
N,
O-ligands
69a,
b and
70, which are derived from carbohydrate via the
n-BuLi-mediated preparation method from 2-bromopyridine. Complex
66 was synthesized with its diastereomeric pair. The catalytic epoxidation of
trans-methylstyrene with
t-BuOOH or cumolhydroperoxide (CHP) in 1.0 mol% of catalysts
66–
68 after 6 h at 50 or 70 °C was investigated. The results are shown in
Table 2.
The influence of the oxidant can be seen at the enantiomeric excesses. With cumyl hydroperoxide, higher product selectivities were obtained, which might be a consequence of π-interactions between substrate and the oxidant. This type of interaction has also been taken into account for the Jacobsen and Cavallo system [
57]. Similar ee values have been observed with a variety of
cis-MoO
22+ epoxidation catalysts bearing chiral ligands, such as
bis-oxazoline,
cis-diol,
cis-8-phenylthiomenthol, and others [
54,
58]. The complex
bis[
N,O-{2′-pyridinylmethanolate}]-dioxomolybdenum (VI) was synthesized and used as catalyst in the epoxidation of 1-octene with ethylbenzenehydroperoxide in liquid phase. Besides giving 1,2-epoxyoctane as the only product, the catalyst resulted in a better yield than its acetylacetonate complex [
46].
Asymmetric epoxidation of
cis-1-propenylphosphonic acid (CPPA) was catalysed by chiral tungsten(VI) and molybdenum(VI) complexes
71a–
d (
Figure 13) [
59]. The complexes were synthesized according to the methods of Herrmann et al. [
53] and Kuhn et al. [
54] The CPPA was converted to 100% of the corresponding epoxides with ee’s varying from 52 to 80% in CH
2Cl
2, which is the better solvent for this specific epoxidation, at reaction temperatures of 0, 25 or 50 °C. Generally, complexes with ligand (+)-campy (which is the pyridinyl alcohol from (+)-camphor) resulted in slightly higher ee’s than (−)-fenpy (the pyridinyl alcohol from (−)-fenchone). Similar product ee’s were observed in the presence of catalysts with similar ligands but different metals. At 0 °C in CH
2Cl
2, complex
71b catalysed the epoxidation reaction to give the product with the highest ee value of 80%. At 50 °C, the ee of the epoxide was reduced to 68% with the same catalyst. The epoxidation mechanism was described as direct oxygen transfer on the interface of the biphasic H
2O-nonprotic system.
Except for VO(acac)
2, oxovanadium compounds have never been employed in oxidation catalysis with molecular oxygen. Oxovanadium(IV) complexes of
bis(aryl)-2-pyridinyl alcohols
72a–
g (
Figure 13) where the ‘R’ is phenyl
72a (73%),
p-(
t-Bu)-C
6H
4 72b (77%),
p-F-C
6H
4 72c (71%), C
12H
8(cyclic)
72d (76%),
m-CF
3-C
6H
4 72e (35%),
p-Cl-C
6H
4 72f (78%) and
p-MeO-C
6H
4 72g (68%) were synthesized by Lobmaier et al. [
60] The ligands were prepared by a nucleophilic attack of 2-lithiopyridine on the corresponding aromatic ketones according to reported procedures [
61,
62,
63,
64]. The catalysts were synthesized via refluxing VO(SO
4)·5H
2O with 2 moles of the pyridinyl alcohol in the presence of CH
3CO
2Na in EtOH. The complex is a square planar pyramid coordination sphere of vanadium with the nitrogens in the pyridine rings
trans to each other. Autoxidation reaction with 1-octene and molecular oxygen resulted in selectivity of approximately 30%, which showed slight improvement from the reaction without any catalyst (23%).
The rhenium pyridinyl alkoxide complexes of the type [ReOCl
3(L)(NBu
4) and [ReOCl(L)
2] (where L = pyridinyl alcohol ligand),
73–
76 (
Figure 14) were synthesized by Lobmaier et al. [
65] The complexes
73a,
b and
74a,
b were synthesized via reacting ReOCl
4−[NBu
4]
+ with the corresponding pyridinyl alcohol in THF at room temperature for one hour. Heating
73a,
b also resulted in
74a,
b, which therefore tells us that complexes
73a,
b are the kinetic, whereas
74a,
b are the thermodynamic products. They also used another method to synthesise complexes
74a,
b, i.e., by reacting ReOCl
4-[NBu
4]
+ with 2 moles of the pyridinyl alcohols (with R = CH
2CH
3, (CH
2)
2CH
3 and Ph) in EtOH at room temperature for three hours. This method is more convenient than the previous one because there is no need for LiCl extraction as the second mole of the pyridinyl alcohol forms a pyridinium chloride salt. It was possible to see the octahedral coordination environment of rhenium with substantial distortion from the single crystal X-ray crystallography of
73. The co-planarity of all chlorines and the
trans coordination of the pyridine ligand to the oxo-ligand of the rhenium are also vividly seen in the X-ray crystallography. Complexes
75a,
b were prepared via reaction of ReOCl
4−[NBu
4]
+ with 4 moles of the pyridinyl alcohol in EtOH at 80 °C for one hour. Complexes
76a,
b were obtained while preparing complex
75a,
b. Palladium (II) and copper (II) complexes of the various pyridinyl alcohols were also synthesized by Lobmaier et al. [
65]; however, as they are inactive towards the epoxidation reaction, we have left them without consideration. The complexes
73a,
b and
74a,
b partially decomposed during testing epoxidation of cyclooctene in TBHP. The complexes
75a,
b, on the other hand, have shown significant catalytic activity in the formation of cyclooctene oxide during the first eight hours.
The epoxidative reactions of both terminal and internal, open chain and cyclic olefins using the common oxidants
t-BOOH, H
2O
2 and TBPH were well investigated by several research groups; the results in both conversion and selectivity were being promising. Herrmann et al.’s [
29] and Lobmaier et al.’s [
60] investigation into the alleviation of the problem pertaining to the oxidation of terminal olefins by molecular oxygen is a good beginning. However, the results obtained are much less both in conversion and selectivity compared to that of epoxidation with oxidants. Therefore, it needs further investigation in order to come up with a better conversion and selectivity.
2.6. Enantioselective Addition of Organozinc Compounds to Aldehydes
Reactions of organozinc compounds to aldehydes follow a nucleophilic addition pathway. The reaction served as one of the very important C-C bond formation reactions that results in chiral secondary alcohols of synthetic importance. The ability to control the enantioselectivity of the product is a value-adding reaction to the synthetic organic chemist. The literature survey of the various investigations that are done so far with special emphasis on the use of pyridinyl and bipyridinyl alcoholato ligands is presented hereunder.
Although dialkylzinc is not often utilised for chiral addition to aldehydes in modern organic synthesis, due to the rise of other fast reacting reagents such as alkyllithium and Grignard, it has served a great deal since its discovery by Frankland [
76] in 1849. The use of a catalytic amount of (
S)-leucinol with diethylzinc, which resulted in 49% ee, is the beginning of research on asymmetric organozinc additions to carbonyl compounds [
77]. According to the reports in literature, coordination of ligands to dimethylzinc converts its linear structure into an approximate tetrahedral structure [
78]. This reduces the bond order of the Zn-C bond and increases the nucleophilicity of the zinc alkyl groups. Consequently, chiral ligands not only control the stereochemistry of the organozinc addition, but also activate the zinc reagents.
In organozinc addition to carbonyls, the stereochemical outcome of the reaction was significantly influenced by the aggregation behaviour of the various zinc-containing species that are involved in the transformation [
79]. The pyridinyl alcohols react with dialkylzinc to produce zinc-based chiral Lewis acid complexes that can further coordinate with both aldehyde and substrates and the dialkylzinc reagent to conduct the catalytic addition. In addition to its action as a Lewis acid to activate carbonyls and also as Lewis base to activate the organozinc reagents, the chiral environment of the ligand controls the stereochemistry of the reaction [
77].
Major methods of enantioselective synthesis of optically-active secondary alcohols are; (i) enantioselective alkylation of aldehydes and (ii) enantioselective reduction of ketones [
5]. Optically-active secondary alcohols are components of many naturally occurring compounds, biologically active compounds, and materials such as liquid crystals [
5]. A number of investigations have been made with the aim of enhancing the nucleophilicity of dialkylzinc (ligands shown in
Figure 18).
The bipyridinyl diol
94 is an efficient enantioselective catalyst for the addition of Et
2Zn to aldehydes (
p-methoxyphenyl,
p-chlorophenyl,
n-hexyl, and benzaldehyde) in the presence of cobalt. Five mol% of the bipyridinyl diol
94 resulted in good c.y.s (65–96%) with high ee (up to 97%) [
5,
80].
The X-ray structure analysis of the Co-complex formed from the ester of the bipyridinyl diol and CoCl2·6H2O confirmed the C2-symmetry of the metal complex; the Co is surrounded tetrahedrally by the two nitrogen atoms of the bipyridinyl diol ligand and the two chlorine atoms. No coordinative bonding of the ether oxygens with the Co is observed.
Chelucci and Soccoloni [
81] synthesized chiral pyridinyl alcohols
57a,
b,
95 and
96 for the catalytic addition of diethylzinc to benzaldehyde, 3-phenylpropanal, and 3-phenylpropynal. They used ligand
96 in order to define whether the enantioselective ability of 2-pyridinyl alcohols could be affected by the presence of another substituent on position 6 of the pyridine ring. The enantioselective addition of ZnEt
2 to the aldehydes was done in the presence of catalytic amount (3 mol%) of
57a,
b,
95 and
96 in hexane/ether at 20 °C and resulted in very high conversion, but moderate to low enantioselectivity (see
Table 5). The lower ee% of
96 in 3-phenylpropynal compared to benzaldehyde and 3-phenylpropanal once again indicated that a predictable improvement of the stereo-differentiating ability of 2-pyridinyl alcohols could be obtained through the introduction of a suitable substituent on the 6-position of the pyridine ring.
Bolm and co-workers [
75,
82] used the pyridinyl alcohols
48 [
70],
79,
84,
85a–
c,
89,
90,
96, and
97 in the enantioselective catalytic addition of diethylzinc to both aromatic and aliphatic aldehydes. All pyridinyl alcohols were used as chiral catalysts and the results obtained are presented in
Table 6 and
Table 7. All products in
Table 6 are the (
R)-enantiomers. For the reduction of benzaldehyde with the pyridinyl alcohols as chiral catalysts the following results were obtained (see
Table 7).
Pyridinyl alcohols 84 and 98 with aryl substituents in 6-position were found to be the most active catalysts. Slow ethylation was observed for pyridines with non-aromatic substituents in 6-position. Catalysts 48 & 89 that bear chelating substituents at 6-position gave good yield with slightly lower ee. The importance of sterically bulky substituent at the chiral centre was shown by the weak enantioselectivity with catalysts derived from 90 and 99 that bear methyl instead of tert-butyl substituent.
A possible mechanistic explanation was given by Bolm et al. [
82] The three possible intermediates that might be present in solution are
100–
102 (
Figure 19). Ethyl transfer might have occurred via a 5/4-bridged intermediate
100 or as a result of six-membered cyclic transition
101 or else through the acetal-like intermediate
102. In all cases, the aldehyde is activated by the formerly coordinated unsaturated zinc alkoxide. The stereochemical outcome of the alkylation is determined by the steric interactions between the aldehyde substituents. The catalyst will be liberated forming stable tetramers of the product zinc alkoxide. The mechanistic approach by Bolm et al. [
82] gave no consideration to the C-6 substituted groups that clearly showed differences in the ee of the product as shown in
Table 7. It is therefore very crucial to further investigate how the C-6-substituted bulky and chelating groups affect the enantioselectivity of the product.
Addition of diethylzinc to benzaldehyde by using bipyridinyl diols (
R,R)-
48 and (
S,S)-
88 as catalysts was also performed by Ishizaki et al. [
83], with the intention to see the effect of the chiral alcohols in 6-position of the bipyridinyl diols. They used 5 mol% of
48 in hexane solvent at room temperature with a reaction time of 16 h and obtained (
R)-1-phenyl-1-propanol (70% c.y. and 68% ee) and the same product with 77% c.y. and 65% ee by using
88 (5 mol%). They also used (
S)-
96 in 10 mol% and four equivalents of ZnEt
2 to obtain (
S)-1-phenyl-1-propanol 70% c.y. and 66% ee. Although this was not the case with Bolm and co-workers’ [
73,
74,
82] results, it is possible to conclude that the chiral alcohol in 6-position of bipyridinyl diol did not play an important role in this reaction.
After synthesising substituted 2-phenyl-5,6,7,8-tetrahydro-6,6-dimethyl-5,7-methanoquinolines
103a,b, Collomb and Von Zelewsky [
84] tested the catalytic activities of the alcohols in the addition reactions of Et
2Zn to benzaldehyde (see
Figure 20).
103a resulted in 75% c.y. and 28% ee, while
103b resulted in 78% c.y. and 91% ee of the tertiary alcohol. The difference in the isomeric alcohols (
R,
R)-
48 and (
S,
S)-
88 did not show a significant change in the ee; however, the change in the pyridinyl alcohol isomers in
103a and
103b has shown an unexpected difference in the enantioselectivity of the product. This leads us to further investigate the effect of the structures of the groups in the chiral alcohol.
Mecedo and Moberg [
15] received similar results to Bolm and co-workers [
73,
74,
82] with respect to the amplification of the enantioselectivity by the chiral alcohol possessing a substituent in 6-position of the pyridine alcohol. They used the pyridinyl alcoholato ligands
104,
105a,
b and
106a,
b (
Figure 20), which they synthesized for this purpose, in the asymmetric addition of diethylzinc to benzaldehyde and
p-chlorobenzaldehyde. The pyridinyl alcohol
104, which does not possess a substituent in 6-position of the alcohol, resulted in low chemical and ee yield. The results are presented in
Table 8.
The influence of the group in the 6-position of the pyridinyl alcohol is seen clearly as all of the 6-substituted pyridinyl alcohols resulted in better c.y.’s and ee’s than those without substituents. Looking once again into the differences only in the isomers of the alcohols 105a versus 106a and 105b versus 106b, it is clearly seen that 105a and 105b resulted in higher c.y.’s and ee’s. Therefore, this needs to be addressed to come up with a complete explanation as to why there existed a significant difference between isomeric alcohols.
With the aim of determining the influence of the catalyst periphery on reactivity and enantioselectivity, the dendrimeric hyperbranched chiral catalysts
107 (90%),
108 (74%), and
109 (84%) (
Figure 21) were synthesized by using Hawker and Frechet’s [
85] convergent approach via the subsequent standard K
2CO
3/18-crown-6 mediated ether formations and alcohol to bromide conversions using CBr
4/PPh
3 [
76].
Compared to the parent alcohol (
S)-
84, the enantiocontrol of the dendrimeric hyperbranched homogeneous chiral catalyst (HCC) was slightly (2–3% ee) lower. The asymmetric amplification phenomena showed that the molecular enlargement (at the level of
107) does not seem to have a significant influence on the aggregation behaviour of the corresponding zinc alkoxide. This result does not agree with Bolm et al. [
73,
74,
82] and Macedo and Moberg’s [
15] result at all. Therefore, the need to further investigate the type, size and electronic character of the pyridinyl alcohols substituted at 6-position is crucial in order to come up with a synonymous conclusion. More emphasis should be placed on the mechanism of the reaction.
Excellent c.y. (99%) with low enantiomeric purity (41%) product was obtained by Genov et al. [
86] upon chiral addition of diethylzinc to benzaldehyde, using the pyridinyl alcohol
57b (in toluene/hexane, 38 h, r.t.). They also used the structurally-related chiral pyridinyl alcoholato ligand
58 that resulted in 0% optical purity and an excellent c.y. (99%) with the same reaction conditions, but in 50 h. According to Genov et al. [
86], the reason for the 0% ee was the insolubility of the zincalkoxide complex. The result obtained using pyridinyl alcohol
57b is similar to that obtained by Chelucci and Soccoloni [
81]. Enantioselective addition of diethylzinc to benzaldehyde in the presence of
110 (see
Figure 22) resulted in 92% c.y. and 47% ee of the (
R)-isomer of benzyl alcohol in 90 h [
87]. The uncomplexed (amino) alcohol resulted in 89% c.y. with only 9% ee of (
R)-alcohol in the same reaction condition. The presence of a tricarbonyl chromium unit, coordinated to the phenyl ring of the aryl pyridinyl alcohol catalyst, induced a significant increase in the enantioselectivity of the catalysed addition of diethylzinc to benzaldehyde. The pyridine diols
48,
111,
112a,b and
113 (
Figure 22) yielded very good chemical yields and showed good enantioselectivities in the nucleophilic addition of diethylzinc to aliphatic and aromatic aldehydes [
88].
Comparable ee’s were obtained for the catalysis in 48 (70% c.y. and 68% ee), 112b ((75% c.y. and 63% ee) and 112a (89% c.y. and 56% ee) during addition to benzaldehyde. The same chemical (87%) and ee (68%) yields were obtained during addition of diethylzinc to n-pentanal and cyclohexanal. All of them yielded the (R)-enantiomer, except 111 and 113, which of course yielded the (S)-isomer plus the least in ee (32% and 38%, respectively). In this specific case, it is not very clear as to why the relatively small groups of CF3 (in 112a) and C2F5 (in 112b) in the chiral position of the pyridinyl alcohols did not decrease the yield and ee of the product, as it is in the pyridinyl alcohols 111 and 113. Comparing the ee obtained by using pyridinyl alcohol 111 with 112a, we can say that electron-withdrawing electronic effect can somehow increase the ee of the product. The electron-withdrawing effect can also be seen comparing the ee’s of the highly electron-withdrawing substituent C2F5 in 112b (63%) with that of the less powerful electron-withdrawing substituent CF3 in 112a (56%).
The chiral oligopyridine derivative ligands
114,
115a,
b and
116 (
Figure 23) were prepared by Kotsuki et al. [
89], with the aim of catalysing the nucleophilic addition of diethylzinc to benzaldehyde. The highest yield was obtained by the chiral catalyst
115a (90% c.y. and 70% ee) in 10 h. Addition of the additive Ti(O
t-Bu)
4 decreased the yield to 84% c.y. and 51% ee. On the other hand, the chiral catalyst
114 has shown increment both in the chemical as well as ee yield upon addition of the additive Ti(O
t-Bu)
4 to the reaction mixture from 64 to 88% c.y. and 2 to 12% ee. The pyridinyl alcohol ligand
115b yielded only 10% c.y. and 3% ee in 48 h. Ligand
116 yielded 100% c.y., but only 6% ee. All of them yielded the (
S)-configuration except
114, which resulted in the (
R)-isomer upon using the additive Ti(O
t-Bu)
4.
A plausible mechanistic explanation of alkylation by Kotsuki et al. [
89] using
115a is similar to Bolm et al.’s [
82] intermediate
101 (see
Scheme 10).
The pyridinyl alcoholate ligands (
S)-
117 and (
R)-
118 (
Figure 23) were applied to the enantioselective addition of diethylzinc to aromatic aldehydes resulting in good yields (82% and 80%, respectively) and moderate enantioselectivities (90% and 87%, respectively) in the case of benzaldehyde [
90]. The chemical yield and enantioselectivity decreased for
p-chlorobenzaldehyde (62%, 49% and 54%, 48%, respectively),
p-methoxybenzaldehyde (51%, 71% and 47%, 50%, respectively) and (
E)-
β-phenylpropenal (76%, 73% and 9%, 2%, respectively). The ee’s obtained by using (
S)-
117 and (
R)-
118 are very close to the ee obtained by the structural analog Collomb and Von Zelewsky’s [
84] pyridinyl alcohol
103b.
Enantioselective addition of diethylzinc to benzaldehyde catalysed by
C2-symmetric chiral bipyridinyl diol ligands
119–
122 (
Figure 24) resulted in 30 to 86% c.y. with 23 to 95% ee [
91]. The enantioselectivity depended on the type of solvent used. For example, in CH
3CN, the alcoholate ligand
120 resulted in the least c.y. (30%) and ee (23%), whereas in toluene a very good c.y. (86%) and moderate ee (95%) were obtained. The absolute configuration of the product in all ligands except in
119 was ‘
R’. Because the alcoholate ligand
120 resulted in a better yield, they used it to catalyse the diethylzinc addition to other aromatic and aliphatic aldehydes, which resulted in good c.y. (52–74%) and low to moderate ee’s (20–80%). The better yield was observed for cyclohexanal (66% c.y. and 80% ee).
With the intention to see whether
C2-symmetry of the bipyridinyl diol ligand is helpful in achieving high enantioselectivity in the alkylation of aldehydes, Kwong and Lee [
91] synthesized the 6-phenyl and 6-(2-pyridinyl) substituted pyridinyl alcohols
79 and
80. All the newly-synthesized ligands were active in catalysing the ethylation of benzaldehyde by diethylzinc. Using the alcoholate ligands
79 and
80, the ee’s obtained were 76% (
S) and 78% (
R), respectively, with complete conversion of benzaldehyde in 1.5 h (for both ligands). Due to the high concentration of diethylzinc in the reaction mixture, the remaining
N,O-part of the ligand coordinated to other zinc atoms during reaction and the two
N,O units probably function independently. On the other hand, further ethylation with the 6-(2-pyridinyl) substituted
123 and
124 (see
Figure 25) gave 21% (
R) and 65% (
R) with complete conversion of benzaldehyde in one and three hours, respectively. Similar results were obtained previously with other pyridinyl alcohols and bipyridinyl diols [
92]. Comparison of the activity and enantioselectivity of ligands
79,
119,
123 and
80,
120,
124 indicated that bipyridinyls
119–
122 function as
N,O-ligands in diethylzinc addition reactions.
Industrial processes with polymer-supported catalysts are common due to the durability and ease of separation of catalysts. One strategy to prepare these catalysts is the attachment of a previously identified catalytically-active compound to a preformed polymeric support. An alternative approach for the generation of polymeric multivalent chiral catalysts
125 and
126 (
Figure 25), which is based on ROMP of strained bicyclic olefins, was reported by Bolm et al. [
93] In both
125 and
126, a chiral hydroxylpyridinyl fragment was the catalytically active subunit.
Enantioselective addition of diethylzinc to benzaldehyde in the presence of
125 and
126 as catalysts resulted in 77 to 88% c.y. with 71 to 73% ee benzyl alcohol. A slight decrease in catalytic activity was observed compared to low molecular weight pyridinyl alcohols (starting material and intermediate) in a similar way to Bolm et al.’s [
79] dendritic hyper branched homogeneous chiral catalysts
107–
109.
The pyridinyl alcohols (
127a,
b and
128a,
b) (
Figure 26) of Cunningham et al. [
94] were tested for their catalytic activity in the nucleophilic addition of diethylzinc to benzaldehyde and resulted in very good chemical yields (80–96%) and poor enantioselectivities (8.5–22.5%). All of them resulted in the (
R)-configuration of the benzyl alcohol. The highest ee (22.5%) was observed during the catalysis of
128a and the least (8.5%) in
128b. Once again, orientation of the bulky group in the pyridinyl alcohol was observed affecting the ee of the product.
The
R-pyridinyl alcohol resulted in high ee% of the product in a similar way to that of Macedo and Moberg’s [
15] pyridinyl alcohols
105a,
b (the
R-isomers) compared to
106a,
b (the
S-isomers) and to Collomb and Von Zelewsky’s [
84] isomeric alcohols
103b (
R-isomer) and
103a (
S-isomer).
The catalytic activities of the tridentate pyridinyl alcohols
88,
119 and
129 were compared to the bidentate pyridinyl alcohols
57a and
130–
132 (
Figure 26) by Goanvic et al. [
95] in the enantioselective alkylation of benzaldehyde with diethylzinc to see the effect on the second arm of pyridine diols. The yields of the benzyl alcohol follows
88 (77% c.y. and 65% ee),
119 (81% c.y. and 55% ee) and
129 (84% c.y. and 4% ee) for the tridentate alcohols and
57a (93% c.y. and 21% ee),
130 (83% c.y. and 47% ee),
131 (79% c.y. and 67% ee (
R)) and
132 (81% c.y. and 35% ee) for the bidentate alcohols. All of them, except
131, resulted in the (
S)-enantiomer of the alcohols. The results show that only one half of the
C2-symmetric ligand is part of the actual complex. The second arm seems to only play a screening role helping in the differentiation of the two enantiotopic faces of the aldehyde within the catalytic complex. Therefore, it is worthwhile to conclude that the beneficial role of a tridentate catalyst compared to bidentate ligand is not as important as expected.
The bipyridinyl diols
133a,
b and the pyridinyl alcohol
134 were tested for their catalytic activities in the enantioselective diethylzinc addition to benzaldehyde by Rahm et al. [
96,
97], following their synthesis (
Figure 27). The bipyridinyl diols
133a,
b resulted in 47% c.y. with 96% ee and 74% c.y. with 83% ee, respectively, of the (
S)-isomer, whereas
134 yielded the (
R)-isomer in 94% c.y. with 28% ee. Very similar concluding results (to that of Rahm et al. [
96,
97]) were obtained by Huang et al. [
98] upon using the bidentate pyridinyl alcohols
135a–
c and the tridentate pyridine diols
136a,
b in the enantioselective addition of diethylzinc to benzaldehyde in toluene at 0 °C (
Figure 27). The enantioselectivities of the bidentate pyridinyl alcohols go up to 25%, while that of the tridentate alcohols is only 15%. On the other hand, they attempted to see the structural influence of the pyridinyl alcohol on the catalytic addition reaction upon synthesising pyridinyl alcohols having one methylene group longer backbone chain between the coordinating
N and
O atoms. The results in the catalytic addition of diethylzinc to benzaldehyde revealed that the pyridinyl alcohols with longer backbone chain resulted in much higher ee (75–83%) compared to the corresponding pyridinyl alcohols
135a–
c (7–25% ee). The possible explanation for the differences in the catalytic activities and enantioselectivities could be made by considering the conformational analysis of the ethylzinc (amino)alkoxide complexes of the pyridinyl alcohols. Ligands with one more CH
2 group longer backbone possibly form the six-membered ring ethylzinc (amino)alkoxide
137, while those with short backbone chain form the complex
138 in situ. It is easier for the benzaldehyde to be coordinated to the
Re-face of
137 than that of the
Re-face of
138 (see
Scheme 11). Moreover, the difference between the two enantiofaces in
138 is less than that of
137; therefore, both the activity and enantioselectivity of catalysts
138 should be lower.
Pyridinyl alcohol
139-catalysed asymmetric alkylation of substituted benzaldehyde using diethylzinc resulted in good chemical yields (31–85%) and moderate enantioselectivities (56–78%) [
99] (
Figure 28). The
ortho- and
para-substituted electron-releasing substituents yielded high enantioselectivities (see
Table 9).
This may happen due to the increased π-π interaction between the pyridinyl group of ligand
139 and the aromatic ring of the benzaldehyde. This, in turn, will fix tightly the aromatic ring of benzaldehyde such that the
si-face of the carbonyl group is preferentially alkylated increasing the enantioselectivity. The reaction was done by using catalytic amount (5 mol%) of the ligand
139 in hexane at room temperature for 5 h. The reaction condition is optimised. Ligand
139, structurally closely related to the Collomb and Von Zelewsky’s [
84] pyridinyl alcohol
103a, resulted in quite different ee’s of the benzyl alcohols.
The presence of the methyl group in
139 might be the reason for the high ee. However, a further study of the same substituents on the benzaldehyde to that used in
139 and performing the diethylzinc addition reaction using
103a would clarify the effect of both the pyridine and the methyl substituents in
103a and
139, respectively. The pyridine diol
140 was used in the enantioselective addition of diethylzinc to benzaldehyde, but resulted in only 8% ee of the 1-phenyl-1-propanol [
100].
Steric energy calculations using the MM2 program indicated that intermediate
141 has lower steric energy (49.322 kcal/mol) than intermediate
142 (54.283 kcal/mol) (
Figure 28). This therefore tells us that the reaction centre might have shifted far way to the second alcohol that cannot induce high enantioselectivity, which is in agreement with Huang et al.’s [
98] proposal.
Capracotta and Comins [
101] tested the catalytic activities of the pyridinyl alcohol
143 in the diethylzinc addition to benzaldehyde; however, the yield obtained was very small (2% ee). They
[ used 20 mol% of catalyst (
55) in toluene at 0 °C for 24 h.
The c.y. was not good either (42%). This might be due to the bulky cycloamino substituent at the 3-position of the pyridine ring that possibly hinders the approach of the carbonyl group towards the pyridinyl alcohol-Zn-alkyl complex via the si-face easily.
Eidamshaus and Reissing [
102,
103] synthesized pyridinyl alcohols
144a–
d and
145a–
d (
Figure 29) via reductive amination of the corresponding pyridinyl aldehyde. The pyridinyl alcohols
144a–
d were used (12 mol%) in the enantioselective addition of diethylzinc to benzaldehyde and gave very good yields (68–93%) with moderate enantioselectivity (69–74%) of the (
R)-configuration product. The electronic properties of the pyridine ligands did not show a significant influence on the chiral induction. The aminomethyl-substituted pyridine derivatives
145a–
d resulted in a relatively high ee (58–88%) and low c.y.’s (48–85%). The highest chemical (87%) and ee (88%) were obtained by ligand
145a and the least ee (58%) by ligand
145d. The reactivity differences of
145a,
145b and
145c cannot be explained by the formerly presented models. In order to explain this fact, Eidamshaus and Reissing [
102,
103] proposed the new transition states
146 and
147. They followed the 5/6-membered ring postulate formed by the pyridine ring chelating an alkyl zinc moiety that acts as Lewis acid and coordinates the reacting aldehyde.
This coordination explains the preferential
re-face attack; however, it was not possible to explain the influence of the C-6 aminomethyl side chain of the pyridinyl alcohol ligands
145a–
d (see
Scheme 12). It was suggested that a third molecule of diethylzinc is involved being coordinated between the amino group and the carbonyl oxygen so that the aminomethyl side chain should be close to the carbonyl centre and leads to the preferred transition state
146. In the transition state
146, the bulky phenyl group of the aminomethyl group points away, whereas the methyl group enhances the asymmetric induction as it points in the same direction as the C-2 phenyl group. In transition state
147, that is the transition state when ligand
145b is used, the phenyl group of the aminomethyl side chain probably points up generating a sterically more crowded transition state. A decreased activation of the carbonyl group and lower enantioselectivity is due to the large distance to the aminomethyl side chain.
2.8. Ring-Opening Reactions of Meso-Epoxides
Lewis acid catalysed reactions are of great interest because of their unique reactivities, selectivities and mild reaction conditions. A large number of Lewis acid catalysts that are used in the synthesis of natural and synthetic compounds have been dealt with in the past decades. The traditionally used Lewis acids, such as AlCl
3, SnCl
4, TiCl
4, and BF
3, were needed in more than the stoichiometric amounts, are moisture sensitive, and cannot be recovered and reused after the reactions are completed [
107]. The catalytic enantioselective ring-opening of
meso-epoxides is an efficient means to convert readily available, inexpensive bulk chemicals into chiral, 1,2-difunctionalised fine chemicals in enantiomerically enriched form [
108]. In the epoxide ring-opening reactions, the catalyst behaves as a Lewis acid, directing the incoming nucleophile into the enantiotopic faces of the epoxide in an S
N2 fashion. To date, the epoxide ring-opening reactions that led to 1,2-azido alcohols, 1,2-halohydrines, 1,2-cyano alcohols, 1,2-diol monoesters and monoethers and 1,2-mercapto alcohols resulted in good to excellent enantioselectivities.
Chiral
β-(amino)alcohols are part of many biologically active compounds and chiral auxiliaries used in the enantioselective reactions. Catalytic enantioselective synthesis of these chiral compounds relies on the asymmetric ring opening of
meso-epoxides [
109]. The major problem in the nucleophilic addition of amines to epoxides catalysed by Lewis acids is the compatibility problems between the Lewis basic amine and the Lewis acid catalyst, which tend to coordinate to one another irreversibly. Crotti et al. [
107,
110,
111] have brought a solution to this problem, in that lanthanide triflates, such as Yb(OTf)
3, are effective catalysts for the aminolysis of 1,2-epoxides, because they are able to exert their exceptional Lewis acidity even in the presence of the amine [
112]. A generalised ring-opening reaction of a
meso-epoxide using alcohol as a nucleophile is shown in
Scheme 16. The nucleophile could also be an amine, a thiol or an indole.
In the catalysis for epoxidation of (1
R)-(+)-
α-pinene using the molybdenum (VI) catalyst
48 with
t-BuOOH, Valente et al. [
55] came across the ring-opening activity of
α-pinene oxide (which is the product by itself) in campholenic aldehyde (8.6%) and epoxy campholenic aldehyde (50.2%). Similarly, using (1
S)-(−)-
α-pinene, the catalyst yielded 9.8% campholenic aldehyde and 58.1% epoxy campholenic aldehyde. The inadvertently obtained reaction products led them to test the activity of catalyst
64 on the ring opening reaction of
α-pinene oxide, which resulted in 20% campholenic aldehyde and 39% epoxy campholenic aldehyde with 81% conversion of the
α-pinene oxide.
Schneider et al. [
108] established the first catalytic enantioselective addition of aliphatic alcohols to
meso-epoxides, employing a novel scandium-bipyridinyl-complex with excellent ee and good c.y.’s. The α,α’-bipyridinyl diol
148 was used together with Sc(OTf)
3 in 10 mol% in the suitable solvent (for their reaction) CH
2Cl
2 at room temperature. They used the
meso-epoxides:
cis-stilbene,
cis-1,2-naphthylethylene,
cis-3,3′-dimethylstilbene, cyclohexene and
cis-2-butene in both alcoholysis and aminolysis reactions. For the alcoholysis reaction; methanol, ethanol,
n-butanol, 3-propenol and
p-methoxybenzyl alcohol and for the aminolysis; aniline,
N-methyl aniline,
p-anisidine and
n-butoxyamine were used. The alcoholysis yielded 75–93% c.y. with 49 to 98% ee, while the aminolysis resulted in 76 to 96% c.y. with 54 to 97% ee. Generally, the ee’s of aromatic epoxides were higher than the aliphatic epoxides in both alcoholysis and aminolysis reactions. Decreasing the catalyst load to 5 mol% decreased the ee from 93 to 90% and the c.y. from 95 to 89% for
cis-stilbene in the aminolysis of the epoxide.
N-methyl aniline showed the highest ee (97%). Conversion of the bipyridinyl diol to its bis-
O-methyl ether led to a complete loss of enantioselectivity both in the alcoholysis and aminolysis of the
meso-epoxides. It is therefore essential to have free hydroxyl groups for the formation of a highly enantioselective scandium-bipyridinyl-complex.
With the focus on important intermediates and building blocks in organic synthesis [
113], Azoulay et al. [
109] used the bipyridinyl diol ligand
148 (
Figure 30) in the catalytic asymmetric ring opening of
meso-epoxides in Sc(DS)
3 using the environmentally friendly solvent water for the first time. The
meso-epoxides of
cis-stilbene,
cis-4,4′-dimethylstilbene,
cis-1,6-diphenyl-3-hexene,
cis-5-decene, and
cis-1,2-dinaphthylethylene were used for the ring-opening aminolysis with aniline. Good c.y.’s (61–89%) with moderate to good ee’s (60–96%) with the highest (89%) c.y. being for
cis-stilbene and
cis-5-decene (in aniline) and the highest ee (96%) for
cis-stilbene in
N-methylaniline were obtained. Aminolysis of
cis-stilbene was also performed with
O-anisidine,
α-naphthylamine and 1-amino-4-bromohaphthalene. The catalyst Sc(DS)
3 was used in 1 mol% and the ligand in 1.2 mol% during the aminolysis reaction. They also demonstrated that using 1 mol% catalyst and 1.2 mol% ligand
148 in the aminolysis of
cis-stilbene, Sc(DS)
3 yielded better (89% c.y. with 91% ee) in water than Sc(OTf) in CH
2Cl
2, which resulted in 85% c.y. and 74% ee of the
β-(amino)alcohol.
Optimisation of the aminolysis of
meso-epoxides with respect to metal, ligand, solvent, catalyst loading, epoxide and amine component was performed by Mai and Schneider [
112]. For the optimisation of the metal, the triflates [M(OTf)
3] of the lanthanide series metals Sc, Y, Ce, Sm and Yb were used in 10 mol% with 12 mol% of the bipyridinyl diol
148 in CH
2Cl
2 at room temperature for
cis-stilbene and cyclohexene oxides. The best results were obtained for Sc(OTf)
3 in both epoxides. For ligand optimisation, the bipyridinyl diol
148 was modified having the intention to increase torsion angles between the pyridine rings by substituting methyl groups in position 2 of the pyridine ring (
148), increasing the steric bulk of the
t-Bu group at the chiral centres (
149 and
150) and also a tripyridine backbone (
151) (
Figure 30). All of them were tested for their catalytic activity with Sc(OTf)
3 10 mol%, 12 mol% of each of the pyridinyl alcohols in CH
2Cl
2 at room temperature in the aminolysis of
cis-stilbene and cyclohexene oxides. However, none of them yielded as excellent results as ligand
148. The solvent optimisation involved CH
2Cl
2, ClCH
2CH
2Cl and CHCl
3, where CH
2Cl
2 was found to be the best solvent in terms of yield. Dimethylformamide (DMF), CH
3CN and THF coordinate to the Lewis acid and shut down the catalytic activity, while toluene and diethyl ether coordinate weakly to give high product yields with no enantioselectivity [
114]
The optimisation of catalyst loading for the reaction with cis-stilbene oxide was done at catalyst loadings of 10, 5, 2 and 1 mol% and the product ee’s were 93, 90, 80 and 71% respectively. The chemical yield did not show a significant change. Therefore, the better catalyst load is 10 mol%. Several epoxides from both aromatic and aliphatic functionalities were tested in the optimised reaction condition and better results were obtained for aromatic meso-epoxides. Sterically hindered anilines such as N-methyl aniline and anilines with electron-withdrawing substituents yielded better ee’s compared to the electron-donating and non-sterically hindered anilines in cis-stilbene.
A combined Lewis acid-Brønsted acid acidity of the indium(III)-bipyridinyl diol complex was proposed by Nandakumar et al. [
115] The indium-bipyridinyl-catalysed enantioselective ring-opening of
meso-epoxides of
cis-stilbene,
cis-1,2-naphthylethylene,
cis-3,3′-dimethylstilbene,
cis-4,4′-dimethylstilbene and
cis-4,4′-dichloro stilbene in benzylthiol, phenylthiol, 4-methyl phenylthiol, ethylthiol,
n-butylthiol and 1-methyl butylthiol resulted in 1,2-mercapto alcohols in good yields (67–91%) and moderate to good enantioselectivities (85–96%). The optimised reaction condition utilised 10 mol% of InBr
3, 11 mol % of the bipyridinyl diol
148 in CH
2Cl
2 at room temperature. The X-ray crystallography revealed that, in the [InBr
2-
148·H
2O]
+ complex, the hydroxyl hydrogens remain intact and the bipyridinyl is four coordinated to the indium metal centre. The hydroxy protons in the pyridine ring make a hydrogen bond to the thiol, leading the metal centre to the thiol for activation (see
152).
Scandium-bipyridinyl-catalysed enantioselective alcoholysis of
meso-epoxides was optimised with respect to metal, ligand, solvent, catalyst loading, alcohol, and epoxide components [
114].
cis-stilbene and cyclohexene oxides with the bipyridinyl diol
148 are used in the optimisation process. The triflates of Sc, Y, La, Ce, Nd, Sm, Tb, Tm, Yb, Cu and Zn were tested for optimisation of the metal in the alcoholysis of
cis-stilbene in
p-methoxybenzyl alcohol (PMBOH) and cyclohexene in allyl alcohol and best yields were obtained for Sc(OTf)
3 (82% c.y. and 97% ee) as in the case of the aminolysis. The bipyridinyl diol ligands
148–
151 were tested for
cis-stilbene oxide ring-opening with allyl alcohol; however, no significant variation in both selectivity and chemical yield was observed, except for
151, which did not give rise to an enantioselective catalyst. 1,2-Dichloroethane was found to be the second choice of solvent after CH
2Cl
2. Optimisation of catalyst loading was performed in 15, 10, 5, 2 and 1 mol% of Sc(OTf)
3, which yielded 98%, 97%, 94%, 90% and 73% within 0.5, 0.5, 5, 8, and 9 days, respectively. Consequently, the appropriate catalyst load is 10 mol%; however, it can be reduced to 5 mol% as there is no significant variation in the yield except prolonged reaction time. Various solvents, including methanol, ethanol, PMBOH, allyl alcohol,
n-butanol,
iso-propanol and 3-propargyl alcohol were used in the optimisation with
cis-stilbene and PMBOH yielded better (82% c.y. and 97% ee) 1,2-diolmonoether. The aromatic epoxides undergo alcoholysis with PMBOH in a better yield than aliphatic epoxides. The
α-branched allylic epoxide resulted in the highest ee in the aliphatic epoxide category.
The X-ray crystallography of Y(OTf)
3-
148 complex shows the bipyridinyl being slightly twisted along its own axis (19.1°) and coordinated in a tetracoordinate fashion with two triflate groups and two water molecules bound to the metal centre. The ESI-MS spectra of the Sc(OTf)
3-
148 complex in CH
2Cl
2/CH
3CN solution confirmed the presence of [M-H]
+ species, which could be taken as proof for the presence of the hydroxy group in the complex. Using a catalytic amount (1 mol%) of Sc(DS)
3 and the chiral bipyridinyl diol ligand (
S,
S)-
148 (1.2 mol%) resulted in ring-opening desymmetrisation of
meso-epoxides with aromatic amines and indole derivatives in water as sole solvent [
116]. The reaction proceeded much faster in water than in dichloromethane. Aromatic epoxides
cis-stilbene,
cis-4,4′-dimethylstilbene,
cis-1,2-dinaphthylethylene oxides resulted in better c.y. (75–89%) and ee (86–96%) compared to the aliphatic epoxides
cis-1,6-diphenyl-3-hexene, and
cis-5-decene oxides (61–89% c.y. and 60–71% ee). The aromatic amines used in the aminolysis include aniline,
o-anisidine,
α-naphthylamine and 1-amino-4-bromo naphthalene.
It was also reported that Sc(DS)3-(S,S)-148 in water resulted in better c.y., ee’s and reactivity than Sc(OTf)3-(S,S)-148 in dichloromethane. Ring-opening desymmetrisation of meso-epoxides cis-stilbene, cis-4,4′-dimethylstilbene, cis-4,4′-dibromostilbene oxides with indole, 5-methoxyindole, 5-methylindole, 2-methylindole and 5-bromo indole in the presence of 5 mol% Sc(DS)3 and 6 mol% ligand (S,S)-148 resulted in good yields (56–62%) and moderate to good ee’s (86–93%). Similarly, the reaction is much faster in water than in dichloromethane.
Aminolysis of aromatic and aliphatic
meso-epoxides was also efficiently catalysed by 10 mol% In(OTf)
3-
148 catalyst in dichloromethane at room temperature to result in good yields and moderate to high enantiomeric excess (up to 98%) of 1,2-(amino)alcohols [
117]. Electron-deficient and sterically hindered anilines gave rise to high ee’s (up to 98%), thereby maintaining the c.y. at a good level. The aliphatic epoxides were ring-opened with low enantioselectivities (31–36%) and high chemical yields (82–91%) just as in the scandium-bipyridinyl catalysed process [
108,
112,
114]. Analysis of the crystal structure of InBr
3-bipyridinyl catalyst (as it was not possible to get crystals of In(OTf)
3-
148 complex) revealed a pentagonal bipyramidal coordination geometry around the metal centre with the hydroxyl protons intact to the complex that closely resembles the related Sc-bipyridinyl complex. This is believed to be the reason for both complexes resulting in the same absolute configuration products.
In the previous sections, we have presented the Sc(OTf)
3 and In(OTf)
3-catalysed process for the addition of alcohols, amines and thiols to
meso-epoxides in the presence of the bipyridinyl diol
148 and its (
S,
S)-isomer, which resulted in 1,2-diol monoethers, 1,2-(amino)alcohols and 1,2-(mercapto)alcohols. Tschöp et al. [
118] further extended this protocol to the addition of phenyl selenol to aromatic
meso-epoxides giving rise to 1,2-seleno alcohols in good yield and up to 94% ee, which complements the Yang et al.’s [
119] process. Stilbene oxide reacted with phenyl selenol in the presence of 10 mol% Sc(OTf)
3 and 10 mol% of ligand
148 in CH
2Cl
2 at room temperature for 12 h to yield 60% 1,2-selenoalcohol with 93% ee and 20% deselenated alcohol. As the deselenated side product was believed to be produced via a radical process removing oxygen from the reaction mixture and undergoing the reaction in the dark increased the 1,2-selenoalcohol yield to 77% after eight hours with no deselenated product. Reactions with and without light and oxygen were done with
meso-epoxides
cis-4,4′-fluorostilbene,
cis-4,4′-chlorostilbene,
cis-4,4′-bromostilbene and
cis-3,3′-meth ylstilbene. The results obtained showed clearly that the deselenated product increased in the presence of oxygen and light in all cases. Based on this result, a deselenation reaction of
cis-4,4′-difluorostilbene and PhSeH via direct
meso-epoxide ring-opening in the presence of Sc(OTf)
3 and bipyridinyl diol
148 in CH
2Cl
2 at room temperature in 24 h resulted in 83% of the deselenated product. The same reaction resulted in 25% yield without catalyst and 81% only in the presence of the bipyridinyl diol ligand
148. Therefore, it is more likely that the deselenation reaction would be assisted by the bipyridinyl diol ligand in the abstraction of the selenol proton that is assumed to be the first step in the radical deselenation process. Cyclohexene oxide, however, did not show a deselenated product even in the presence of both oxygen and light.
The square pyramidal Cu(DS)
2-(
S,
S)-
148 complex and the pentagonal bipyramidal structure Sc(DS)
3-(
S,
S)-
148 complex resulted in opposite yet nearly similar enantioselectivities in asymmetric ring-opening reactions of
meso-epoxides;
cis-stilbene oxide,
cis-4,4′-dimethyl stilbene oxide,
cis-4,4′-dibromostilbene oxide,
cis-1,2-dinaphthylethylene oxide and with indoles 5-methoxyindole, 5-methylindole, 5-bromoindole, 2-methylindole and indole [
120]. 10 mol% of catalyst and 12 mol% of ligand were used in water as sole solvent at room temperature for 22 h to result in 48 to 82% c.y. and 85 to 96% ee of the (
S,
S)-product by Cu(DS)
2 and 46 to 80% c.y. and 84 to 95% ee of the (
R,
R)-product by Sc(DS)
3. Using organic solvents such as CH
2Cl
2, THF, toluene etc. and catalyst Cu(OTf)
2 or Sc(OTf)
3-(
S,
S)-
148 resulted in much lower yields of the product. This is therefore one way of obtaining the two enantiomers of the alcohol by using the appropriate metal with the bipyridinyl diol ligand (
S,
S)-
148 complex.
The ring opening reactions of
meso-epoxides with aniline and indole derivatives in the presence of Zn(II) and Cu(II) surfactant-type catalysts resulted in the corresponding products in moderate to high ee’s and good to excellent c.y.’s were reported by Kokubo et al. [
121] It is well known that most catalytic organic reactions take place in organic solvents as a result of the insolubility of most organic compounds in water. However, surfactant-type catalysts were becoming solutions to such problems. Alkyl epoxides and stilbene derivative oxides were made to react with aniline derivatives in the presence of 5 mol% of Cu(DS)
2 and/or Zn(DS)
2 catalysts and 6 mol% bipyridinyl diol ligand (
S,
S)-
148 in water at room temperature to yield product in moderate to good (44–95%) selectivity and good to excellent chemical yield (43–100%), respectively. The same reaction was made in CH
2Cl
2 using Zn(DS)
2 that resulted in 70 to 90% ee with 10 to 92% c.y. Cu(DS)
2 was not as effective in CH
2Cl
2. The Zn(DS)
2 and Cu(DS)
2 with (
S,
S)-
148 in water were highly reactive and with higher enantioselectivity yields than the Zn(OTf)
2 and Cu(OTf)
2 with (
S,
S)-
148 in CH
2Cl
2.
The indole derivatives resulted in good to excellent ee’s in water using Sc(SD)
3 and Cu(SD)
2 catalysts and (
S,
S)-
148. In CH
2Cl
2, only Cu(OTf)
2 resulted in product in moderate yield and high selectivity. The rate in CH
2Cl
2 and water was nearly the same; however, the selectivity was higher in water than in CH
2Cl
2. In all cases, Sc(III) resulted in the opposite enantiomer than Cu(II) or Zn(II) catalysts. The X-ray structure of [CuBr
2·(
S,
S)-
148] shows that the bipyridine coordinated with only one of the hydroxyl groups and the two nitrogen atoms with the copper metal. The second hydroxyl group therefore is most likely used to coordinate to the substrate as it is shown in complex
153 (
Figure 31).
Careful examination of the aminolysis or alcoholysis of meso-epoxides by the transition metal complexes supported by the bipyridinyl diol 148, its isomer (S,S)-148, and the modified versions of 148 reveals the use of limited or a few bipyridinyl diols only. Even though the yield obtained by the Mo(VI) complex 64 is low, it indicated the fact that pyridinyl alcohols other than the bipyridinyl diol 148 or structurally-related diols could be used in the catalysis of the reaction. In addition to this, complex 153 revealed that the second alcohol function on the bipyridinyl diol does not coordinate to the metal, and therefore there seems to be a high possibility to use pyridinyl mono alcohols with varied structures having different steric and electronic effects to get the best ee and c.y.’s in this homogeneously catalysed reaction.
2.9. Aldol Reaction
Carbon-carbon forming reactions are very useful in organic synthesis. An asymmetric aldol reaction is one of the most useful carbon-carbon bond-forming reactions to afford optically active β-hydroxy carbonyl compounds. Recently, several asymmetric aldol reactions have been developed to pursue efficiency in obtaining these chiral compounds [
122]. In this section, we shall discuss the asymmetric aldol reactions that involve the pyridinyl alcohols or their derivatives.
The major problem with catalytic enantioselective aldol reactions of silicon enolates in aqueous media is the competitive ligand exchange between a chiral ligand and water, which promotes achiral free Lewis acid-catalysed pathways [
122]. However, the water-tolerance nature of the lanthanide trifluoromethanesulfonates (Ln(OTf)
3) made them useful Lewis acid catalysts in aqueous media [
123]. In addition, the rare earth triflates are strong Lewis acids because of the hard character of their metal cations and the strong electron-withdrawing effect of the trifluoromethanesulfonyl group [
124]. The advantage of these reactions compared to the conventional Lewis acid-mediated reactions in dry organic solvents is the ease to remove water from the solvents, substrates and catalysts [
125]. As water is used as a ‘solvent’ in the metabolic process of living organisms, studies on organic reactions in aqueous media may lead to a real understanding of life and nature; particularly, chiral catalysts, which work well in aqueous media may become models of enzymes [
124].
The first demonstration of this kind of reaction was made by Kobayashi in Yb(OTf)
3-catalysed aldol reactions of silyl enol ethers with formaldehyde in aqueous THF [
126]. A generalised aldol reaction is shown in
Scheme 17.
The use of pyridino-crown-ether
154 (
Figure 32), synthesized from 2,6-pyridine dimethanol, in the catalytic asymmetric aldol reactions catalysed by lanthanide triflates in aqueous media was reported by Kobayashi et al. [
125] An adduct of aldol with 85% yield in 82% (
syn/
anti) and 78% ee was obtained by reacting benzaldehyde with silylenol ether by using the catalyst 10 mol% Pr(OTf)
3 and 12 mol%
154 in H
2O/EtOH (1/9, v/v) at 0 °C in 18 h.
The same reaction in pure EtOH resulted in 51% yield with 70% de and 23% ee. In DCM (dichloromethane), the yield decreased by as much as 3% with 28% de and 22% ee. Using the reaction condition in
Scheme 17, the triflates of the lanthanide series metals Sc, Yb, Ho, Y, Dy, Tb, Gd, Eu, Sm, Nd, Pr, Ce and La were used for catalysis. The yield obtained is correlated to the diameter of the cation. The large cation diameter metals such as La, Ce, Pr and Nd resulted in high enantio- and diastereo-selectivity, while the small diameter cations Sc and Yb did not show enantioselectivity at all. Maximum enantio- and diastereo-selectivity was obtained for Ce (86% de and 82% ee). The reason for the difference in the selectivity is the sufficient coordination ability of those cations with large diameters with the crown ether in the proposed mol% ratio of metal to crown ether.
Manabe et al. [
122] achieved chiral Lewis acid catalysed enantioselective hydroxymethylation of silicon enolates for the first time. The reaction of 10 equivalents of formaldehyde and the silicon enolate (see
Scheme 17) in the presence of 20 mol % Pr(OTf)
3 catalyst and 24 mol% of the crown ether ligand
154 in H
2O/THF (THF as co-solvent) at 30 °C in 18 h yielded 91% c.y. with 48% ee of the hydroxymethyl. Using the same reaction condition, a series of
mono-,
di- and
tri-substituted aromatic and aliphatic silicon enolates were tested and moderate to very good yields (40–92%) with poor to moderate ee’s (14–54%) were obtained. Using ethanol as co-solvent showed the best enantioselectivity, although a slight decrease in the chemical yield is observed.
Detailed study and further improvement of the asymmetric aldol reactions in aqueous media are described by Hamada et al. [
124]. They further investigated the effects of the metal cation diameter, the better solvent for the reaction, effect of electron-withdrawing and electron-donating substituents on the
para-position of the pyridine ring in the crown ether and structure of complex. In addition to this, the design of the crown ether and a model transition state for the complex Pr
3+-crown ether were proposed. They began their study with the effect of metals on the catalysis of silyl enol ethers and came across with similar results as before, i.e., large diameter cations La, Ce, Pr, and Nd resulted in the aldol adduct with high diastereo- and enantioselectivities, while the small diameter cation metals Dy, Y, Ho, Yb and Sc gave lower selectivities. Their investigation revealed that the large diameter cations strongly bind to the crown ether, while the small diameter cations bind weakly.
The effect of the electron-donating (MeO) and electron-withdrawing substituents (Br) like in the para-position of the crown ether was revealed by decreasing the reactivity without affecting the selectivity of the catalyst significantly. The same level of diastereo- and enantioselectivities for the large diameter cations La, Ce and Pr was observed in the electron-withdrawing substituted crown ethers. This is because the electron-withdrawing bromine will decrease the binding ability of the crown ether and a couple of cations will be available in the reaction solution. This therefore leads to an achiral catalysis and therefore low selectivity. In the case of electron-donating substituent, the high electron density on the nitrogen of the crown ether will increase the binding ability of the metal cations and therefore a decrease in the Lewis acidity of the crown ether-metal complex [
124]. Regardless of the ratios between 1/3–1/27, H
2O/EtOH resulted in good selectivities, even though the increase in the amount of water decreased the selectivity slightly. In relation to this, they examined the effect of water and came up with the most probable suggestion that water aids in creating an effective chiral environment for the Pr
3+-
154 complex. They also investigated whether the structure of the crown ether-rare earth complexes affects the selectivity; however, they found that almost all complexes have similar structure and the structure of the complex does not affect the selectivity. They studied the structure by considering the crystal of the complex [Pr(NO
3)
2-
154]
3 [Pr(NO
3)
6] as it was not possible to get the crystal of Pr(OTf)
3. The selectivity differences depended on the binding ability and catalytic activity of complexes. Therefore, in order to attain high selectivity in the asymmetric aldol reaction, the design of a chiral crown ether, which complexes strongly with RE(OTf)
3 and does not decrease the Lewis acidity of RE(OTf)
3, is essential (RE = rare earth). The model transition state for the aldol reaction is shown in
Scheme 18 [
124].
A series of aldol reactions of aromatic and aliphatic
α,
β-unsaturated aldehydes with silyl enol ethers were performed by Hamada et al. [
124], which resulted in good to high yields and diastereo- and enantioselectivities. In the aldol reactions using the silyl enol ether derived from a thioester, the addition of 2,6-di-
tert-butylpyridine effectively enhanced the yields of aldol adducts in aqueous media.
The use of chiral pyridinyl
N-oxide ethers
155–
157 (
Figure 33) as catalysts in the enantioselective aldol additions to ketones was reported by Denmark and Fan [
127]. The very reactive nature of trichlorosilyl enolate of methyl acetate with aldehydes resulted in the less enantioselective product.
The strong chelation of the trichlorosilyl fragment renders the enol addition essentially irreversible. In spite of all these challenging features of the silyl enolate, Denmark and Fan [
127] made the aldol addition of trichlorosilyl enolate of methyl acetate with acetophenone using the chiral pyridinyl
N-oxide ethers as catalysts, which they believe will function as Lewis basic promoters by coordinating and ionising the chloride ions. The reaction took place at room temperature using 10 mol% of the catalysts (
155–
157) in CH
2Cl
2, followed by treatment with aqueous NaHCO
3. The result of the reaction is presented in
Table 10. The yields of the reaction clearly reveal the fact that the sense of asymmetric induction is dominated by the configuration of the chiral axis and that the 6,6′-stereocenters play a secondary role. This is seen clearly by the negligible influence of the bulky phenyl substituted catalyst
156b. A study of a broad substrate generality on the catalytic activity was made on aromatic, heteroaromatic, olefinic, acetylenic, aliphatic, branched and linear ketones.
The aromatic ketones gave the highest selectivities (80–86% ee), while the olefinic ketones were the poorest performers, even less than the aliphatic ketones (20–41% ee). Therefore, the bipyridinyl N-oxide ethers function as catalysts in the enantioselective aldol additions of hindered and enolisable ketones, as well as those with multiple reactive sites.
Due to the moderate yield and enantioselectivity of the previous catalytic asymmetric hydroxy methylation of silicon enolates [
122] in aqueous solution, Ishikawa et al. [
128] further continued their investigation and came up with a new catalyst that resulted in a better yield and selectivity. Optimisation of the reaction condition was made by the reaction of the commercially available formaldehyde and the trimethylsilyl enolate of phenyl propanone. The better result (80% c.y. and 90% ee) was obtained by using five equivalents of formaldehyde in 0.36 M concentration, 10 mol% of the Sc(OTf)
3 catalyst and 12 mol% of the bipyridinyl diol (
S,
S)-
148 in H
2O/DME (1,2-dimethoxyethane) in 1:9 ratio at −20 °C in 24 h. Using 5 mol% of the catalyst reduced the yield to 67% c.y. and 86% ee. Substrate generalisation was made by reacting formaldehyde (using the optimised reaction condition) with aliphatic and aromatic silyl enolates, some of them possessing ethyl or siloxy groups at
α-position. The substrates resulted in poor to excellent yields (24–90%), with moderate to good enantioselectivities (60–94% ee). In some cases, addition of 2,6-di-
tert-butylpyridine slightly improved the yield. Formaldehyde reacted to the enolates that possess
α-substituent from the same face regardless of the substituents at
α-potion. The X-ray crystal structure of the ScBr
3-(
S,
S)-
148 revealed a pentagonal bipyramidal structure in which the hydroxy groups of the bipyridinyl diol coordinate to Sc
3+ in a tetradentate manner, which is in agreement with Nakamura et al.’s [
115] [InBr
2-
148·H
2O]
+ crystal structure
152. This structure is perhaps a key for the high enantioselectivity.
Kobayashi et al. [
129] added the water-unstable Bi(OTf)
3 in a “water compatible Lewis acid” category upon using the basic bipyridinyl diol ligand (
S,
S)-
148. Bi(OTf)
3 became an active and enantioselective catalyst in the asymmetric hydroxymethylation of silicone enolates with an aqueous solution of formaldehyde. During optimisation of the reaction conditions, by reacting aqueous formaldehyde with trimethyl silyl enolate of phenyl propanone, the highest yield (92%) and enantioselectivity (93%) were obtained upon using 10 mol% of Bi(OTf)
3 and 30 mol% of (
S,
S)-
148 in H
2O/DME (1/9) at 0 °C in 4 h. Decreasing the catalyst loading to 1 mol%, the ligand to 3 mol% and undergoing the reaction for 12 h at 0 °C, increased the yield to 93%, while decreasing the enantioselectivity to 91%. Further reduction of the catalyst and ligand load to 0.5 mol% and 1.5 mol% resulted in 76% c.y. and 90% ee, respectively. The substrate generalisation reactions were performed using 1 mol% of Bi(OTf)
3 and 3 mol% of (
S,
S)-
148 in H
2O/DME (1/9) at 0 °C . Substrate generalisation was made for the silyl enolates of aliphatic and aromatic compounds possessing
α-substituted ethyl and siloxy groups that resulted in good to excellent yields (59–93%) and moderate to good enantioselectivities (77–95% ee). The addition of 6,6′-
tert-butyl bipyridine reduced the rate of decomposition of some silicon enolates. The highly water-unstable Bi(OTf)
3 catalyst was stabilised by the presence of the basic ligand. The pentagonal bipyramidal structure with the tetradentate ligand occupying four of the equatorial sites of BiBr
3-(
S,
S)-
148 was found to be similar to the single crystal X-ray structure of the complexes Sc(III)Br
3 and [InBr
2-
148·H
2O]
+ (
152). It is quite interesting to see Sc and Bi having different ionic diameters forming similar complexes with (
S,
S)-
148.
The relentless effort of Kobayashi et al. [
130] made them undergo catalytic asymmetric hydroxy methylation of silicone enolates using formaldehyde in water as sole solvent. The use of 10 mol% of Sc(DS)
3 and 12 mol% of the bipyridinyl diol (
S,
S)-
148 in Triton X-705, at room temperature, resulted in good to high yield (81–81%) and high enantioselectivity (>90% ee) in most cases.
The use of 2 or 1 mol% Sc(DS)
3 led to the same range in yield and enantioselectivity to that of 10 mol% Sc(DS)
3. Aliphatic and substituted aromatic silicon enolates with
α-ethyl, -methyl, -
tert-butyl and -
iso-propyl substituted silicon enolates were involved in the substrate generalisation test. The relatively highly sensitive ketone-derived silicon enolates in water (compared to silyl enol ethers) reacted smoothly under the reaction conditions to afford the desired hydroxymethylated adducts in good yield and high enantioselectivity. Kobayashi et al. [
130] applied this method in the synthesis of the artificial odorant (
S)-2-(hydroxymethyl)-2,5-dimethylindane.
2.10. Allylic Alkylation
The significant contribution made by Kobayashi et al. [
130] in this area is so valuable that it has shown the advantages of using bipyridinyl diols in the aldol reactions. However, the pyridinyl alcohol pool is so vast that there are still more alternative pyridinyl diols with varying substituents on the pyridine ring with respect to both steric and electronic factors. It is therefore worthwhile to consider more pyridinyl alcohols and further investigate the catalytic activity to come up in the aldol reaction with excellent c.y. and ee.
The allylic alkylation reaction is a particularly useful catalytic process that enables the synthetic organic chemist to form carbon-carbon bonds, which is the backbone of organic synthesis. A significant number of catalysed allylic alkylations that involve the use of a variety of
P,
P-,
P,
N-, and
N,
N-based ligands, with
C1 as well as
C2 symmetry, have proven to induce high selectivity in the process [
131,
132]. In this section, we discuss the
N,O-based pyridinyl alcoholato ligands in the catalytic enantioselective allylic alkylation reactions. A general equation for the catalytic asymmetric allylation of aldehydes is presented in
Scheme 19.
Palladium-catalysed allylic alkylation using pyridinooxazolines (compounds with
158 as backbone) as ligands was reported by Bremberg et al. [
131] Racemic 1,3-diphenyl-2-propenyl acetate reacted with dimethyl malonate through the addition of 2 mol% of [(η
3-C
3H
5)PdCl]
2 catalyst, 6 mol% of pyridinooxazolines
105b,
106b and
158 in the presence of
N,O-
bis(trimethylsilyl)acetamide (BSA) with a catalytic amount of KOAc and resulted in 90% ee (with
105b), 95% ee (with
106b) and 88% ee of the (
R)-isomer (with
158) [
97]. The reason for having similar selectivities is probably that in the preferred conformation of the ligand, the
t-Bu group is remote from the allyl group and therefore only has a minor influence on its position.
The following year, Hallman et al. [
133] prepared polymer-supported chiral pyridinooxazolines
159a–
c, using ligands
158,
105b and
106b respectively, and assessed these in the palladium catalysed substitution of
rac-1,3-diphenyl-2-propenyl acetate (
Figure 34). Reagents bound to these new types of resins can be used in solvents with a wider range of polarity and are more easily analysed by spectroscopic methods in the solvent-swollen state. Using the same reaction conditions and catalyst as before, and the polymer-supported pyridinooxazolines
159a–
c ligands, they [
133] obtained the (
R)-configuration product in 60–100% c.y. with 80% ee in
159a, 4% c.y. with 66% ee in
159b and 3% c.y. with 61% ee in
159c. For comparison purposes
160 was also investigated, 76–98% c.y. with 77% ee was obtained. In contrast to previous investigations [
131,
134,
135], only minor differences in the enantioselectivity exhibited by diastereomeric ligands were observed in the present study. However, the low yields of products obtained using ligands
159b and
159c do not allow definite conclusions regarding the selectivity, which has previously been shown to depend on the conformational preferences of the ligands.
With regard to the mechanism of the Pd-catalysed enantioselective allylic alkylation, two steps are very important in order to induce enantiodiscrimination: (i) the activation of the allylic substrate by the Pd
0-ligand complex with formation of the Pd
0-olefin intermediate; and (ii) the attack of the nucleophile on the Pd-(η
3-allyl) cation [
132].
With the aim of gaining an insight into the possible correlation between the intermediate palladium and allyl isomer containing the chiral bidentate ligand and the enantiodiscrimination of the nucleophilic attack, Arena et al. [
132] prepared pyridine-phosphinite ligand
161a (from pyridinyl alcohols) and tested in the palladium(II)-catalysed enantioselective allylic alkylation of 1,3-diphenyl-1,2-propenyl acetate in dimethyl malonate (
Figure 35). 1,3-Diphenyl-1,2-propenyl acetate was added to a dichloromethane solution of [Pd(η
3-C
3H
5)
161a]CF
3SO
3 and then added dimethyl malonate,
N,O-bis(trimethylsilyl)acetamide and a catalytic amount of KOAc to obtain 95% of the racemic product. The lack of enantiomeric excess using
161a was believed to be a consequence of the presence of the relatively small pyridine instead of the sterically bulky PPh
2 that did not sterically hinder the allylic intermediates. Based on the NMR spectroscopy, it was possible to see the low steric demand of the pyridine moiety leading to the fast exchange of all the four possible configurational isomers of the Pd-allyl complexes present in solution.
Therefore, the number and the relative concentrations of the Pd-allyl conformers present in solution as intermediates in the allylic alkylation are factors determining the enantioselectivity of the process.
In light of their earlier results, Arena et al. [
136] have increased the steric hindrance of the pyridine-phosphite ligands by introducing different substituents on the 6-position in the pyridine moiety (
161b–
d) (
Figure 35). Furthermore, with the aim of obtaining a greater conformational rigidity, starting from 2-aminopyridine, they have also synthesized chiral
P,N-ligands, which form a five-membered chelate ring with the metal centre. The synthesized ligands
161b–
d were tested in the enantioselective Pd-catalysed allylic substitution of 1,3-diphenylprop-2-enyl acetate with dimethyl malonate according to Trost and Murphy’s procedure [
137]. The results showed that the phenyl group in
161d greatly accelerated the reaction and yielded 95% in four hours, but the enantioselectivity remained low (7%). The ligands
161b and
161c yielded 91% and 94% respectively with low enantioselectivities 7% ee. Therefore, it is possible to conclude that enantioselectivity of the ligands
161b–
d poorly depended on the size of substituent in position-6 of the pyridine ring. The most likely reason would be that the Me, Br and Ph groups at position-6 on the pyridine ring are not sufficient to exert a steric control on the Pd-catalysed allylic alkylation of 1,3-diphenylprop-2-enyl acetate. The NMR spectra of the allylic intermediates [Pd(PhCHCHCHPh){(
S)-
161b-(
S)-
161d}]
+ proved that all four possible isomeric forms are present in the same concentration ratio and undergo fast exchange processes [
136]. This is the most likely reason for the modest enantioselectivity observed. A chelate ring size became apparent as ligands
161b–
d form a six-membered chelate ring, showing less asymmetric induction than those with five-membered chelate rings.
Nordstrom et al. [
134] investigated the usefulness of their previously prepared 2-(1-hydroxyalkyl)-6-(4,5-dihydro-2-oxazolyl)pyridines
105,
106,
162–
166 in the palladium catalysed allylation of
rac-1,3-diphenyl-2-propenyl acetate (
Figure 36). This type of ligand has the advantage that large structural variations are possible in that the substituents in the oxazoline ring and in the alcohol part of the molecule as well as the relative configuration of the two stereogenic centres can be varied; in addition, the alcohol group can be alkylated using different electrophiles. A catalytic amount of KOAc, 2.5 mol% [(η
3-C
3H
5)PdCl]
2, 5 mol% ligand, (MeOCO)
2CH
2 and BSA were added to a CH
2Cl
2 solution of
rac-1,3-diphenyl-2-propenyl acetate at room temperature and made to react for four days.
The yields and enantioselectivities of the substitution product in relation to each ligand are provided in
Table 11. Even though it took four days to be completed, the reaction proceeded smoothly with most of the ligands, except with
165, to provide high yields of the product. Ligands with (
S)-configuration at stereogenic centre in oxazoline ring resulted in the preferred formation of the (
S)-isomer of the product, while those with (
R)-configuration resulted in the (
R)-isomer of the product. Ligands with the same absolute configuration at the two stereogenic centres became even more efficient when changing the
tert-butyl group to a neomenthyl (comparing ligands
105b and
106b,
Table 11), while those with the opposite configuration at the two centres became less efficient (comparing ligands
105a and
106a,
Table 11).
According to the conformation of the complex deduced from a Nuclear Overhauser Experiment (NOE), a nucleophilic attack took place
trans to the oxazoline ring, as shown in
Scheme 20 [
134].
In order to examine whether the stereochemical control could be extended to other ligand systems, Stranne and Moberg [
138] investigated the substitution of
rac-1,3-diphenylpropenyl acetate with malonate employing 2-pyrrolidinopyridines carrying hydroxyalkyl (
167a,
b), methoxyalkyl (
168a,
b) and siloxyalkyl (
169a,
b) substituents in the 6-position of the pyridine ring (
Figure 37). Having this aim, they performed allylic substitution of
rac-1,3-diphenyl-2-propenyl acetate in CH
2Cl
2 at room temperature in the presence of a (π-allyl)palladium-ligand complex generated in situ from 2 mol% of
bis[(π-allyl)palladium chloride] and 6 mol% of the appropriate ligand. The nucleophile was generated from dimethyl malonate in the presence of
N,O-
bis(trimethylsilyl)acetamide (BSA) and a catalytic amount of KOAc. The reactions were run for four days to ensure complete conversion for the less reactive systems. The results of the reaction are presented in
Table 12.
The absolute configuration of the product depended on the absolute configuration at the benzylic centre rather than on the configuration of the pyrrolidine ring. Ligands with (
R)-configuration in the benzylic centre resulted in the (
S)-product and vice versa and also ligands with (
S)-configuration at the benzylic centre yielded high ee, which is in complete agreement with Nordstrom et al.’s [
134] result. However, the dichotomy previously observed with analogous oxazoline ligands [
139] was not observed in this case. Those diastereomers of the ligand assumed to have the bulky groups on the same side of the coordination plane in the intermediate π-allyl palladium complex resulted in high enantioselectivity (80–84% ee), whereas for those assumed to adopt
pseudo-
C2 conformation, the enantioselectivity is dependent on the nature of the substituent.
In metal complexes containing ligands with two different donor atoms, nucleophilic attack usually takes place
trans to the better π-acceptor (see
Scheme 20). In ligands with donor atoms having similar properties, steric congestion directs attack of the nucleophile [
138].
In order to expand the scope of bipyridinyl ligands, Kwong et al. [
140] investigated the synthesis, spectroscopic characterisation and X-ray crystal structure of Zn(L)(OTf)
2 (L is the methyl ether of
148). The application of the zinc-complex
170 (
Figure 38) as catalyst for allyl addition reactions of aldehydes was also tested. Using 10 mol% of the Zn(L)(OTf)
2 in the allylation reactions of aromatic, substituted aromatic, aliphatic and heterocyclic aldehydes and allyltributyltin at room temperature in CH
2Cl
2 resulted in excellent chemical yield (82–98%) and poor to moderate enantioselectivity (36–59% ee). From the yields of the chemical reaction, it is possible to see that enantioselectivities are independent of the electronic properties of the substituents (electron-withdrawing and electron-donating) in the substituted aromatic aldehydes. However, straight chain and heterocyclic aldehydes showed better enantioselectivities than aromatic aldehydes. It was believed that the chiral Zn-bipyridinyl complex,
170, functions as Lewis acid in this allylation reaction. Generally, chiral methyl ether of ligand
148 is more efficient than the chiral
bis(oxazoline) ligand. The X-ray crystal structure of the chloride complex of Zn helped them propose the most likely structure of the Zn(L)(OTf)
2 complex (
170). In addition to the enantioselective addition of diethylzinc to benzaldehyde, Eidamshaus and Reissing [
103] used the
N-oxide of pyridinyl alcohol ligand
144b (10 mol%) in the allylation of benzaldehyde using three equivalents of EtN(
i-Pr)
2 and allylsilyltrichloride in CH
3CN at room temperature that yielded 81% with 24% ee. They also used the TBS (tributylsilyl) protected alcohol of the
N-oxide
144b in the same reaction and obtained 12% c.y. with 47% ee. The use of
144b in the alkynylation of acetylbenzene in the presence of 1M (in hexane) of diethylzinc at room temperature and benzaldehyde resulted in 66% c.y. with 24% ee of the β-substituted acetylbenzene alcohol. Once again, the ligand
144b (0.4 equiv.) was used in the preparation of β-hydroxy-substituted nitroalkanes (Henry reaction) from benzaldehyde in the presence of CH
3NO
2 (10 equiv.), Cu(OTf)
2 (0.3 equiv.) and one equivalent of EtN(
i-Pr)
2 in
i-PrOH at −50 °C in 18 h. The β-hydroxy-substituted nitroalkane was obtained in good yield (81%) and moderate enantioselectivity (70% ee).


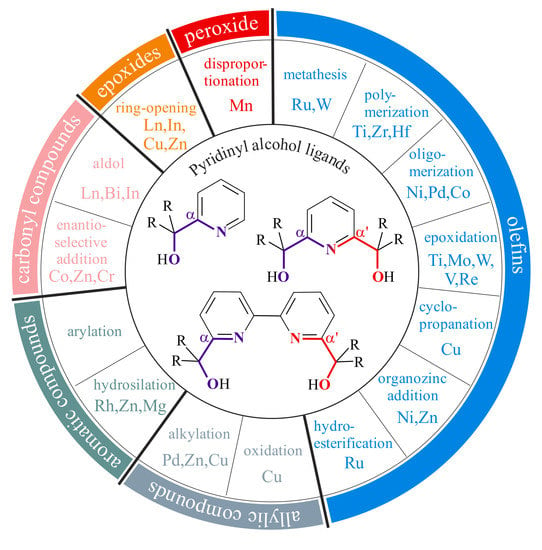


































































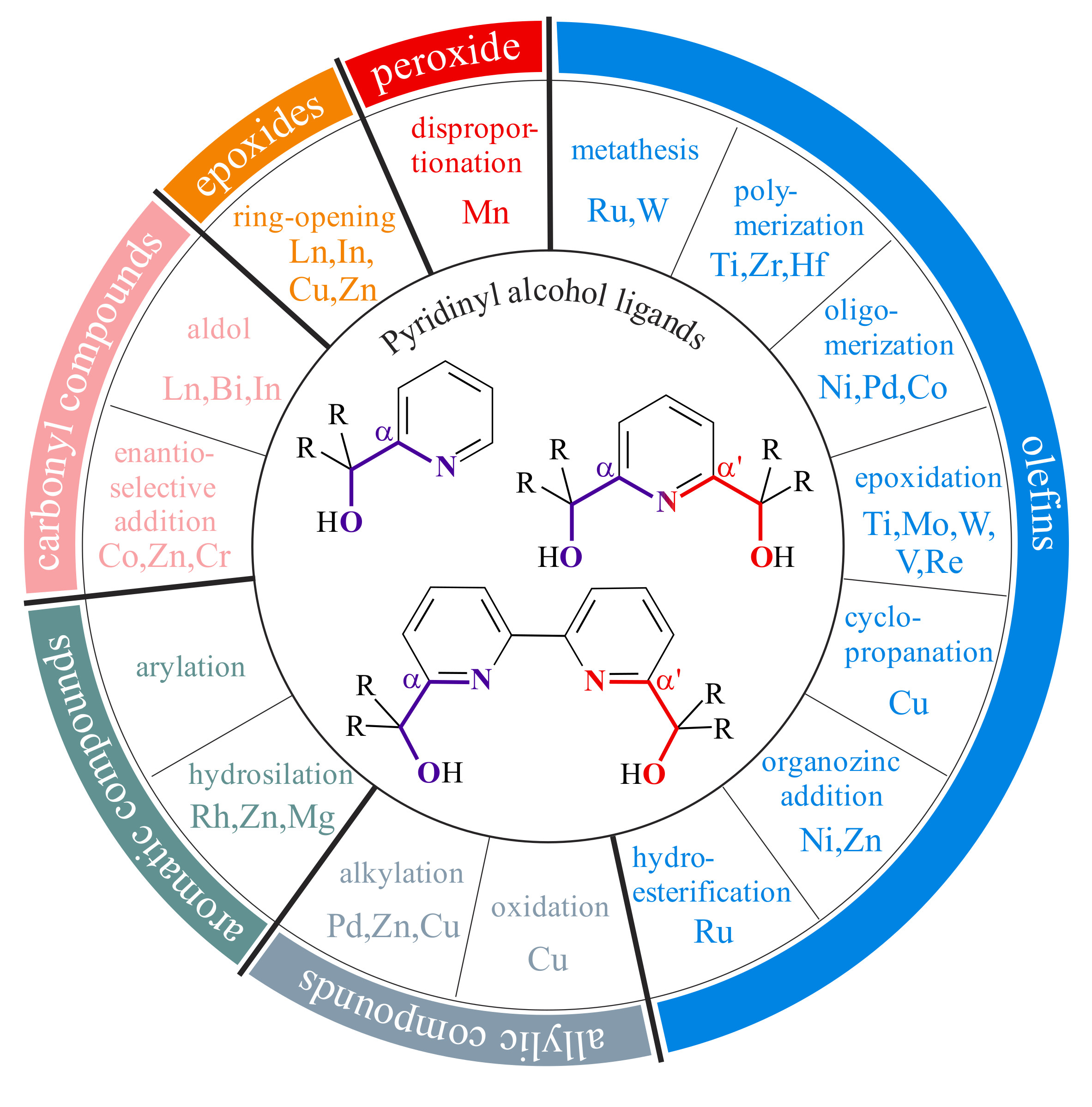{kind=link}
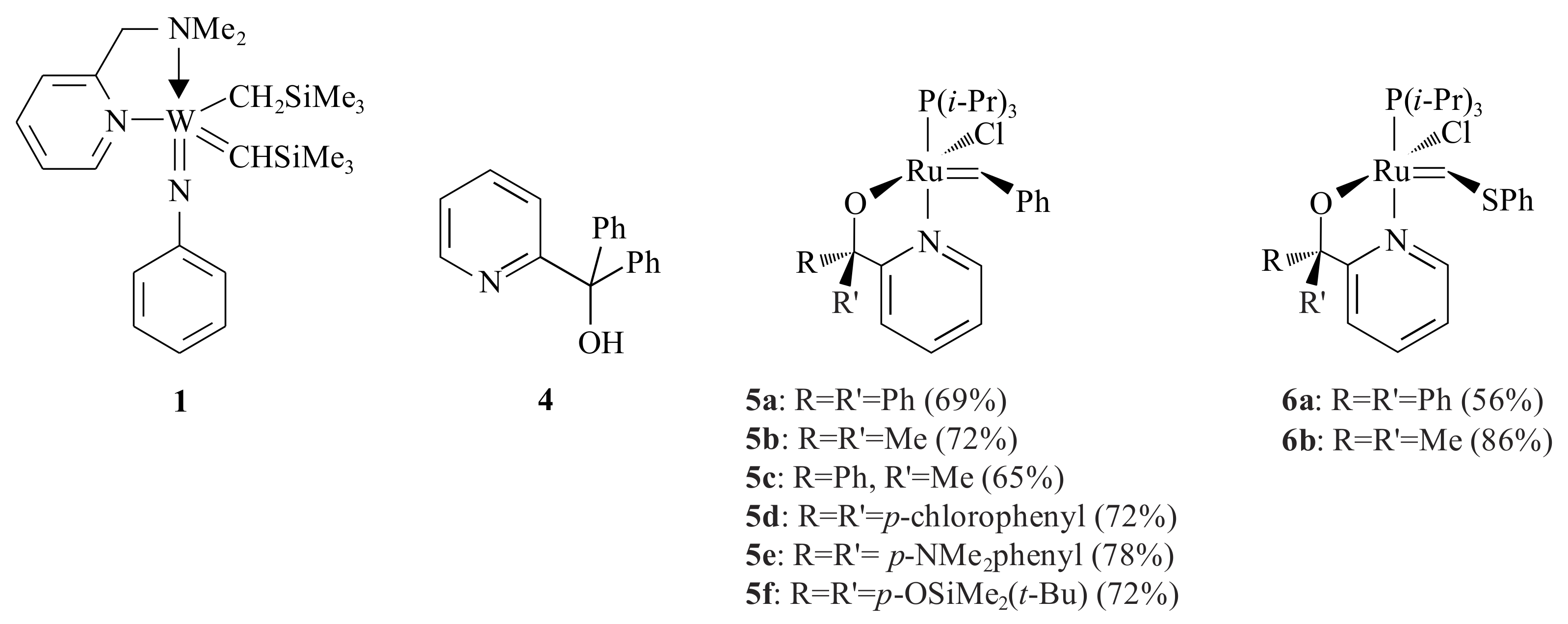{kind=link}
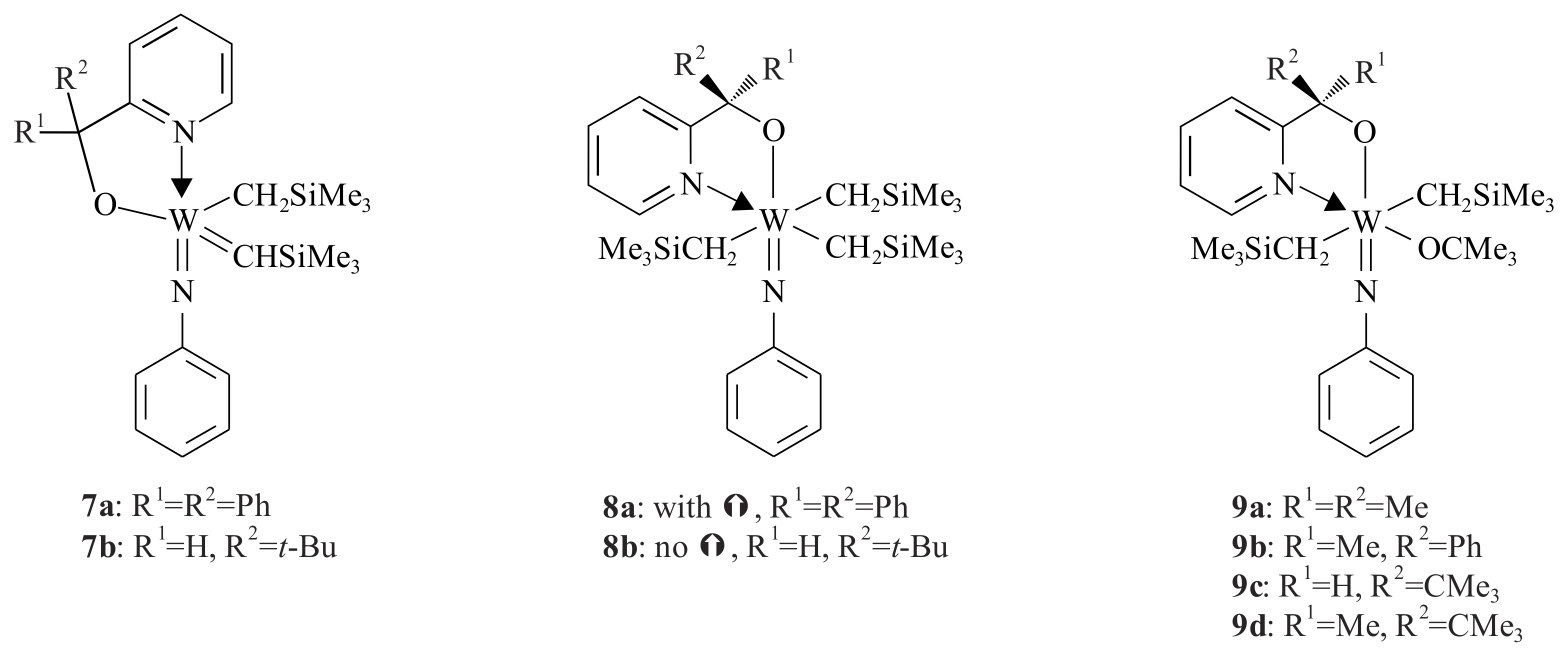{kind=link}
{kind=link}
{kind=link}
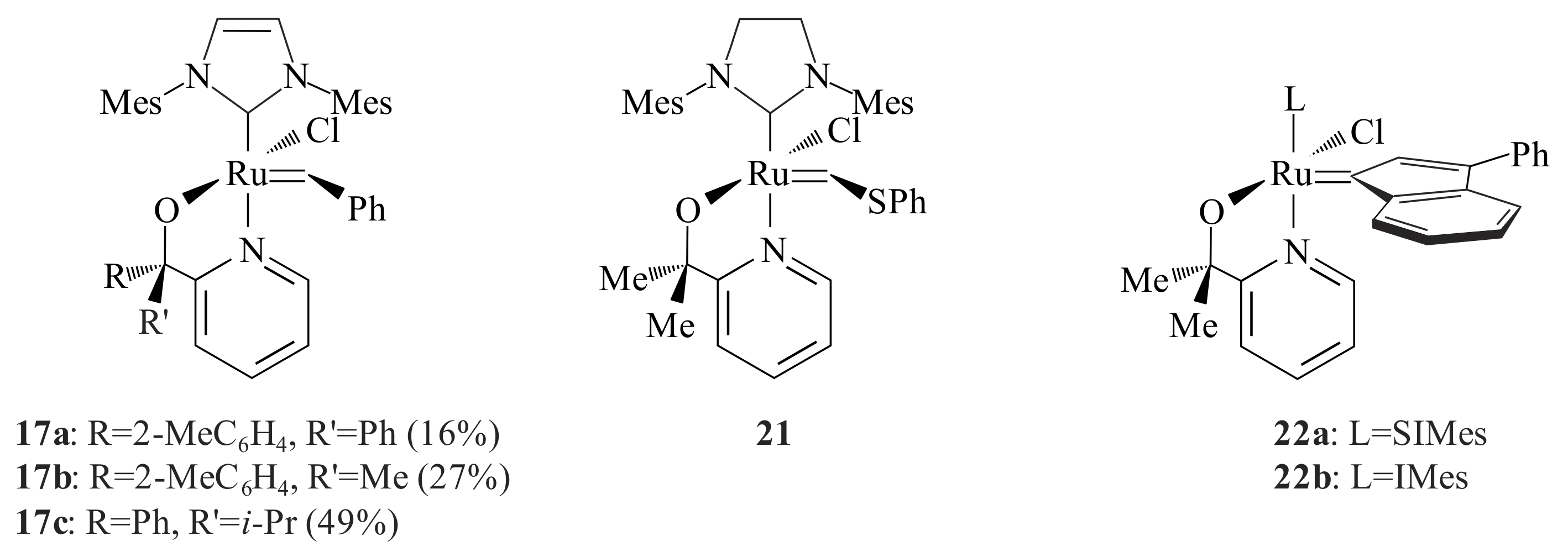{kind=link}
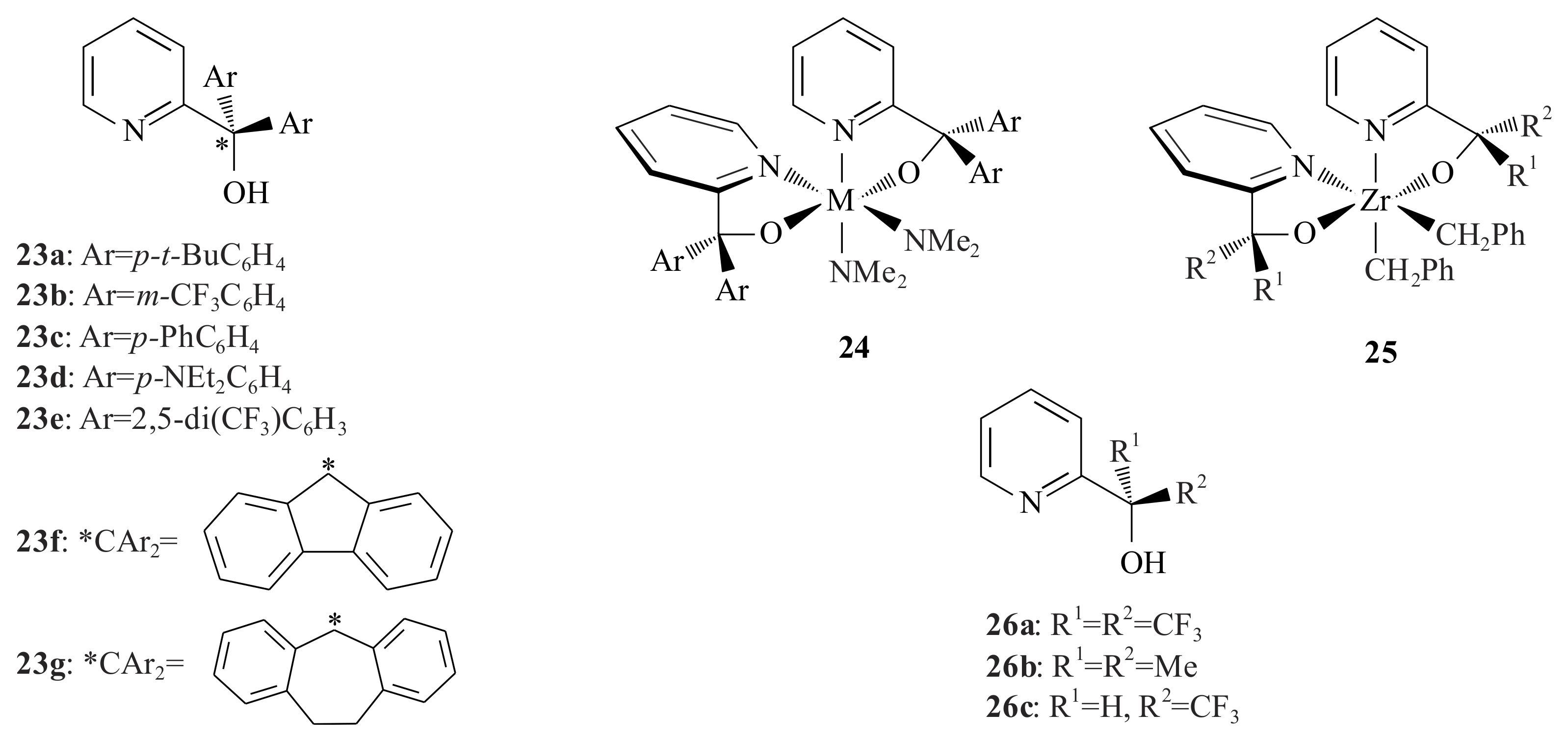{kind=link}
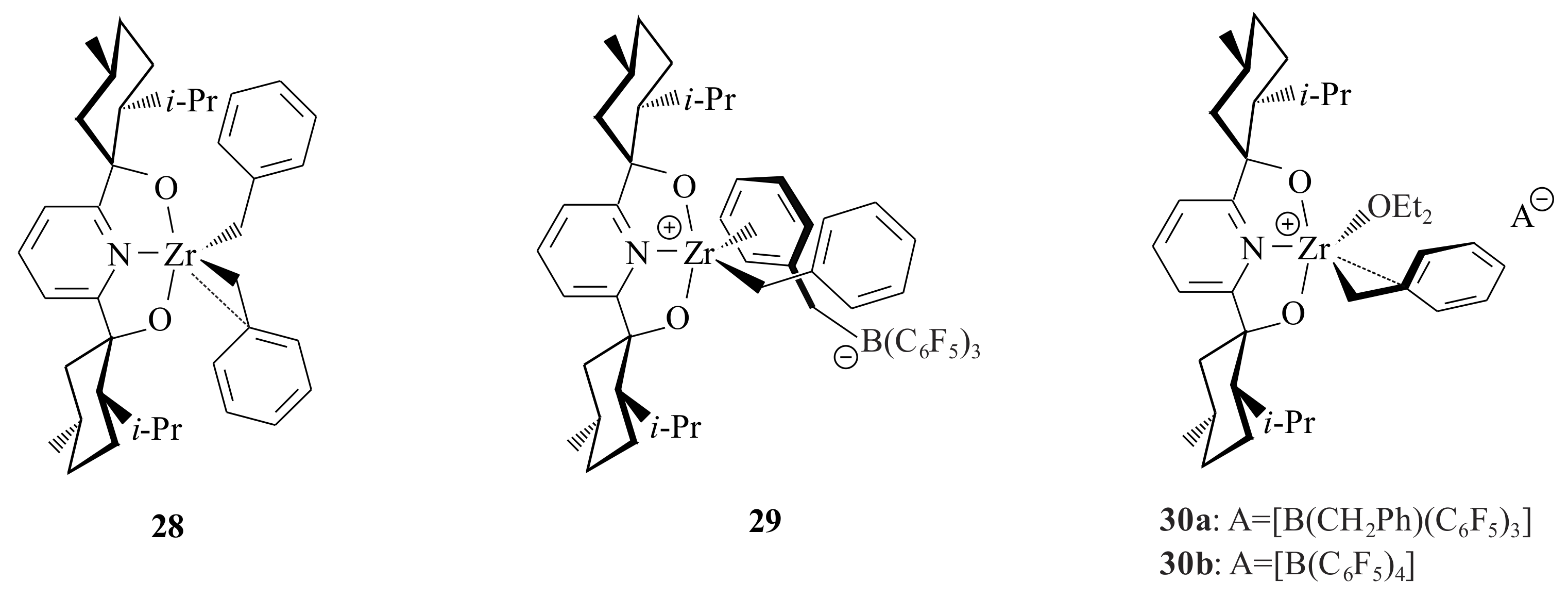{kind=link}
{kind=link}
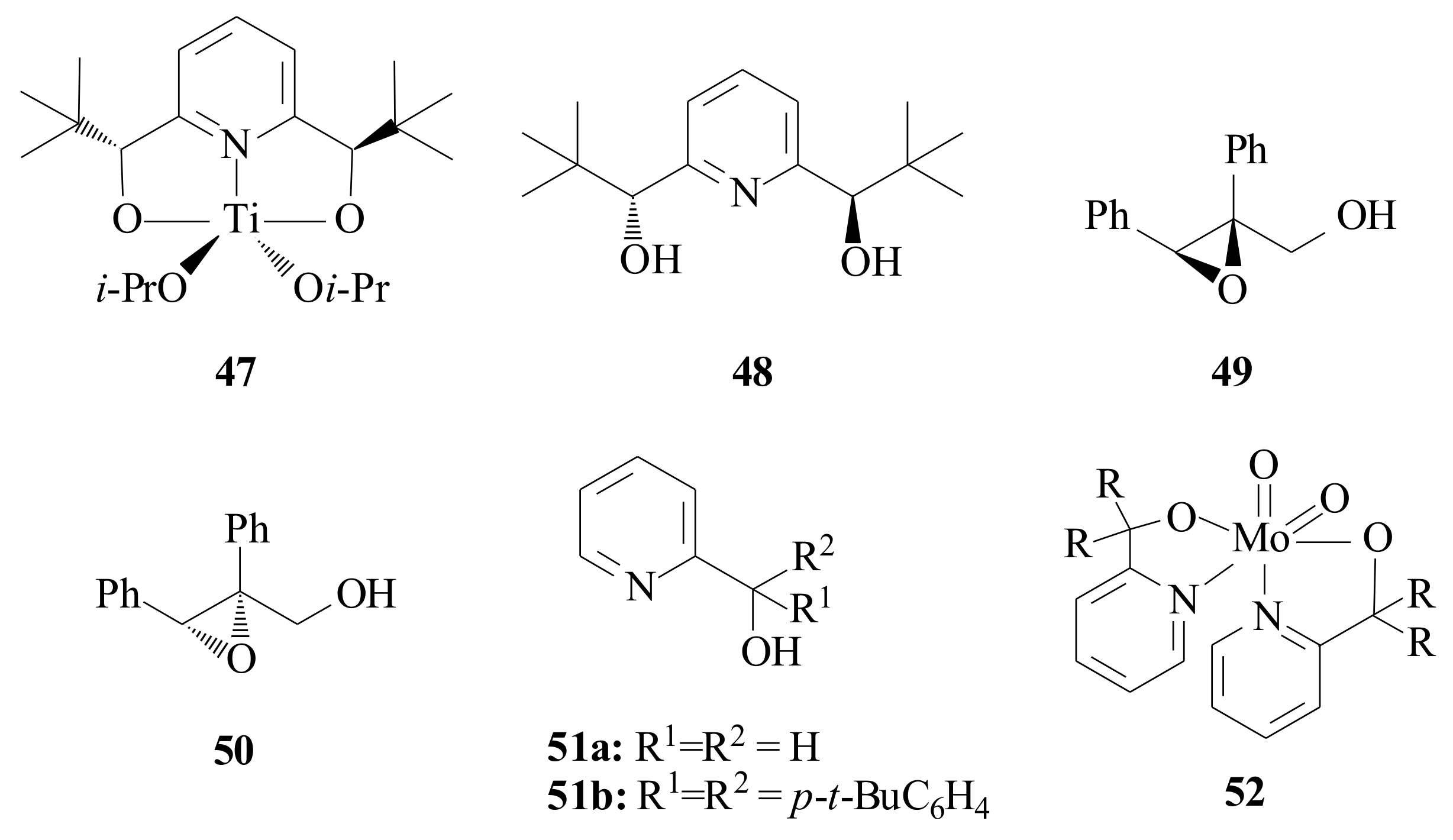{kind=link}
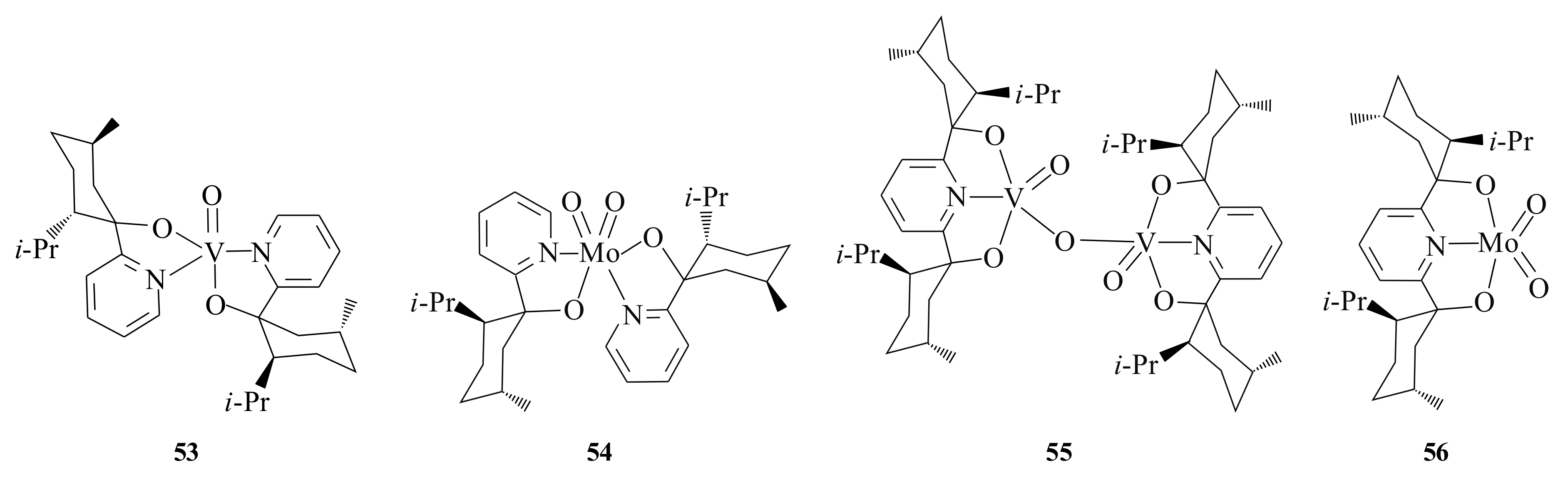{kind=link}
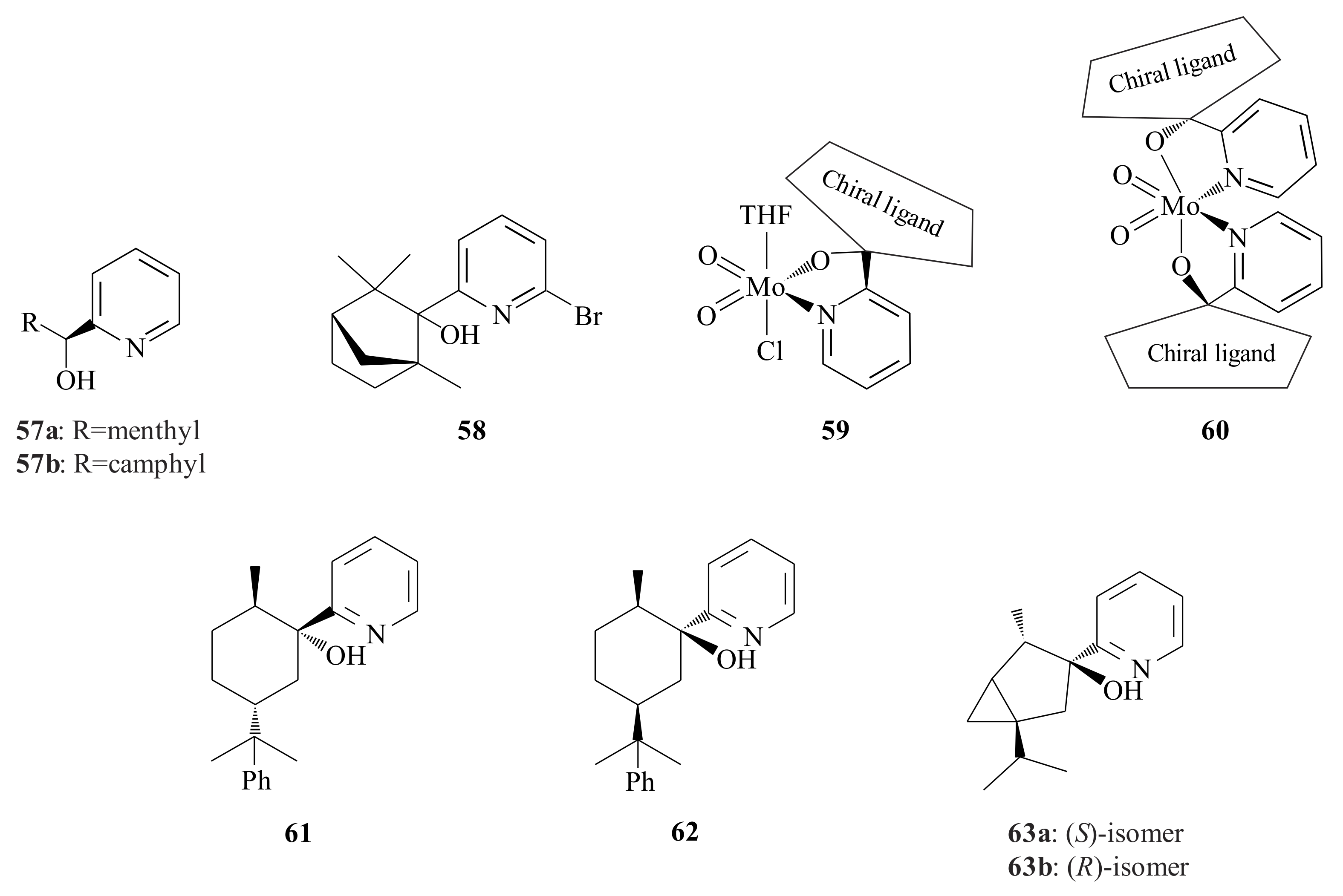{kind=link}
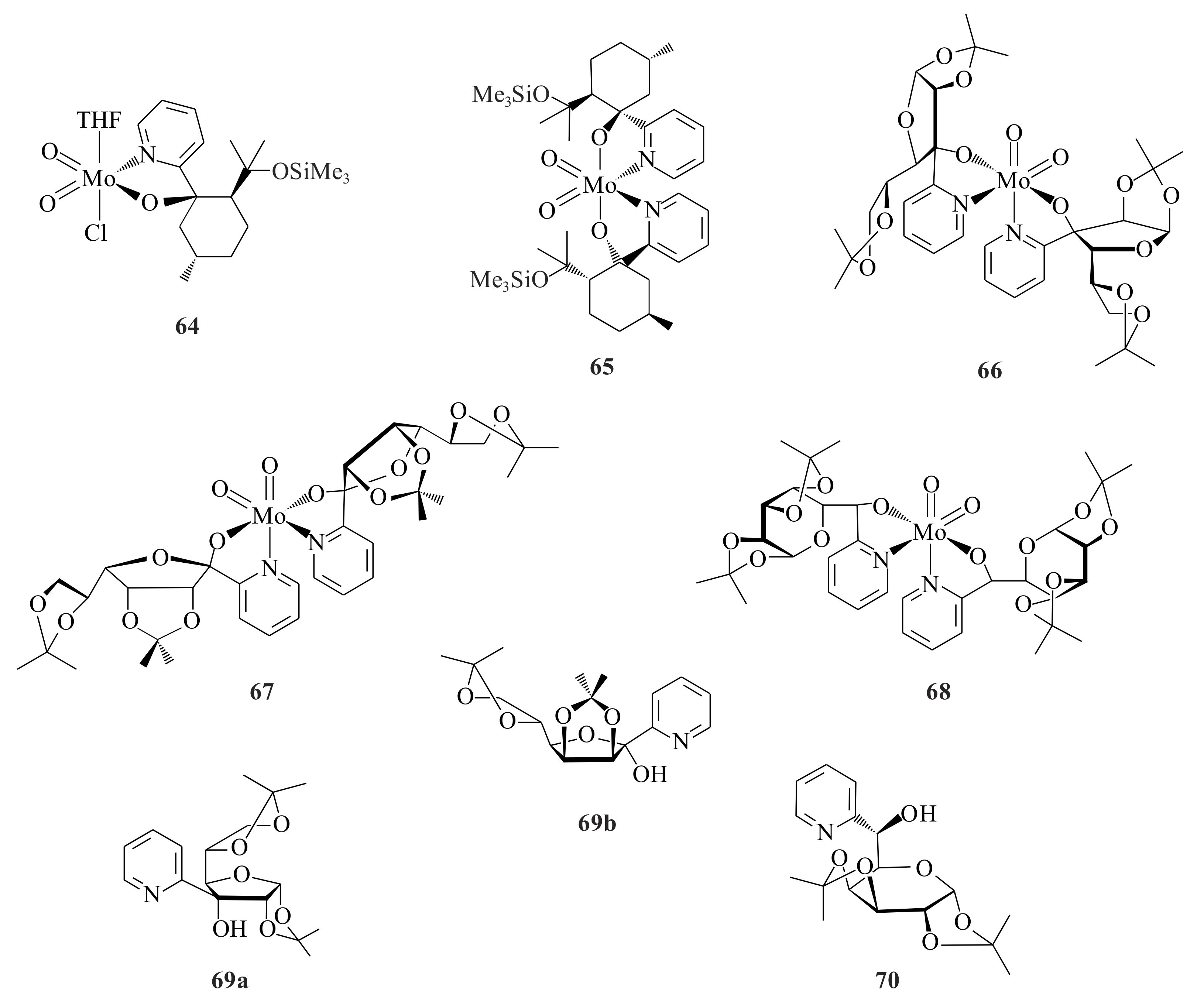{kind=link}
{kind=link}
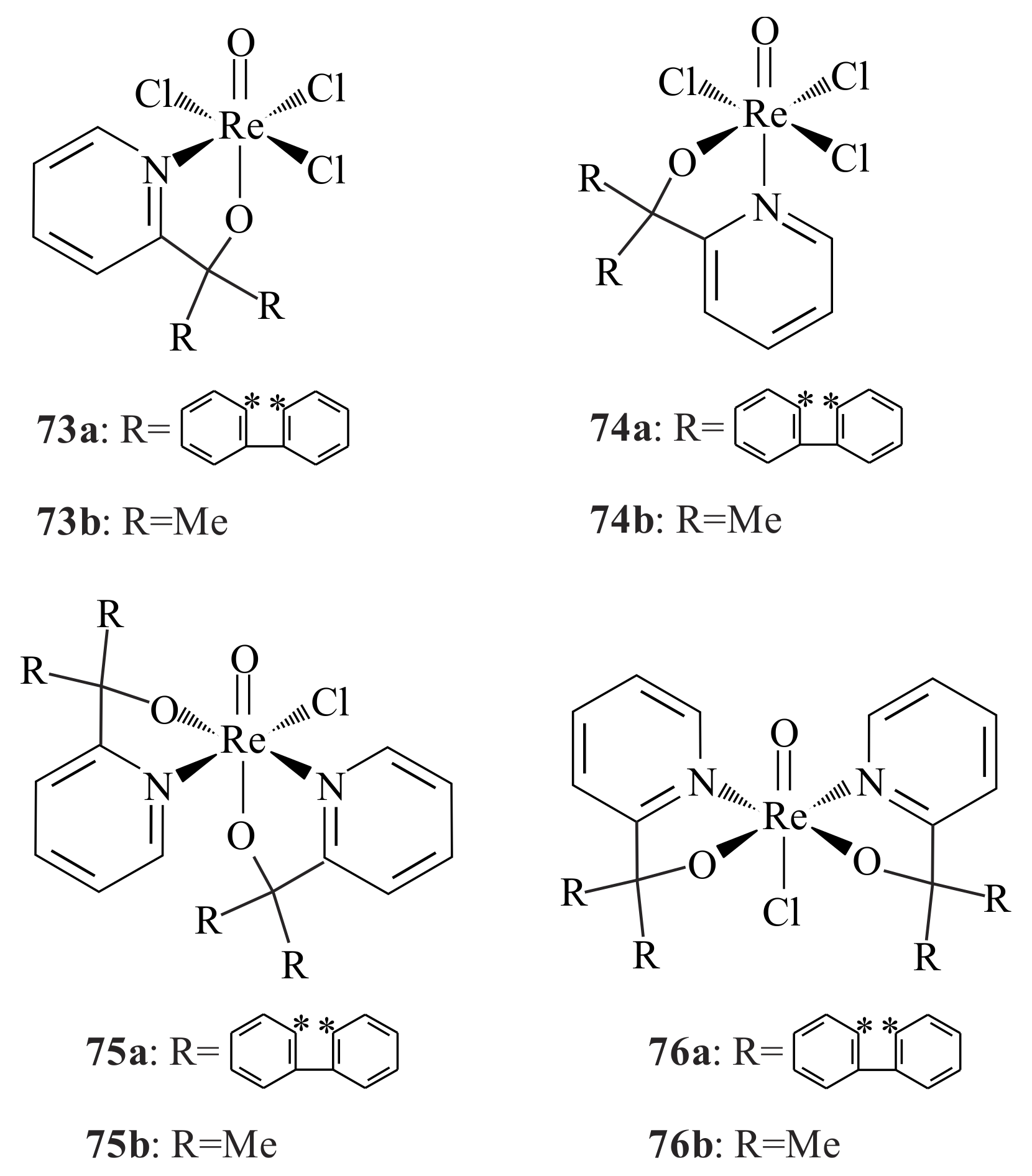{kind=link}
{kind=link}
{kind=link}
{kind=link}
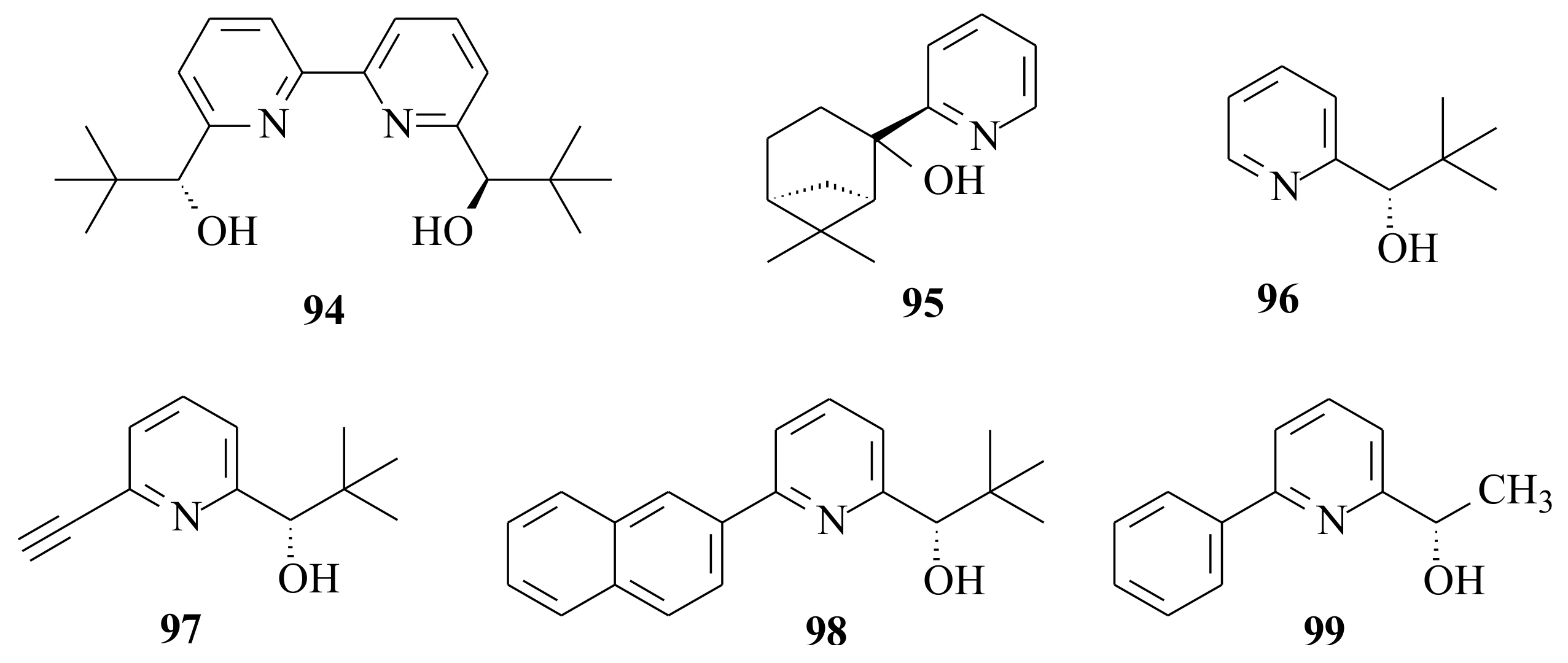{kind=link}
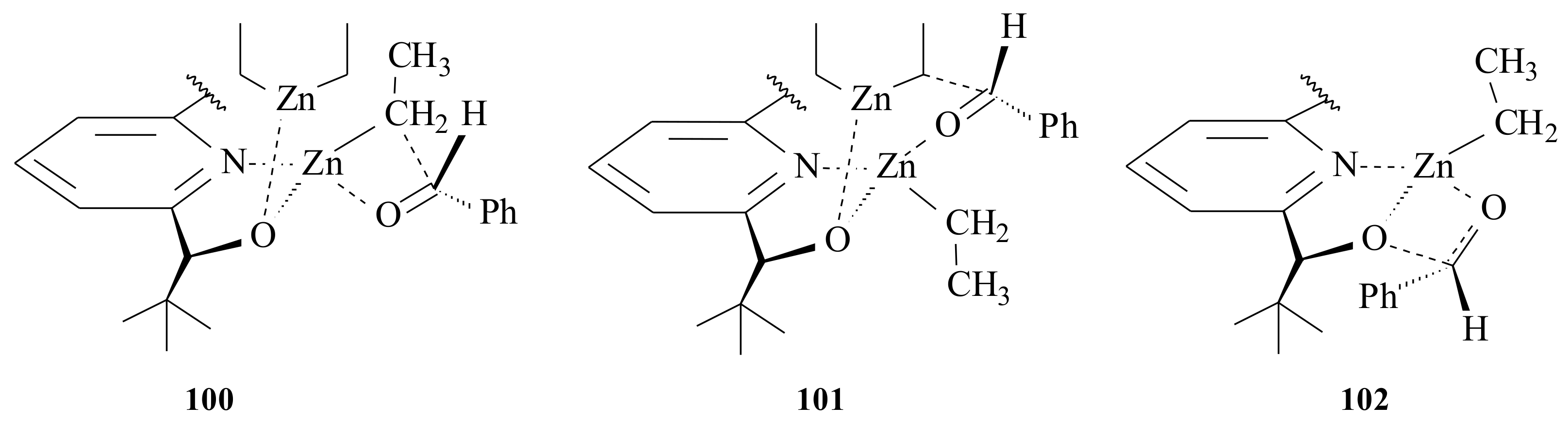{kind=link}
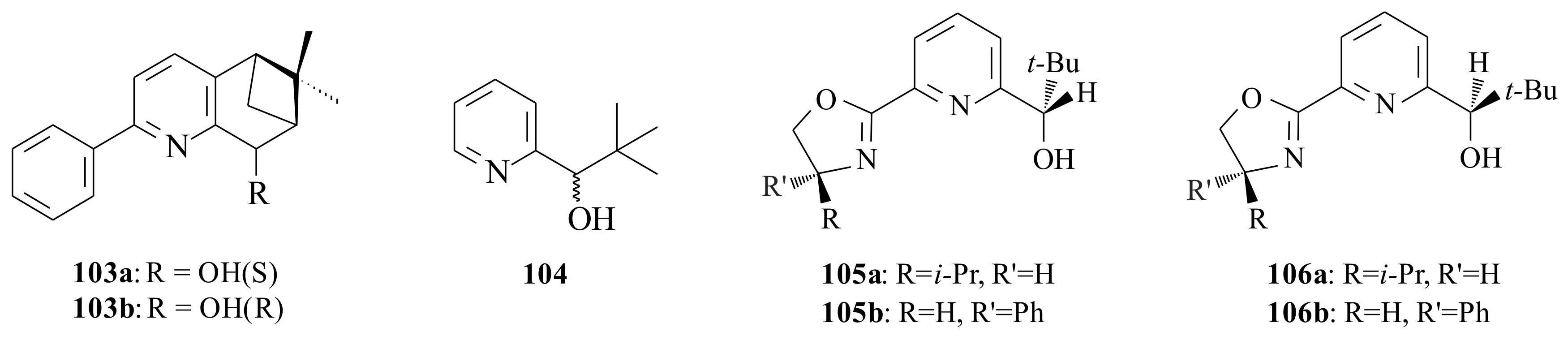{kind=link}
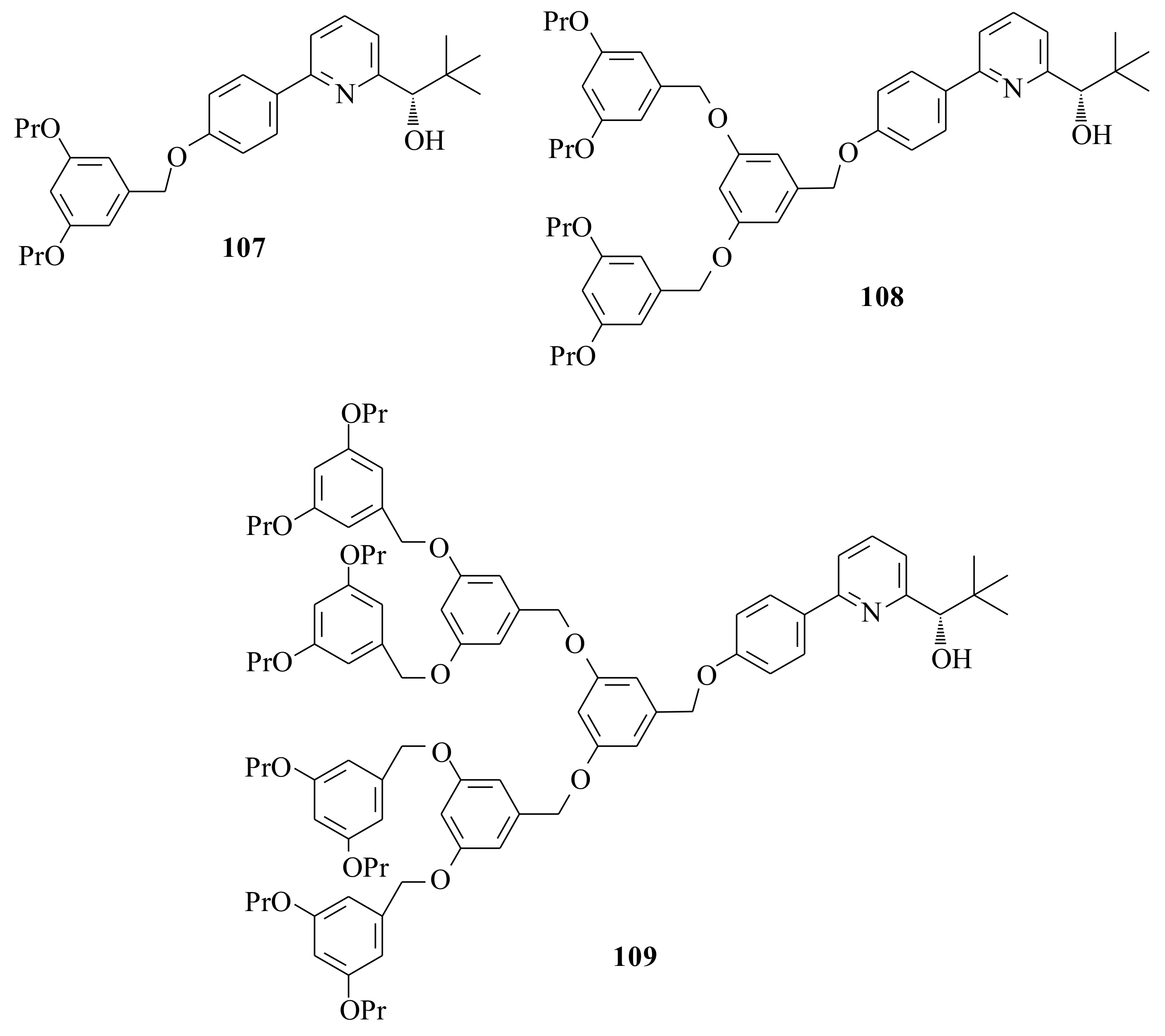{kind=link}
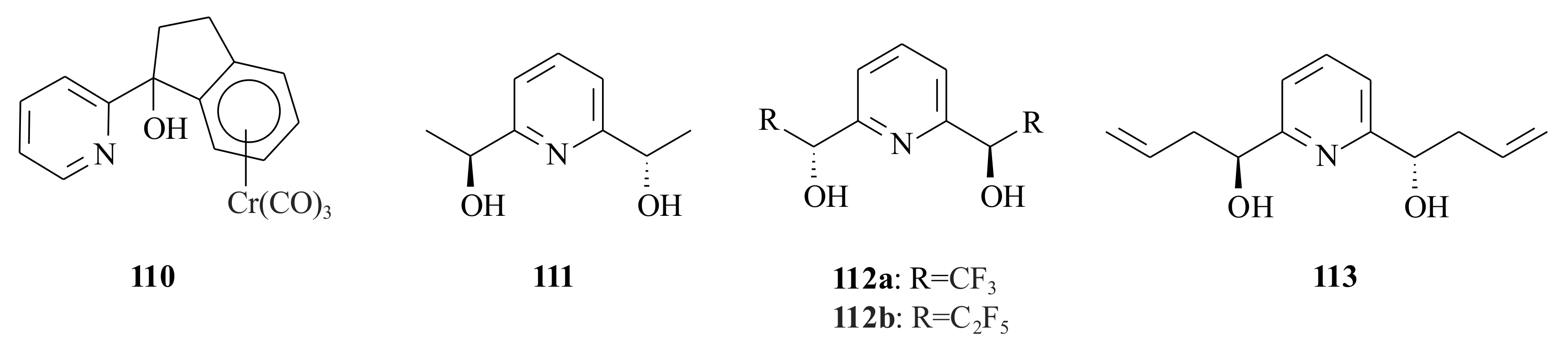{kind=link}
{kind=link}
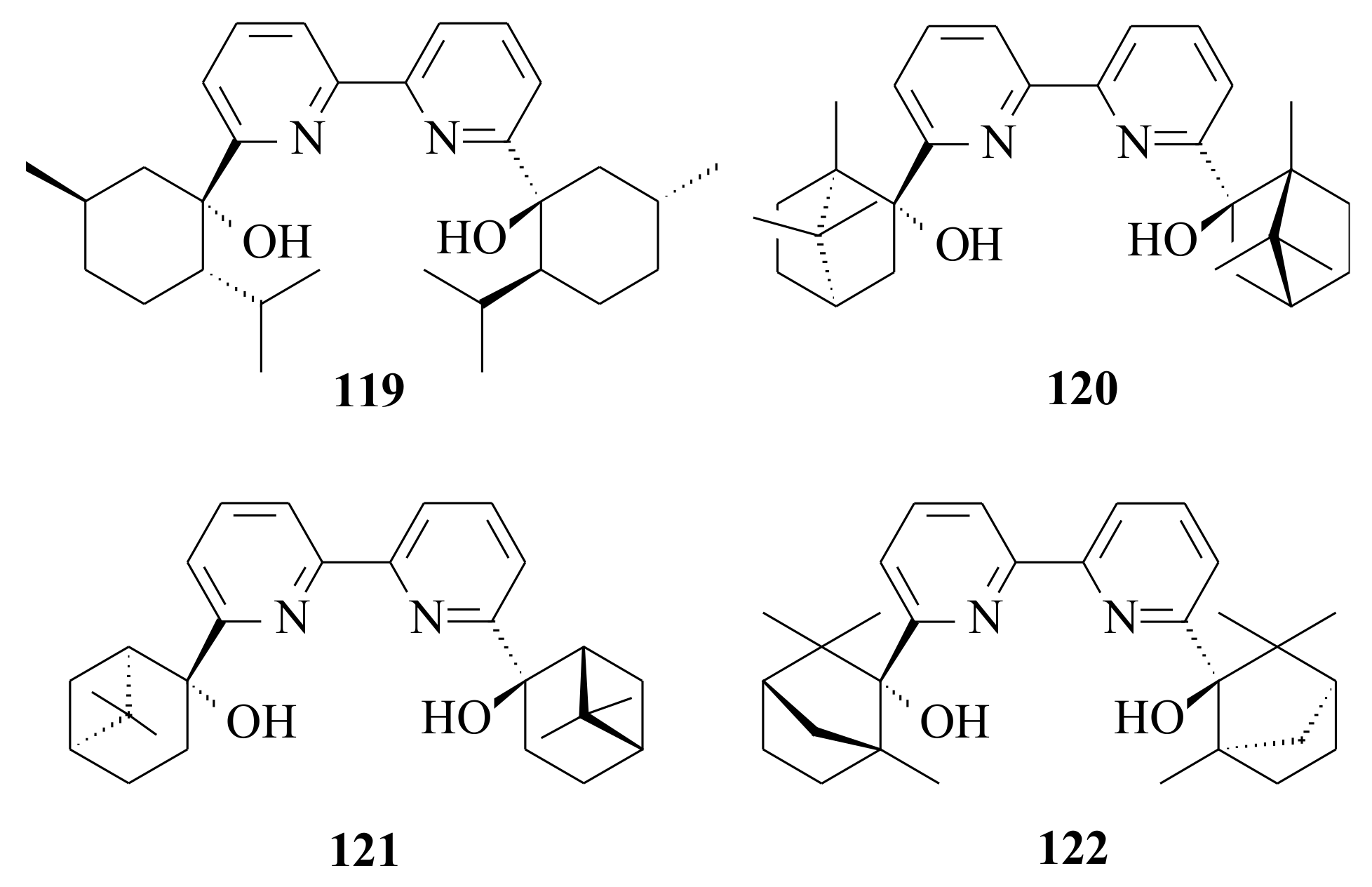{kind=link}
{kind=link}
{kind=link}
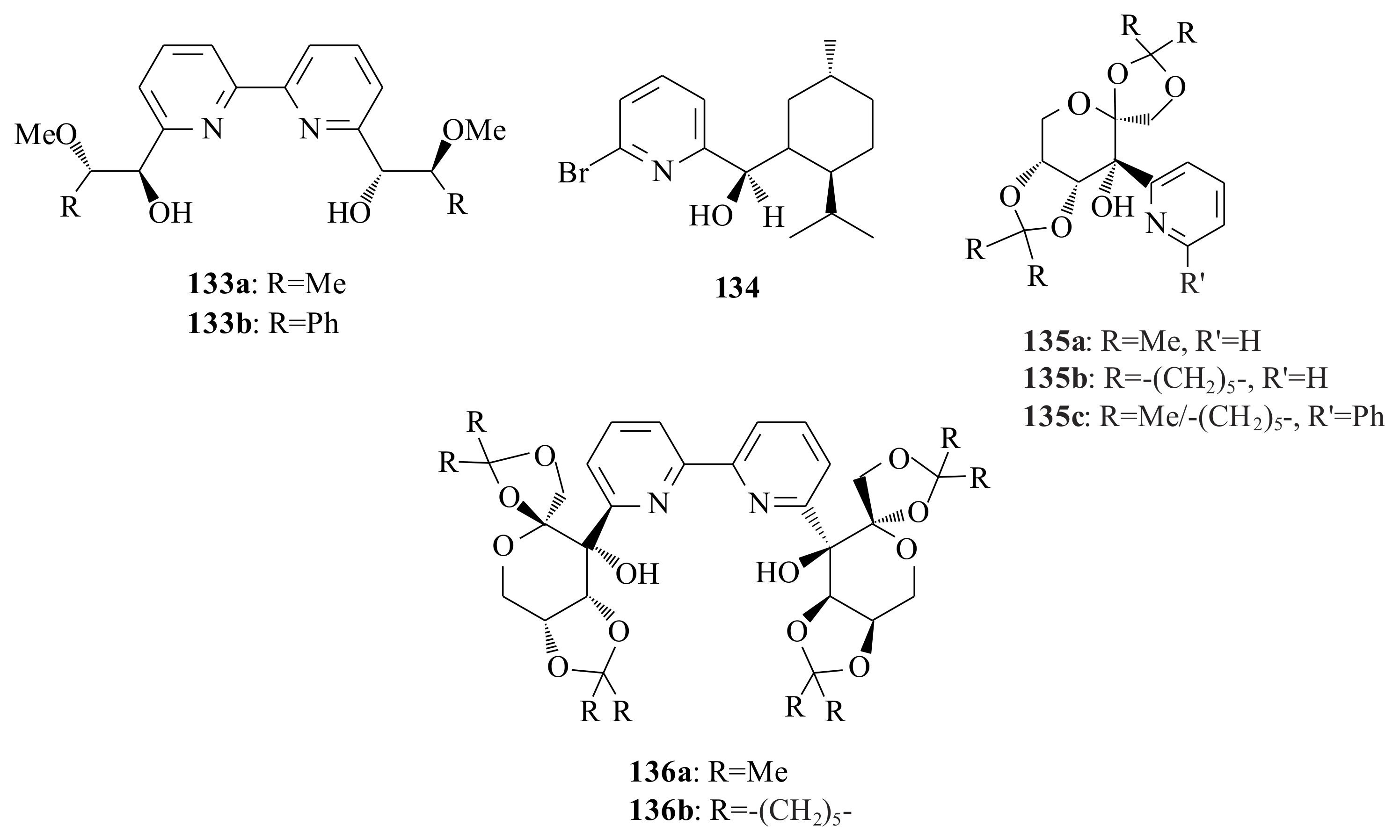{kind=link}
{kind=link}
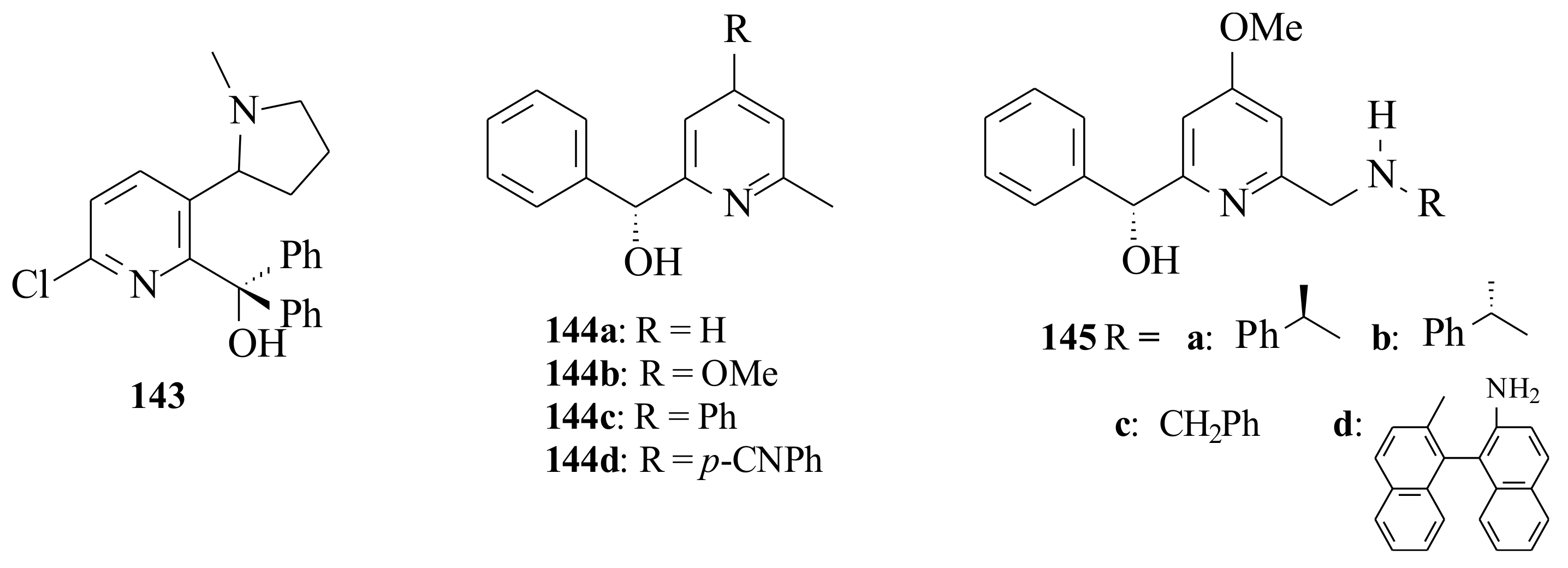{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
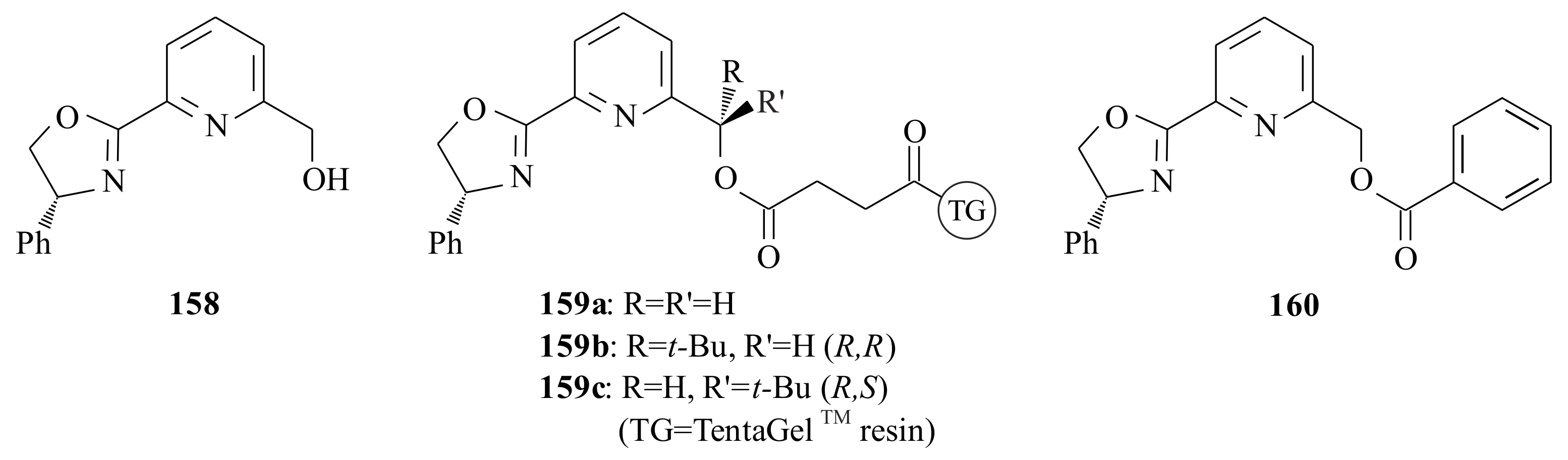{kind=link}
{kind=link}
{kind=link}
{kind=link}
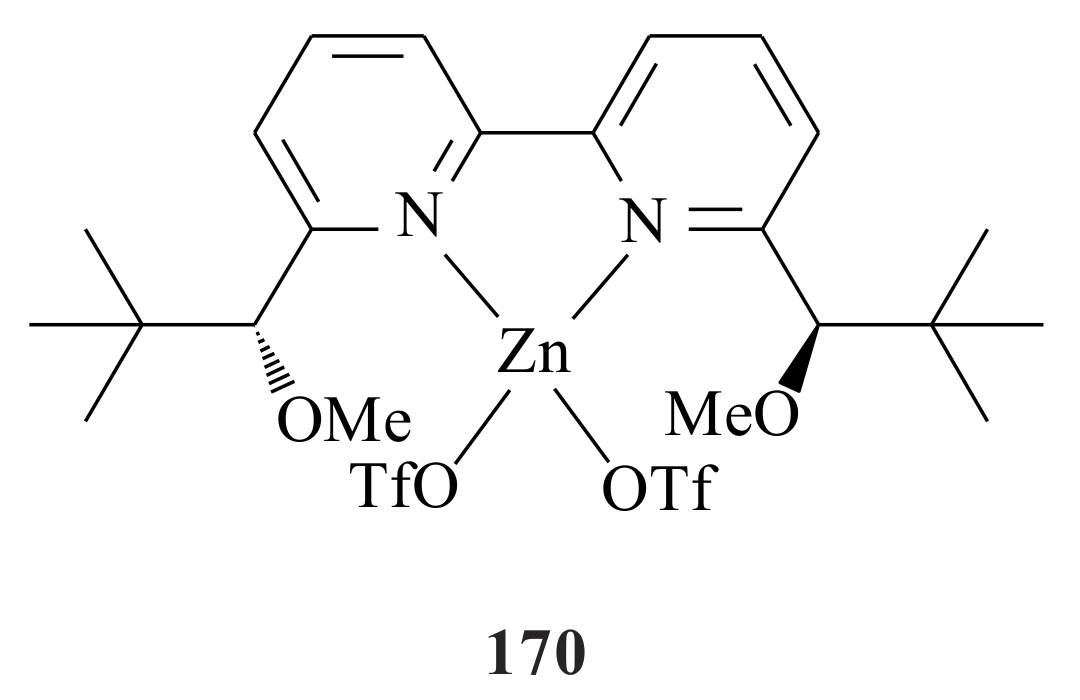{kind=link}
{kind=link}
{kind=link}
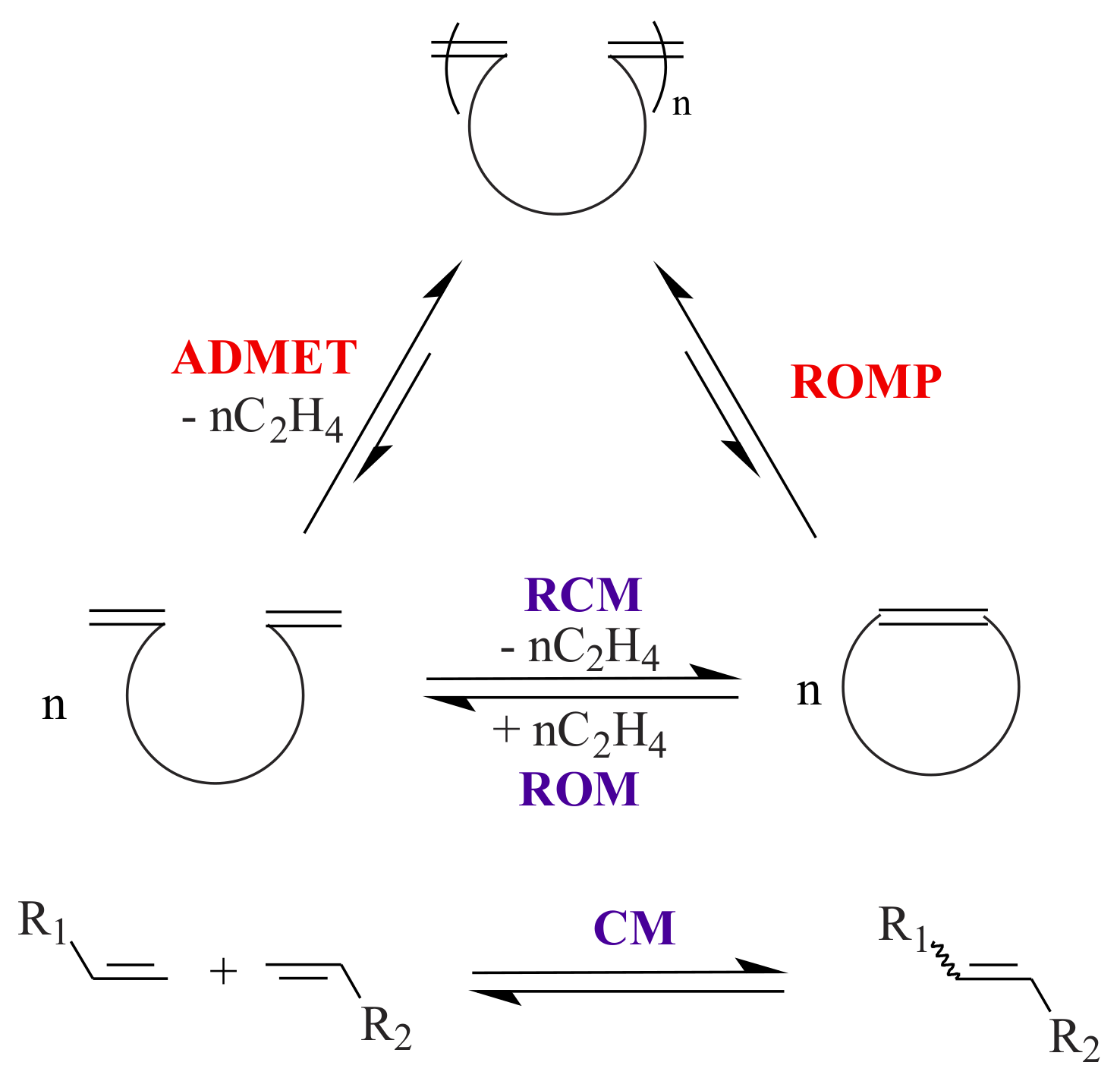{kind=link}
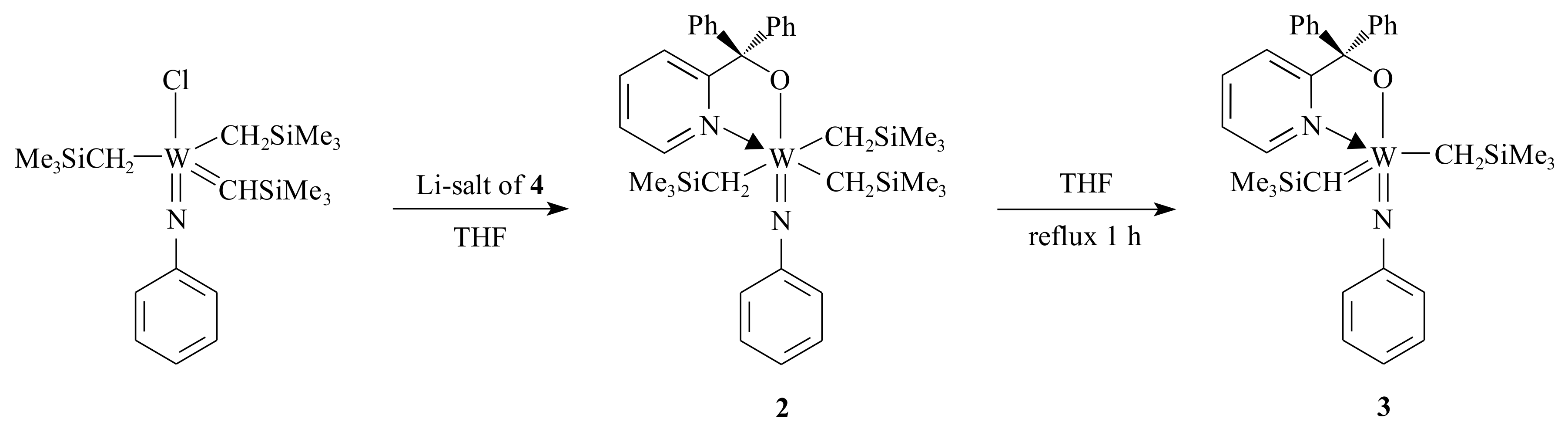{kind=link}
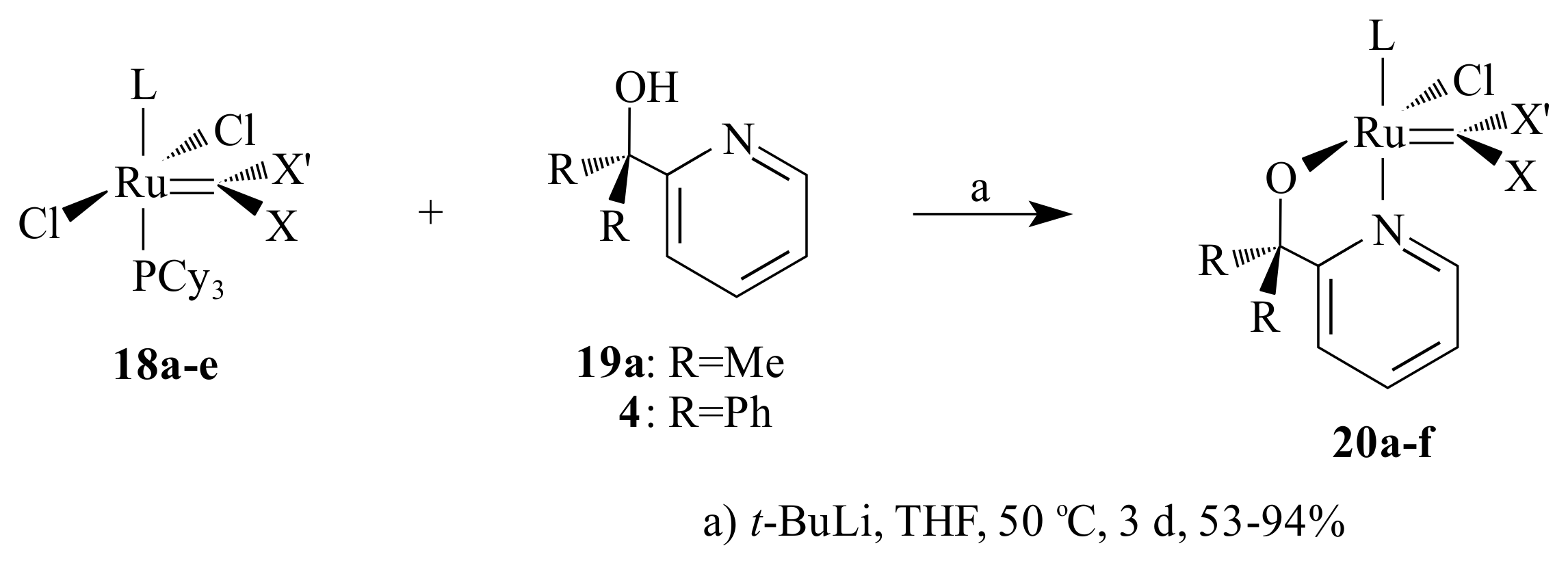{kind=link}
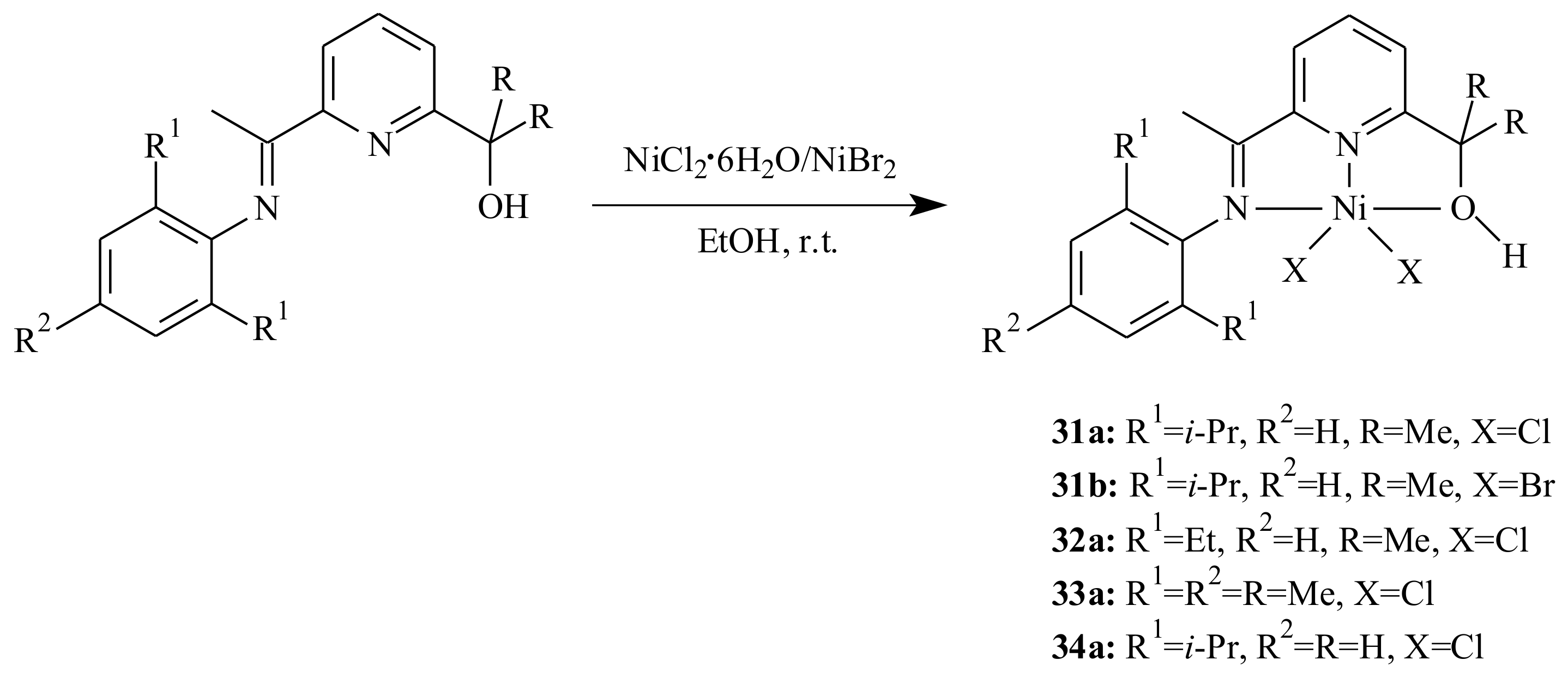{kind=link}
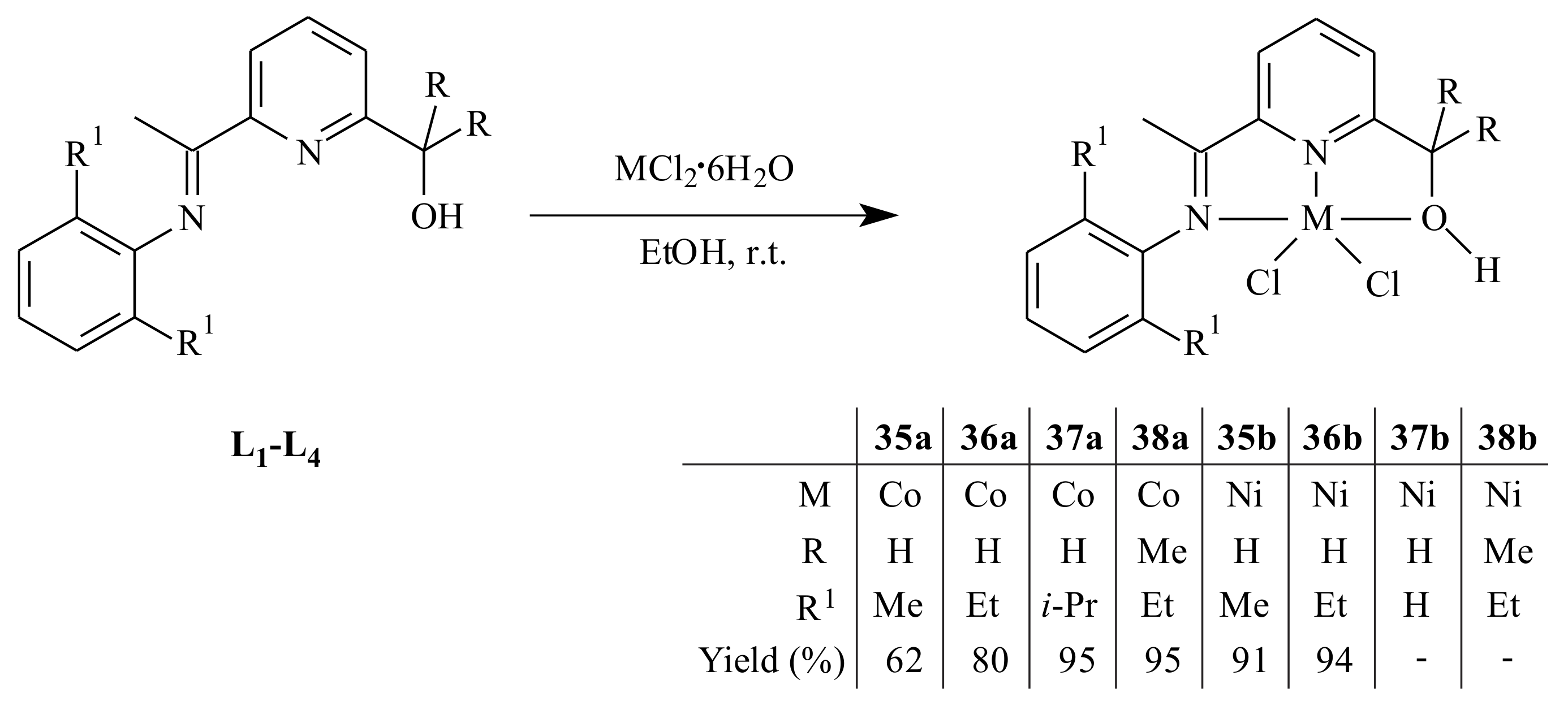{kind=link}
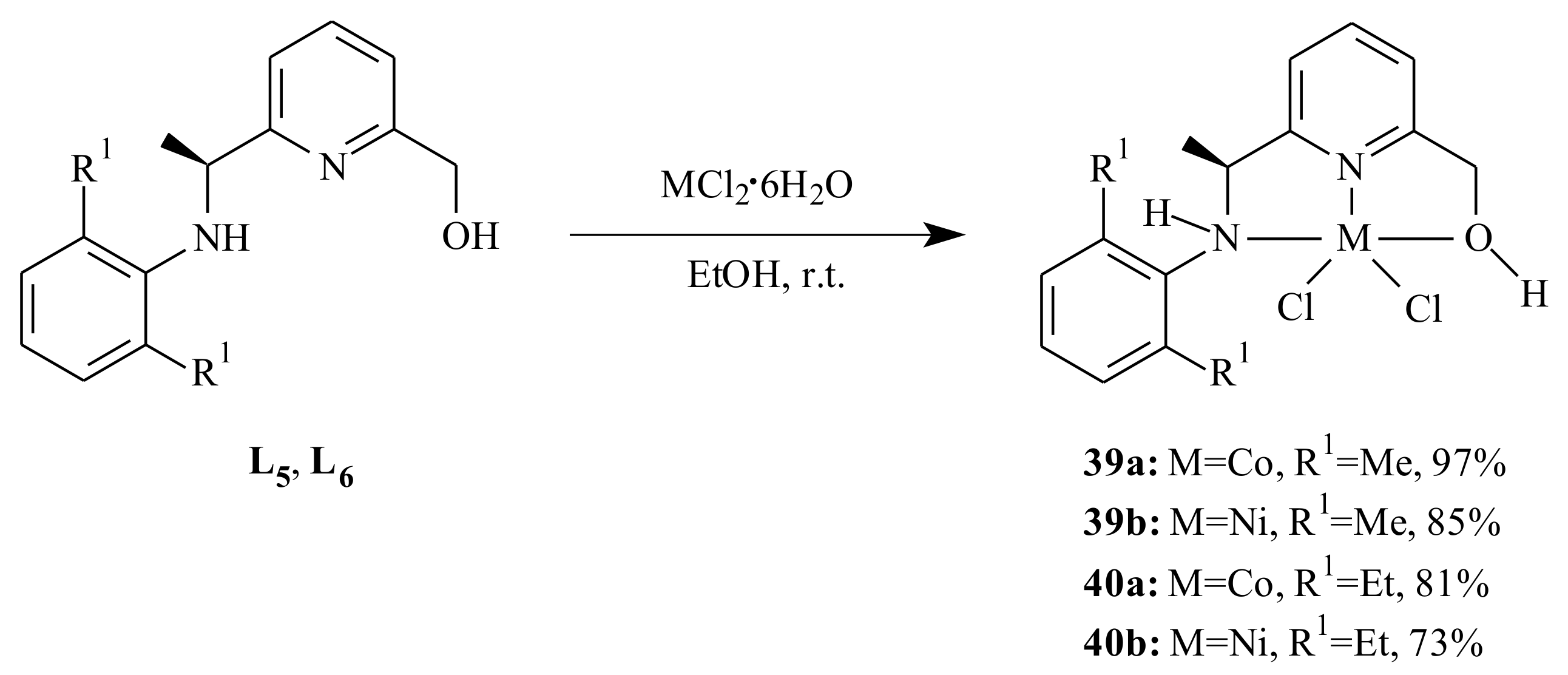{kind=link}
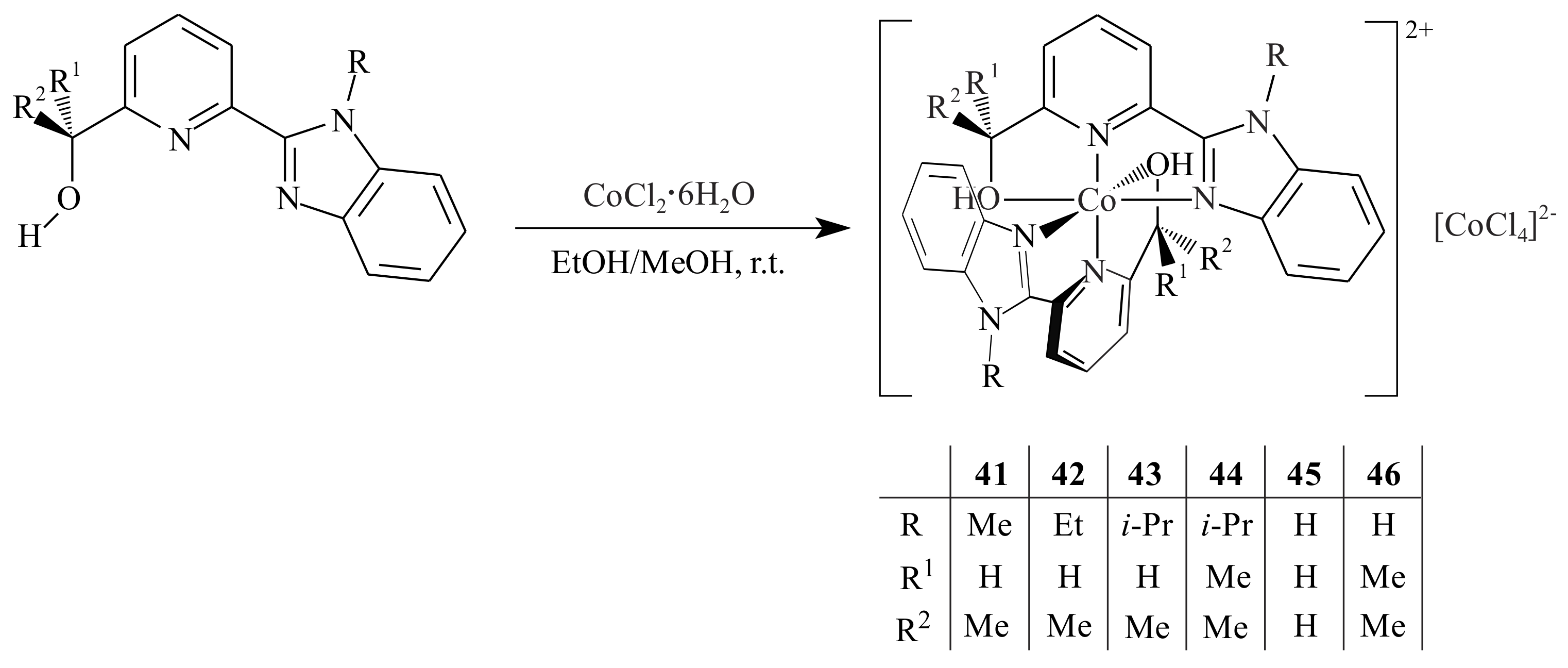{kind=link}
{kind=link}
{kind=link}
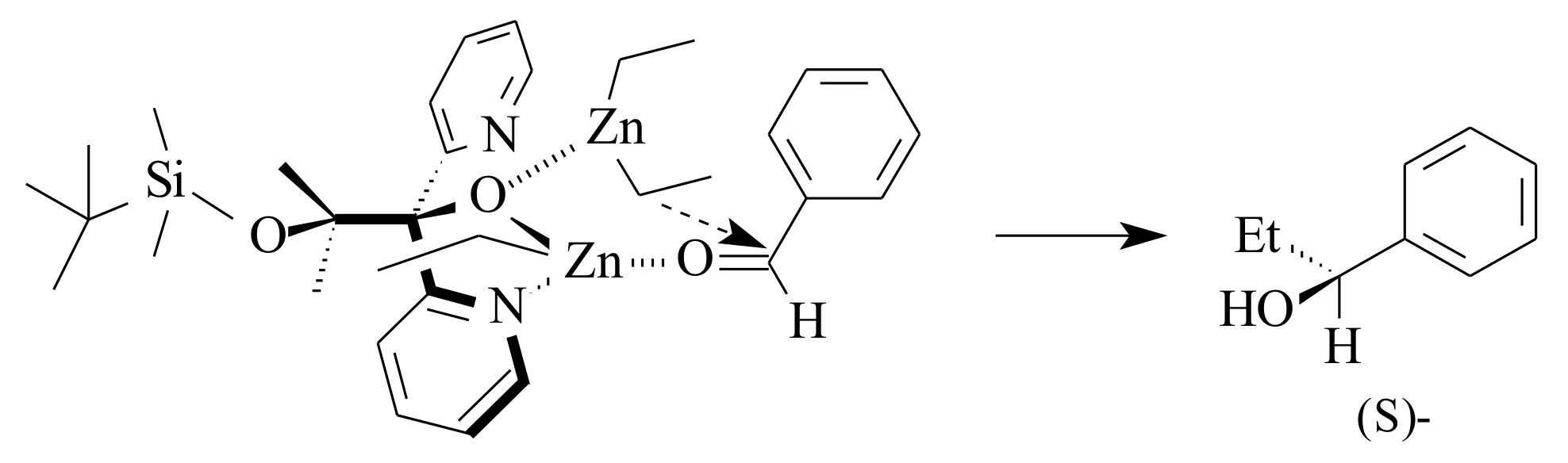{kind=link}
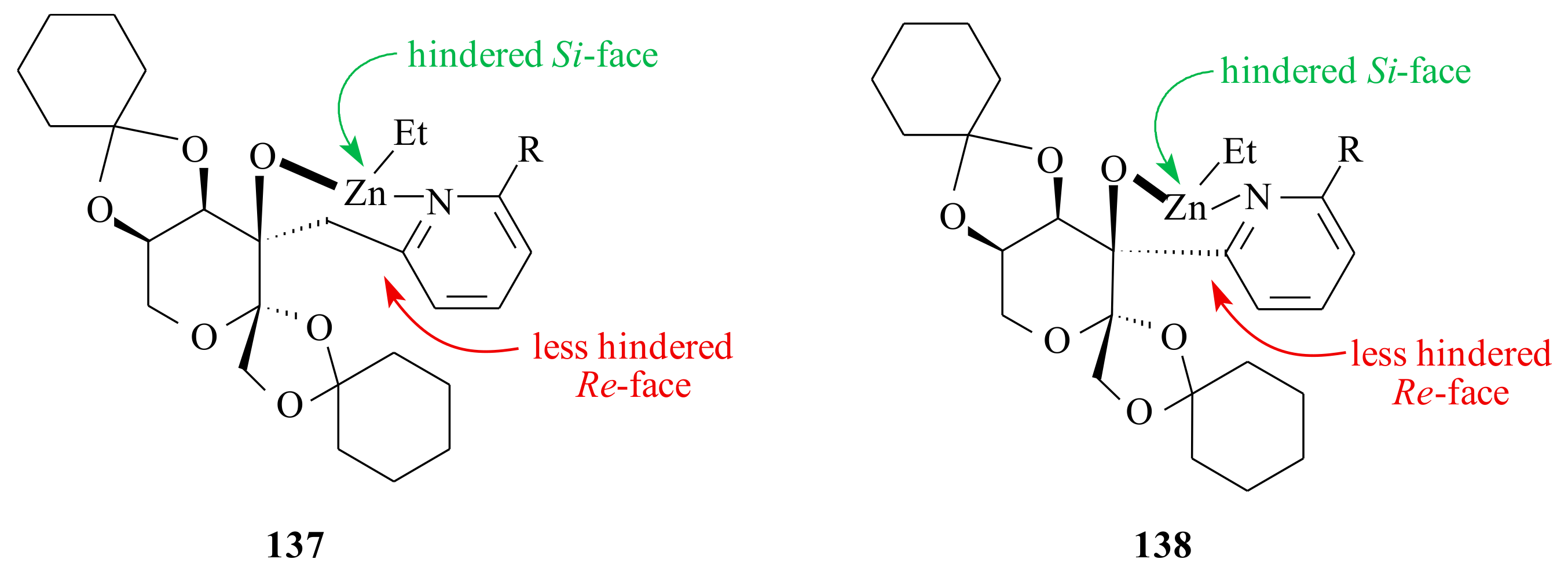{kind=link}
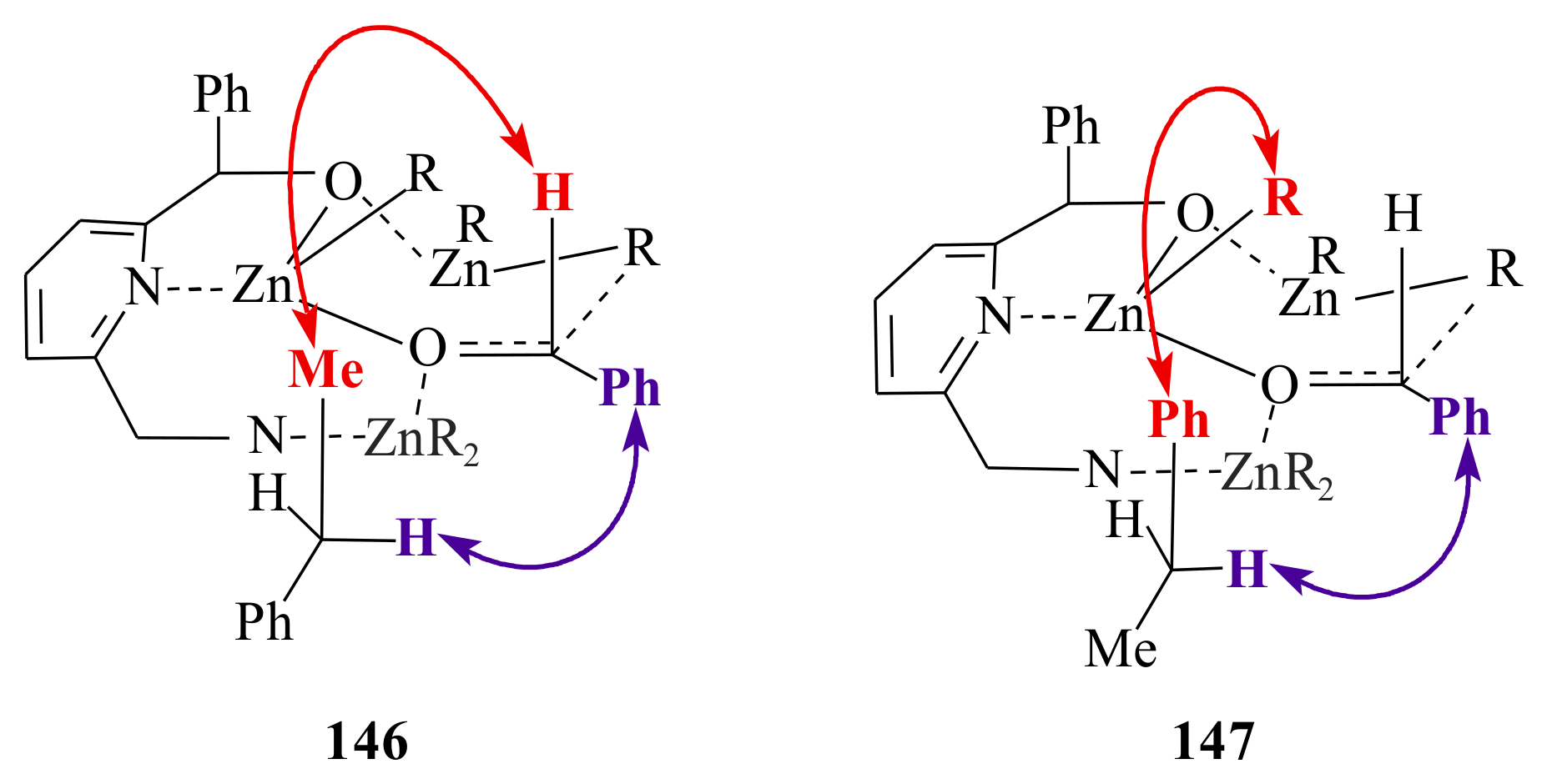{kind=link}
{kind=link}
{kind=link}
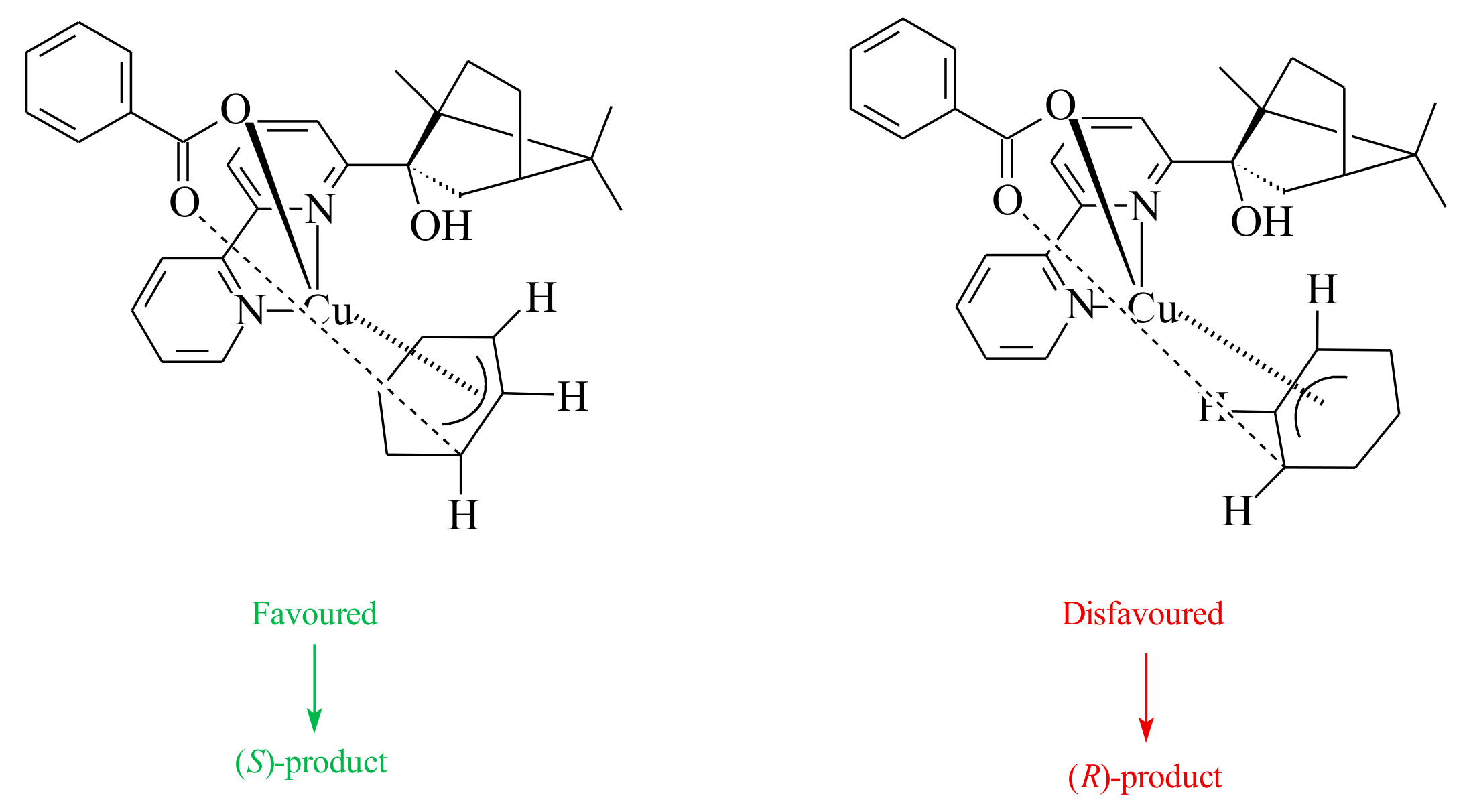{kind=link}
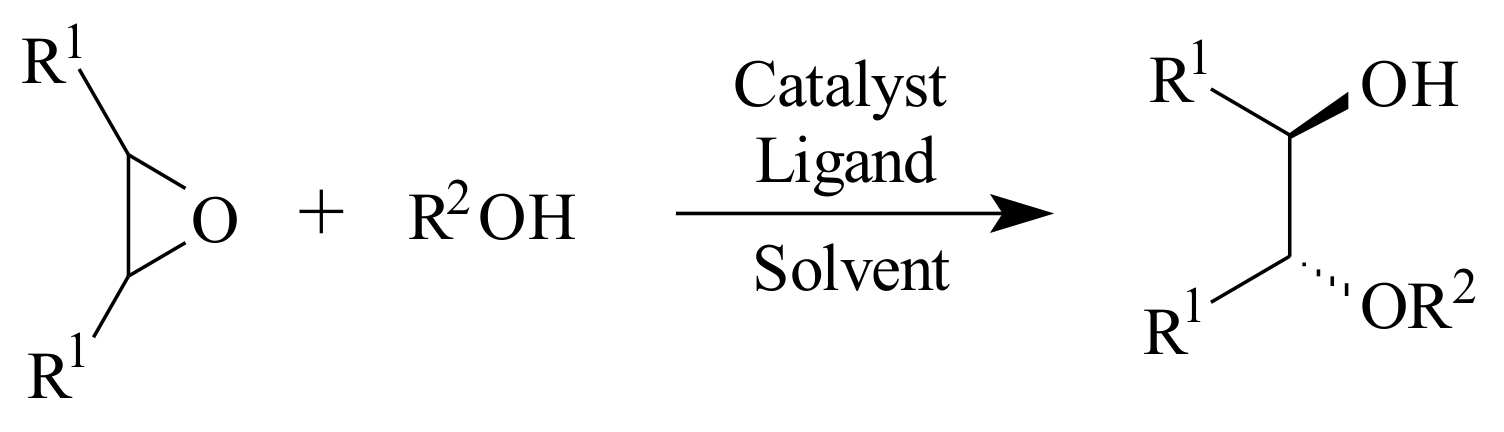{kind=link}
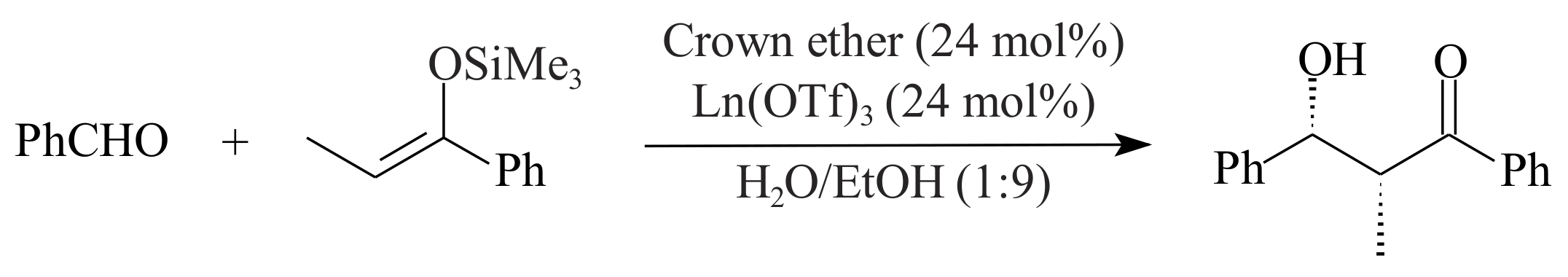{kind=link}
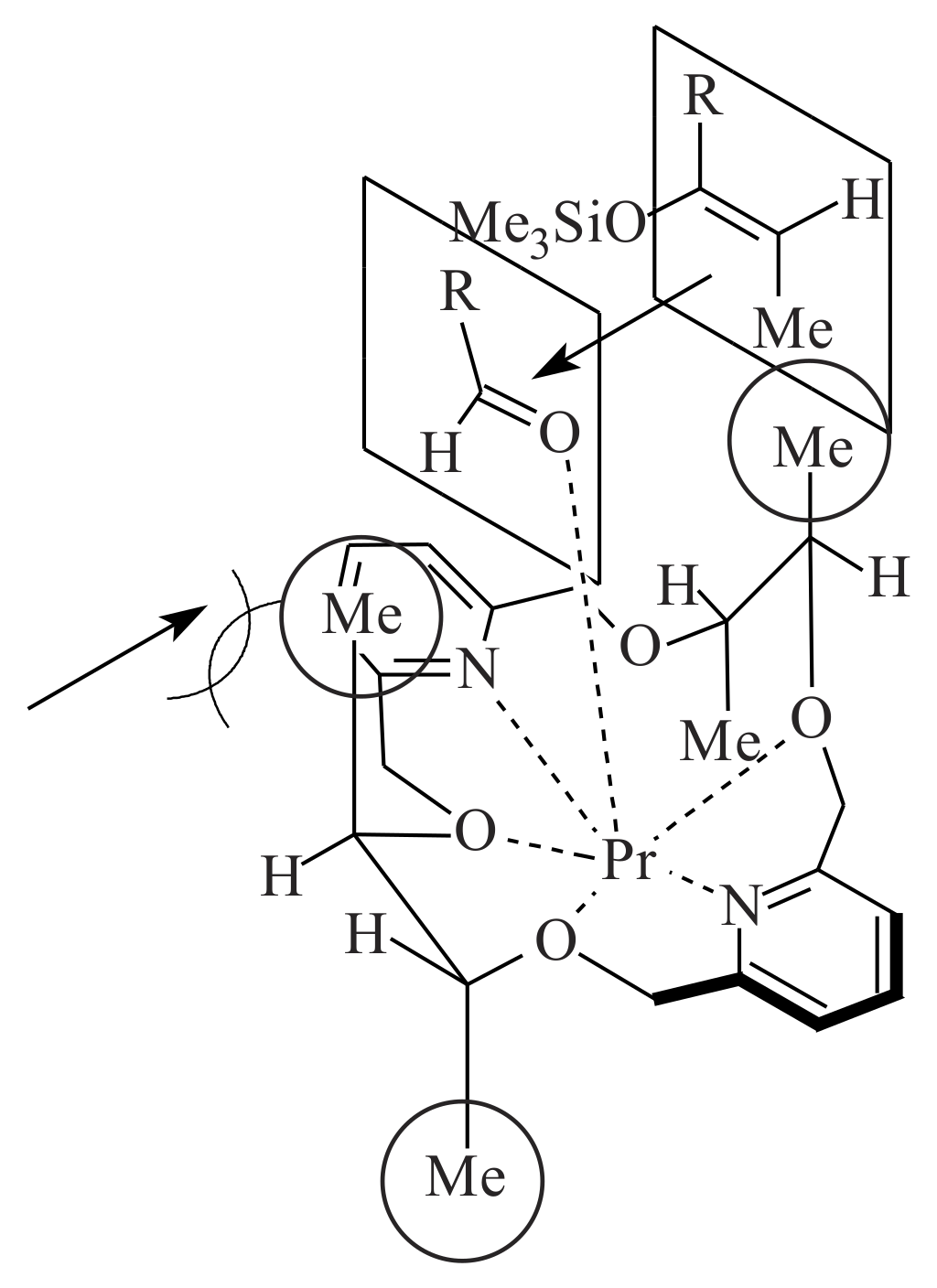{kind=link}
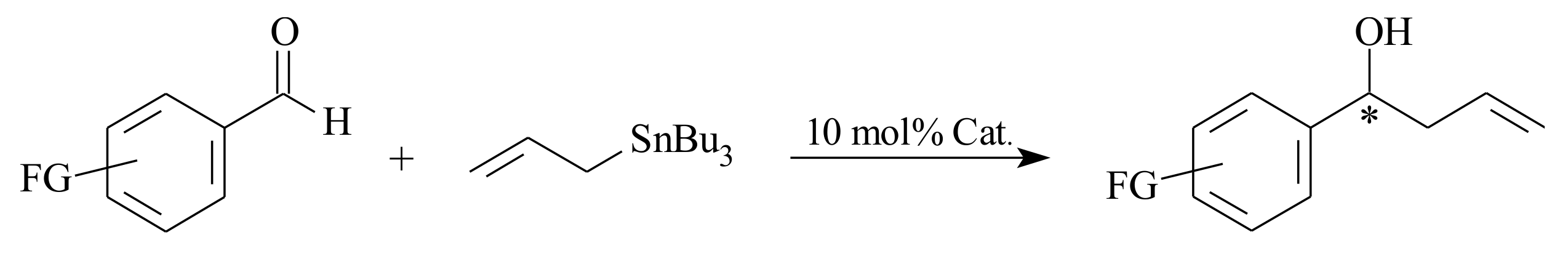{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}