
Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol

 , , ,
, , ,  ,
, 
Abstract

1. Introduction
2. Results
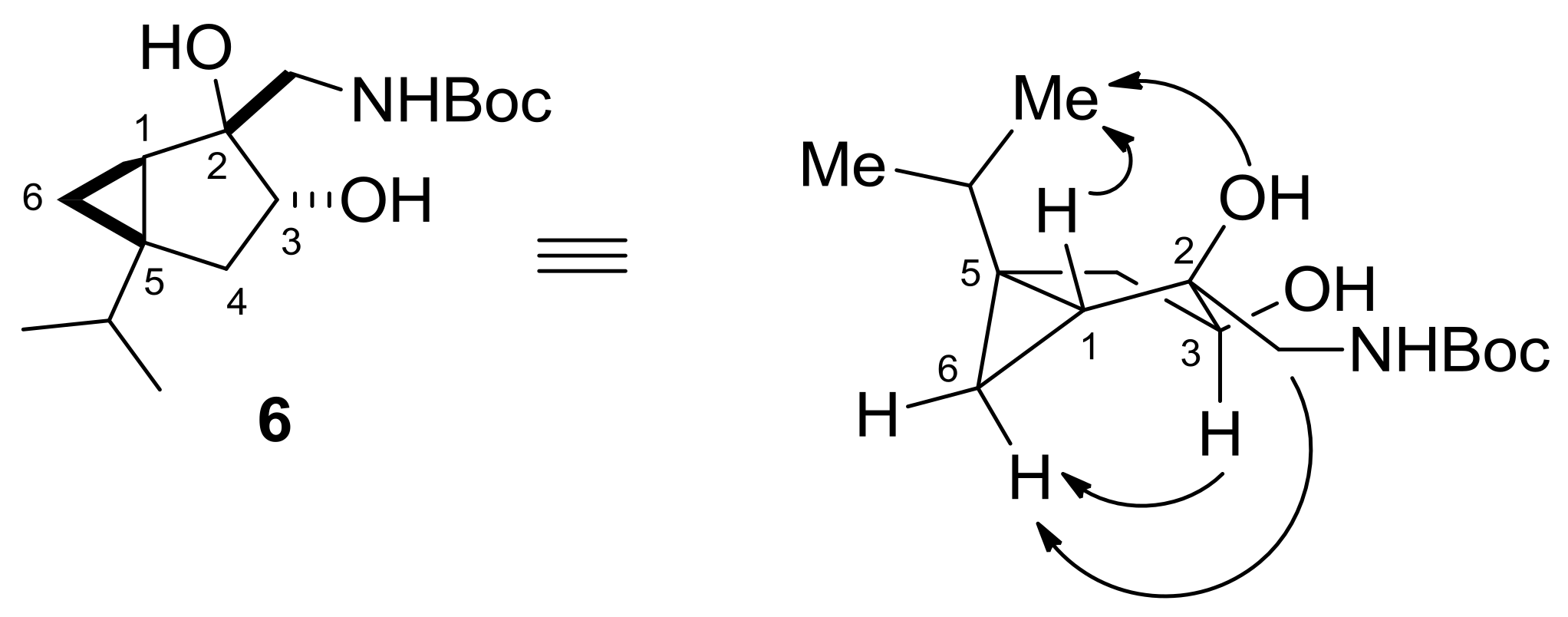
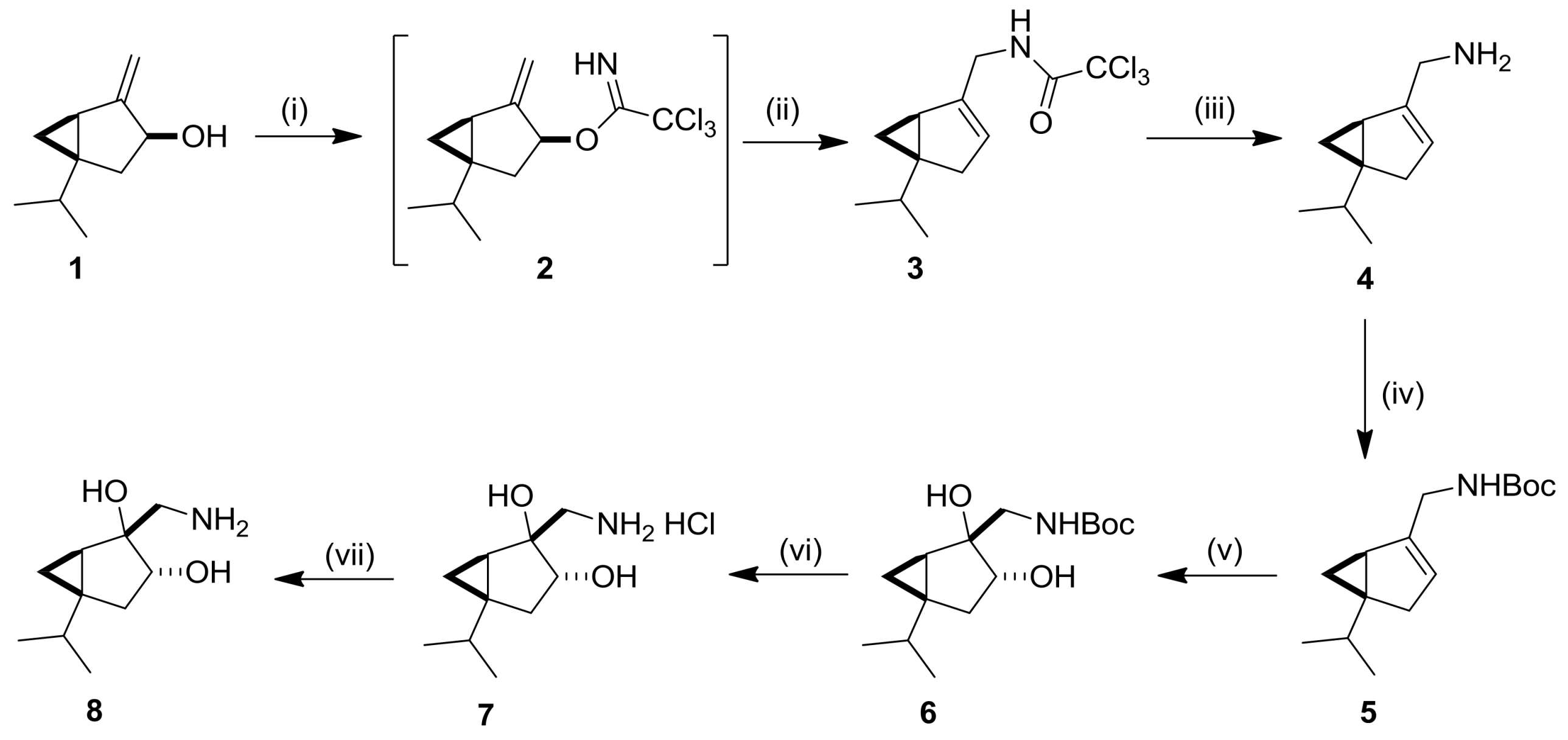
2.1. Synthesis and Transformations of Sabinol-Based 3-Amino-1,2-Diols
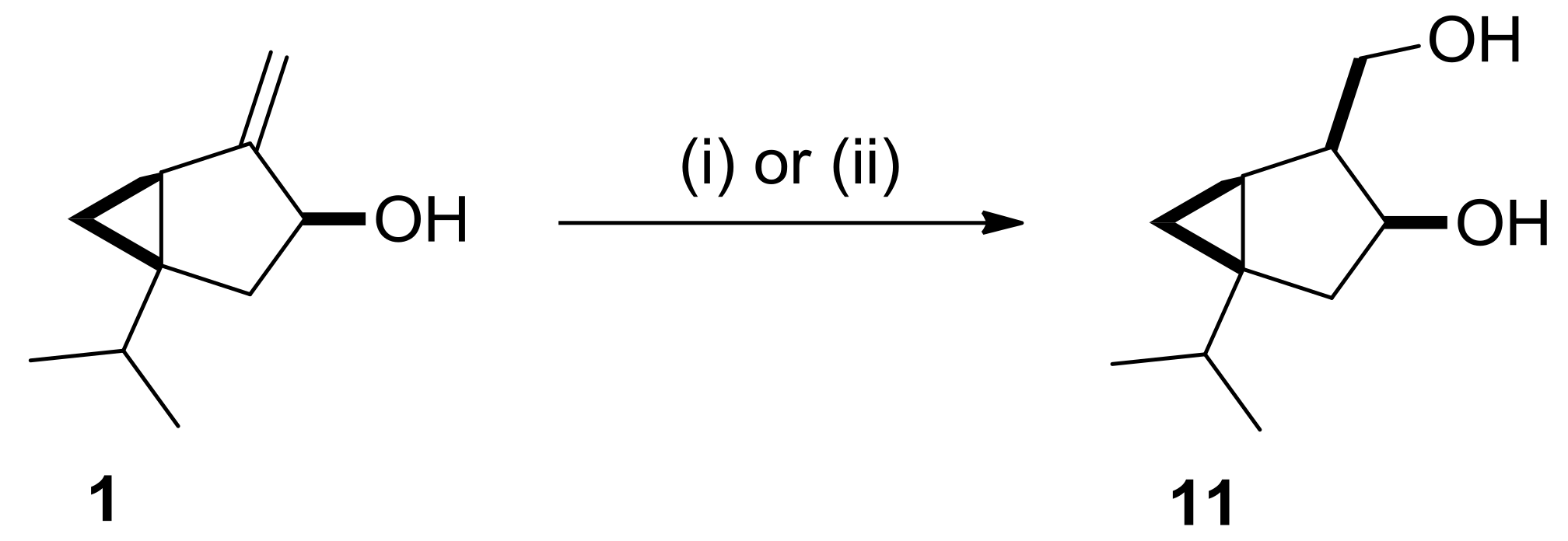
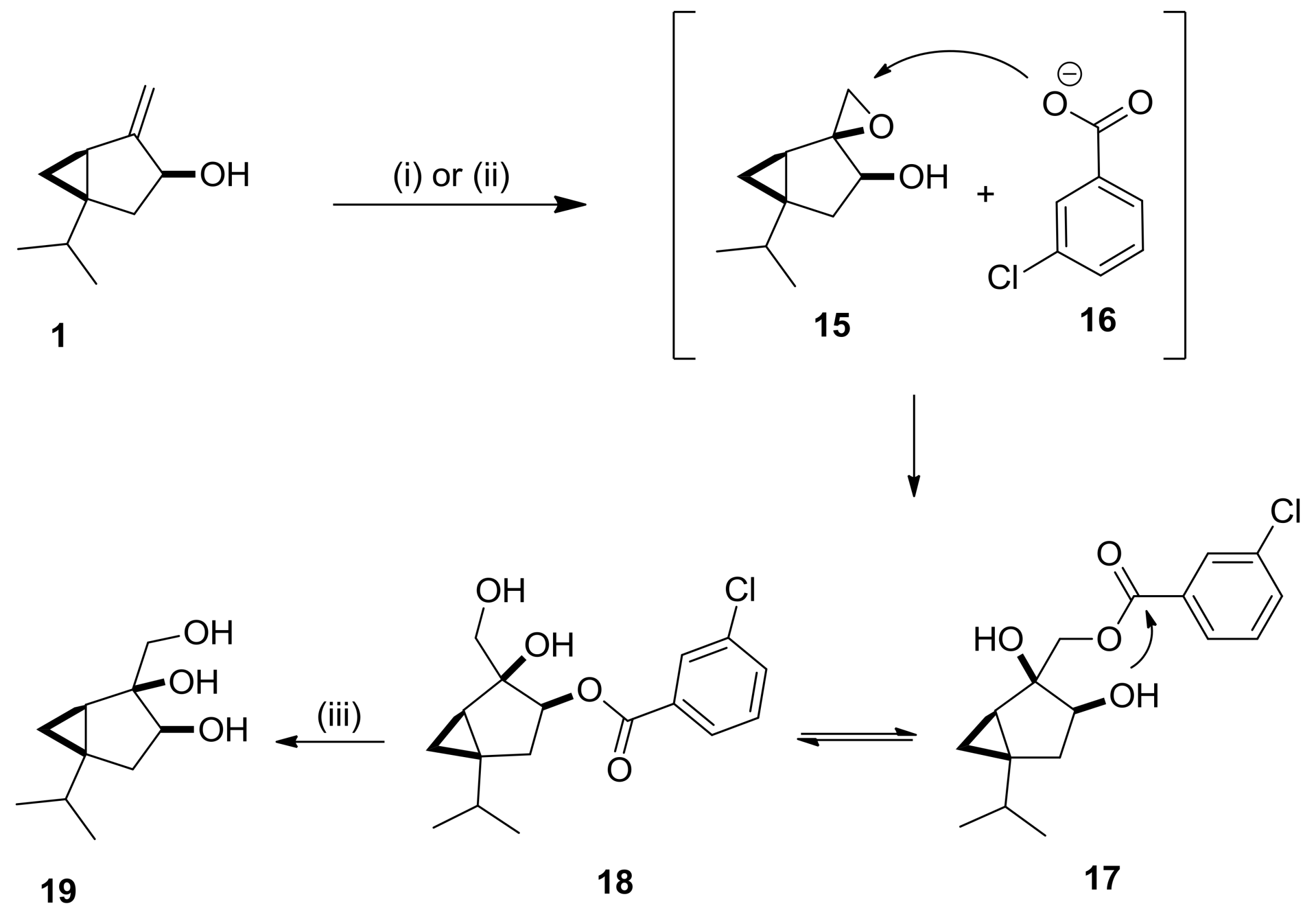
2.2. Synthesis and Transformations of Sabinol-Based Diols and Triols
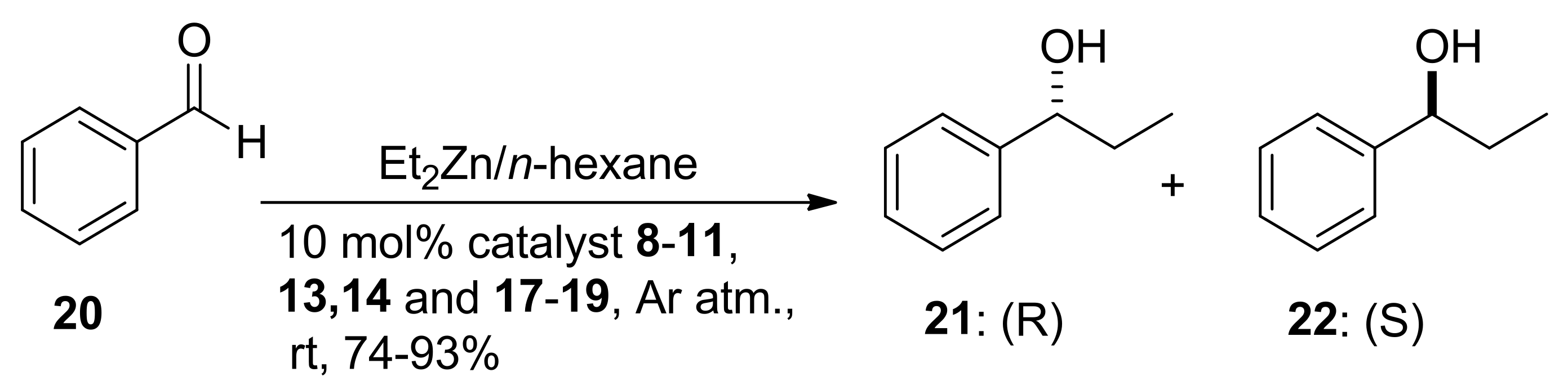
2.3. Application of Sabinol-Based Aminodiol, Diol and Triol Derivatives as Chiral Ligands in the Enantioselective Addition of Diethylzinc to Benzaldehyde
3. Experimental Section
3.1. Materials and Methods
3.2. General Procedure for the Preparation of 13 and 14
3.3. General Procedure for the Preparation of 17 and 18
3.4. General Procedure for the Reaction of Aldehydes with Diethylzinc in the Presence of Chiral Catalyst
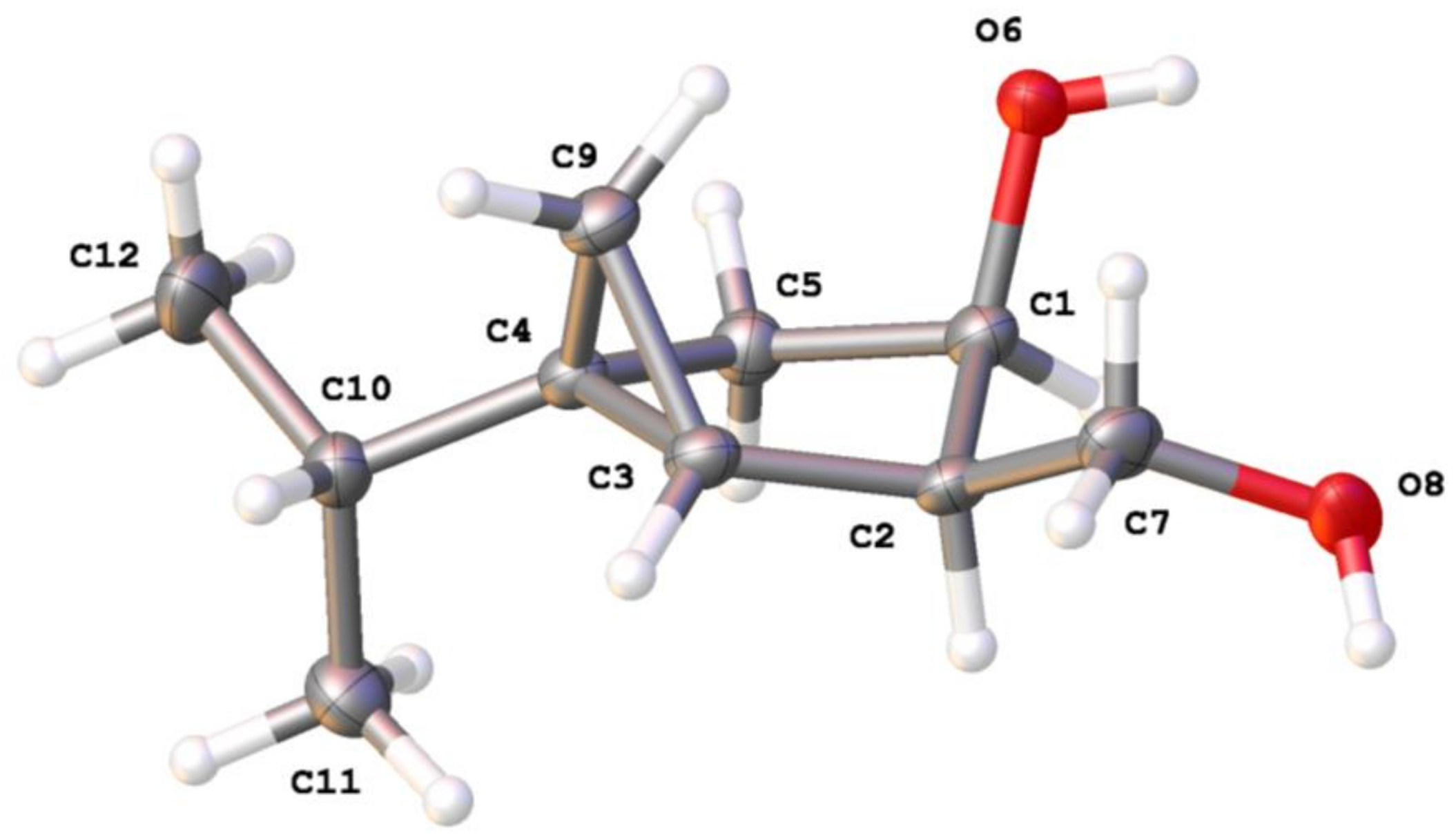
3.5. X-ray Structure Determination of 11
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005; ISBN 978-3-527-30517-9. [Google Scholar]
- Dalko, P.I. (Ed.) Enantioselective Organocatalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; ISBN 978-3-527-61094-5. [Google Scholar]
- Lin, G.-Q.; You, Q.-D.; Cheng, J.-F. (Eds.) Chiral Drugs: Chemistry and Biological Action; Wiley: Hoboken, NJ, USA, 2011; ISBN 978-0-470-58720-1. [Google Scholar]
- Carreira, E.M.; Yamamoto, H. Comprehensive Chirality; Elsevier: Oxford, UK, 2012; ISBN 978-0-08-095168-3. [Google Scholar]
- El Alami, M.S.I.; El Amrani, M.A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chiral Ligands Derived from Monoterpenes: Application in the Synthesis of Optically Pure Secondary Alcohols via Asymmetric Catalysis. Chem. Eur. J. 2015, 21, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Andrés, C.; González, I.; Nieto, J.; Rosón, C.D. Lewis acid mediated diastereoselective keto-ene cyclization on chiral perhydro-1,3-benzoxazines: Synthesis of enantiopure cis-3,4-disubstituted 3-hydroxypyrrolidines. Tetrahedron 2009, 65, 9728–9736. [Google Scholar] [CrossRef]
- Andrés, C.; Infante, R.; Nieto, J. Perhydro-1,3-benzoxazines derived from (−)-8-aminomenthol as ligands for the catalytic enantioselective addition of diethylzinc to aldehydes. Tetrahedron Asymmetry 2010, 21, 2230–2237. [Google Scholar] [CrossRef]
- Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Stereoselective synthesis and application of tridentate aminodiols derived from (+)-pulegone. Tetrahedron Asymmetry 2016, 27, 480–486. [Google Scholar] [CrossRef]
- Hobuß, D.; Hasenjäger, J.; Driessen-Hölscher, B.; Baro, A.; Axenov, K.V.; Laschat, S.; Frey, W. Novel α-pinene-derived mono- and bisphosphinite ligands: Synthesis and application in catalytic hydrogenation. Inorg. Chim. Acta 2011, 374, 94–103. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Balázs, Á.; Martinek, T.A.; Fülöp, F. Enantioselective addition of diethylzinc to aldehydes catalyzed by γ-amino alcohols derived from (+)- and (−)-α-pinene. Tetrahedron Asymmetry 2006, 17, 199–204. [Google Scholar] [CrossRef]
- Csillag, K.; Németh, L.; Martinek, T.A.; Szakonyi, Z.; Fülöp, F. Stereoselective synthesis of pinane-type tridentate aminodiols and their application in the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 2012, 23, 144–150. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Rewucki, P.; Walenczak, S. Sulfur-containing derivatives from (1R)-(−)-myrtenal designed as chiral ligands. Tetrahedron 2016, 72, 3851–3857. [Google Scholar] [CrossRef]
- Yang, J.; Xu, H.; Xu, X.; Rui, J.; Fang, X.; Cao, X.; Wang, S. Synthesis, optical properties, and cellular imaging of novel quinazolin-2-amine nopinone derivatives. Dyes Pigment. 2016, 128, 75–83. [Google Scholar] [CrossRef]
- Yang, J.; Xu, X.; Rui, J.; Wang, Z.; Zhang, Y.; Wang, S.; Wu, L. Synthesis, optical properties and application of a set of novel pyrazole nopinone derivatives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 183, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Dickmu, G.C.; Smoliakova, I.P. Preparation and characterization of cyclopalladated complexes derived from l-(−)-fenchone. J. Organomet. Chem. 2014, 772–773, 42–48. [Google Scholar] [CrossRef]
- De las Casas Engel, T.; Maroto, B.L.; Martínez, A.G.; de la Moya Cerero, S. N/N/O versus N/O/O and N/O amino isoborneols in the enantioselective ethylation of benzaldehyde. Tetrahedron Asymmetry 2008, 19, 269–272. [Google Scholar] [CrossRef]
- Sánchez-Carnerero, E.M.; de las Casas Engel, T.; Maroto, B.L.; de la Moya Cerero, S. Polyoxygenated ketopinic-acid-derived γ-amino alcohols in the enantioselective diethylzinc addition to benzaldehyde. Tetrahedron Asymmetry 2009, 20, 2655–2657. [Google Scholar] [CrossRef]
- García Martínez, A.; Teso Vilar, E.; García Fraile, A.; de la Moya Cerero, S.; Lora Maroto, B. Synthesis and catalytic activity of 10-(aminomethyl)isoborneol-based catalysts: The role of the C(2)-group on the asymmetric induction. Tetrahedron Asymmetry 2003, 14, 1959–1963. [Google Scholar] [CrossRef]
- Stoyanova, M.P.; Shivachev, B.L.; Nikolova, R.P.; Dimitrov, V. Highly efficient synthesis of chiral aminoalcohols and aminodiols with camphane skeleton. Tetrahedron Asymmetry 2013, 24, 1426–1434. [Google Scholar] [CrossRef]
- Lait, S.M.; Rankic, D.A.; Keay, B.A. 1,3-Aminoalcohols and Their Derivatives in Asymmetric Organic Synthesis. Chem. Rev. 2007, 107, 767–796. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, F.; Bernáth, G.; Pihlaja, K. Synthesis, Stereochemistry and Transformations of Cyclopentane-, Cyclohexane-, Cycloheptane-, and Cyclooctane-Fused 1,3-Oxazines, 1,3-Thiazines, and Pyrimidines. Adv. Heterocycl. Chem. 1997, 69, 349–477. [Google Scholar]
- Lázár, L.; Fülöp, F. 1,3-Oxazines and their Benzo Derivatives. Compr. Heterocycl. Chem. III 2008, 373–459. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Hetényi, A.; Fülöp, F. Synthesis of enantiomeric spirooxazolines and spirooxazolidines by the regioselective ring closure of (–)-α-pinene-based aminodiols. Arkivoc 2007, 33–42. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csillag, K.; Fülöp, F. Stereoselective synthesis of carane-based aminodiols as chiral ligands for the catalytic addition of diethylzinc to aldehydes. Tetrahedron Asymmetry 2011, 22, 1021–1027. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Stereoselective Synthesis and Modelling-Driven Optimisation of Carane-Based Aminodiols and 1,3-Oxazines as Catalysts for the Enantioselective Addition of Diethylzinc to Benzaldehyde. Chem. Eur. J. 2016, 22, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, H.; Rosenberg, S.; Baker, W.; Stein, H.; Klinghofer, V.; Barlow, J.; Spina, K.; Polakowski, J.; Kovar, P.; Cohen, J.; et al. Discovery of a peptide-based renin inhibitor with oral bioavailability and efficacy. Science 1992, 257, 1940–1943. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Figueroa, S.; Greenhouse, R.; Padilla, F.; Dillon, M.P.; Gever, J.R.; Ford, A.P.D.W. Discovery and synthesis of a novel and selective drug-like P2X1 antagonist. Bioorg. Med. Chem. Lett. 2005, 15, 3292–3295. [Google Scholar] [CrossRef] [PubMed]
- Grajewska, A.; Rozwadowska, M.D. Stereoselective synthesis of cytoxazone and its analogues. Tetrahedron Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Narina, S.V.; Kumar, T.S.; George, S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone via Rh-catalyzed diastereoselective oxidative C–H aminations. Tetrahedron Lett. 2007, 48, 65–68. [Google Scholar] [CrossRef]
- Kakeya, H.; Morishita, M.; Koshino, H.; Morita, T.; Kobayashi, K.; Osada, H. Cytoxazone: A Novel Cytokine Modulator Containing a 2-Oxazolidinone Ring Produced by Streptomyces sp. J. Org. Chem. 1999, 64, 1052–1053. [Google Scholar] [CrossRef]
- Paraskar, A.S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone, novel cytokine modulators via Sharpless asymmetric epoxidation and l-proline catalyzed Mannich reaction. Tetrahedron 2006, 62, 5756–5762. [Google Scholar] [CrossRef]
- Zhu, W.; Burnette, A.; Dorjsuren, D.; Roberts, P.E.; Huleihel, M.; Shoemaker, R.H.; Marquez, V.E.; Agbaria, R.; Sei, S. Potent Antiviral Activity of North-Methanocarbathymidine against Kaposi’s Sarcoma-Associated Herpesvirus. Antimicrob. Agents Chemother. 2005, 49, 4965–4973. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roy, C.D.; Brown, H.C. A Study of Transesterification of Chiral (−)-Pinanediol Methylboronic Ester with Various Structurally Modified Diols. Monatshefte Für Chem. Chem. Mon. 2007, 138, 747–753. [Google Scholar] [CrossRef]
- Cherng, Y.-J.; Fang, J.-M.; Lu, T.-J. Pinane-Type Tridentate Reagents for Enantioselective Reactions: Reduction of Ketones and Addition of Diethylzinc to Aldehydes. J. Org. Chem. 1999, 64, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Ardashov, O.V.; Pavlova, A.V.; Il’ina, I.V.; Morozova, E.A.; Korchagina, D.V.; Karpova, E.V.; Volcho, K.P.; Tolstikova, T.G.; Salakhutdinov, N.F. Highly Potent Activity of (1R,2R,6S)-3-Methyl-6-(prop-1-en-2-yl)cyclohex-3-ene-1,2-diol in Animal Models of Parkinson’s Disease. J. Med. Chem. 2011, 54, 3866–3874. [Google Scholar] [CrossRef] [PubMed]
- Radulović, N.S.; Mladenović, M.Z.; Randjelovic, P.J.; Stojanović, N.M.; Dekić, M.S.; Blagojević, P.D. Toxic essential oils. Part IV: The essential oil of Achillea falcata L. as a source of biologically/pharmacologically active trans-sabinyl esters. Food Chem. Toxicol. 2015, 80, 114–129. [Google Scholar] [CrossRef]
- Tuberoso, C.I.G.; Kowalczyk, A.; Coroneo, V.; Russo, M.T.; Dessì, S.; Cabras, P. Chemical Composition and Antioxidant, Antimicrobial, and Antifungal Activities of the Essential Oil of Achillea ligustica All. J. Agric. Food Chem. 2005, 53, 10148–10153. [Google Scholar] [CrossRef] [PubMed]
- Rudloff, E.V. Gas–Liquid Chromatography of Terpenes: Part ix. the Volatile Oil of the Leaves of Juniperus Sabina L. Can. J. Chem. 1963, 41, 2876–2881. [Google Scholar] [CrossRef]
- Banthorpe, D.; Davies, H.; Gatford, C.; Williams, S. Monoterpene Patterns in Juniperus and Thuja Species. Planta Med. 1973, 23, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.; Pages, N.; Fournier, C.; Callen, G. Contribution to the Study of the Essential Oil of Various Cultivars of Juniperus sabina. Planta Med. 1991, 57, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Suleimenov, E.M.; Raldugin, V.A.; Shakirov, M.M.; Bagryanskaya, I.Y.; Gatilov, Y.V.; Kulyjiasov, A.T.; Adekenov, S.M. [4+2]-Cyclodimer of sabinone: Formation, crystal structure, and NMR spectra. Russ. Chem. Bull. 2003, 52, 1210–1212. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Kattanguru, P.; Gholap, S.P.; Chaudhari, D.A. Recent advances in the Overman rearrangement: Synthesis of natural products and valuable compounds. Org. Biomol. Chem. 2017, 15, 2672–2710. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Martínez, E.; Ayala-Mata, F.; Velázquez-Ponce, P.; Medina, M.E.; Jiménez-Vazquez, H.A.; Joseph-Nathan, P.; Zepeda, L.G. Nucleophilic additions on acetyldioxanes derived from (−)-(1R)-myrtenal used as chiral auxiliaries: Substituent effects on the stereochemical outcome. Tetrahedron Asymmetry 2017, 28, 1350–1358. [Google Scholar] [CrossRef]
- Chen, X.; Gu, W.; Jing, X.; Pan, X. A New Approach for Synthesis of Erythro 8-O-4′ Neolignans. Synth. Commun. 2002, 32, 557–564. [Google Scholar] [CrossRef]
- Fang, W.; Wei, Y.; Tang, X.-Y.; Shi, M. Gold(I)-Catalyzed Cycloisomerization of ortho-(Propargyloxy)arenemethylenecyclopropanes Controlled by Adjacent Substituents at Aromatic Rings. Chem. Eur. J. 2017, 23, 6845–6852. [Google Scholar] [CrossRef] [PubMed]
- Hurlocker, B.; Hu, C.; Woerpel, K.A. Structure and Reactivity of an Isolable Seven-Membered-Ring trans-Alkene. Angew. Chem. Int. Ed. 2015, 54, 4295–4298. [Google Scholar] [CrossRef] [PubMed]
- Garside, P.; Halsall, T.G.; Hornby, G.M. Action of peracetic acid on (+)-sabinol. J. Chem. Soc. C Org. 1969, 716–721. [Google Scholar] [CrossRef]
- CrysAlis PRO, Agilent Technologies UK Ltd.: Oxfordshire, UK, 2012.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 3–17 are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst (10 mol %) | Yield 1 (%) | ee 2 (%) | Config. of Major Product 3 |
|---|---|---|---|---|
| 1 | 8 | 88 | 44 | S |
| 2 | 9 | 98 | 66 | S |
| 3 | 10 | 98 | 58 | S |
| 4 | 11 | 73 | 6 | S |
| 5 | 13 | 88 | 12 | S |
| 6 | 14 | 87 | 17 | S |
| 7 | 17 | 73 | 9 | S |
| 8 | 18 | 95 | 4 | S |
| 9 | 19 | 81 | 8 | S |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashenov, Y.; Daniels, M.; Robeyns, K.; Van Meervelt, L.; Dehaen, W.; Suleimen, Y.M.; Szakonyi, Z. Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol. Molecules 2018, 23, 771. https://doi.org/10.3390/molecules23040771
Tashenov Y, Daniels M, Robeyns K, Van Meervelt L, Dehaen W, Suleimen YM, Szakonyi Z. Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol. Molecules. 2018; 23(4):771. https://doi.org/10.3390/molecules23040771
Chicago/Turabian StyleTashenov, Yerbolat, Mathias Daniels, Koen Robeyns, Luc Van Meervelt, Wim Dehaen, Yerlan M. Suleimen, and Zsolt Szakonyi. 2018. "Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol" Molecules 23, no. 4: 771. https://doi.org/10.3390/molecules23040771
APA StyleTashenov, Y., Daniels, M., Robeyns, K., Van Meervelt, L., Dehaen, W., Suleimen, Y. M., & Szakonyi, Z. (2018). Stereoselective Syntheses and Application of Chiral Bi- and Tridentate Ligands Derived from (+)-Sabinol. Molecules, 23(4), 771. https://doi.org/10.3390/molecules23040771