
Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors


 ,
,  ,
, 
Abstract
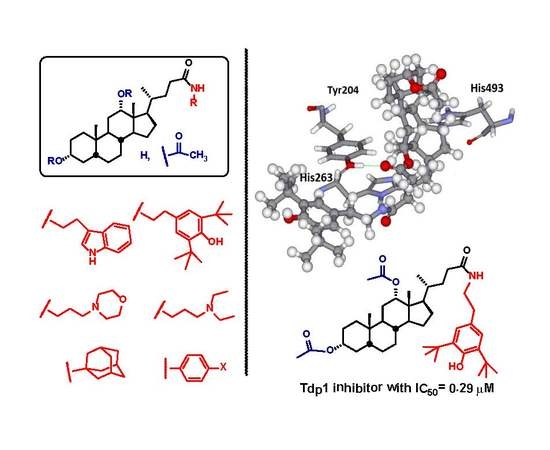
1. Introduction
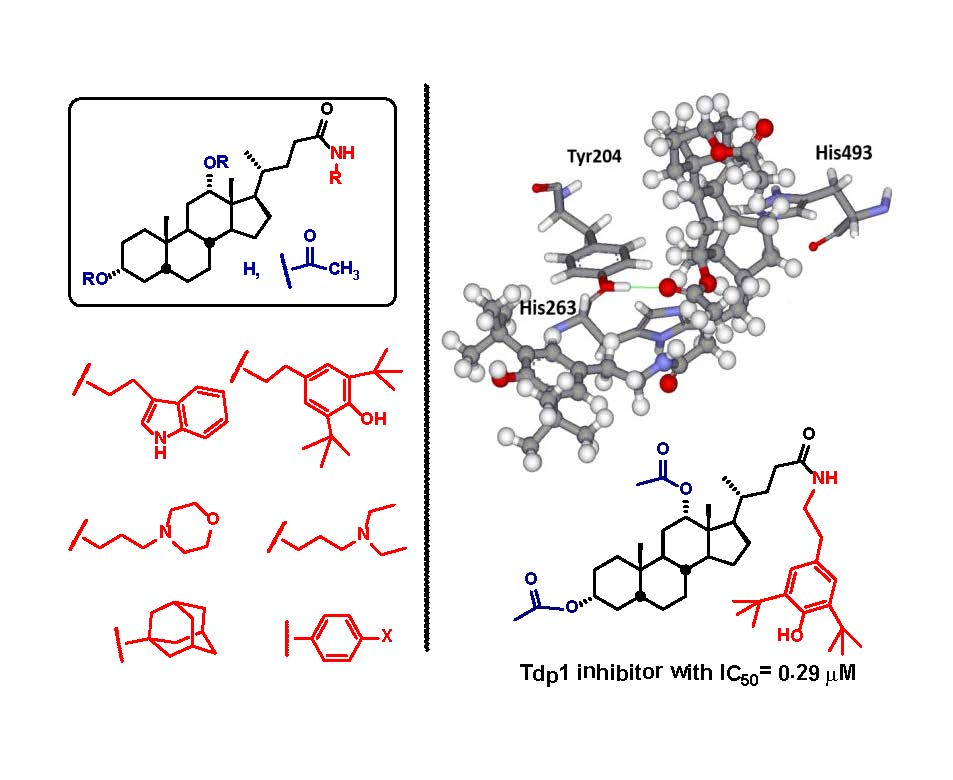
2. Results and Discussion
2.1. Virtual Screening
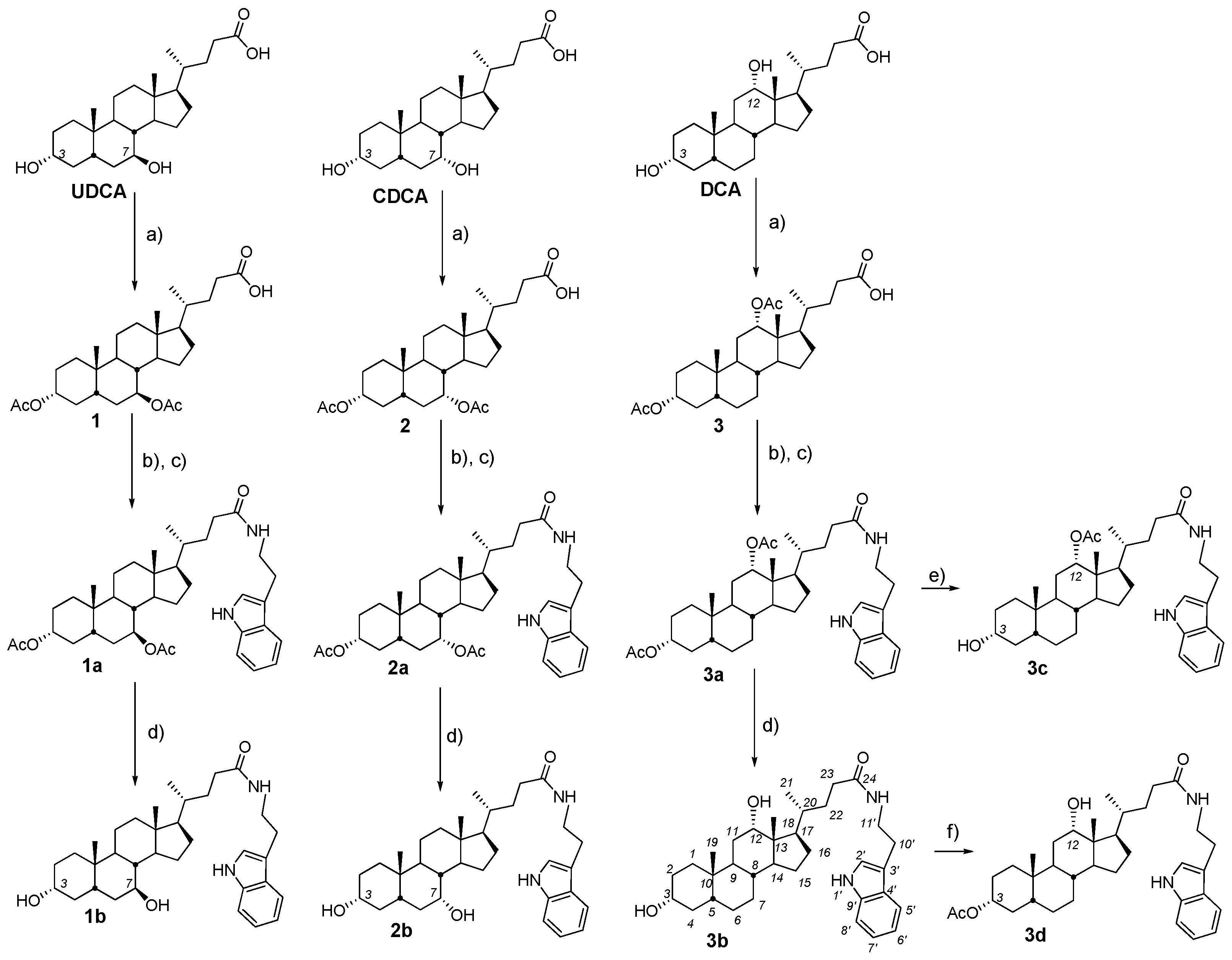
2.2. Synthesis and Testing of Bile Acids Tryptamides
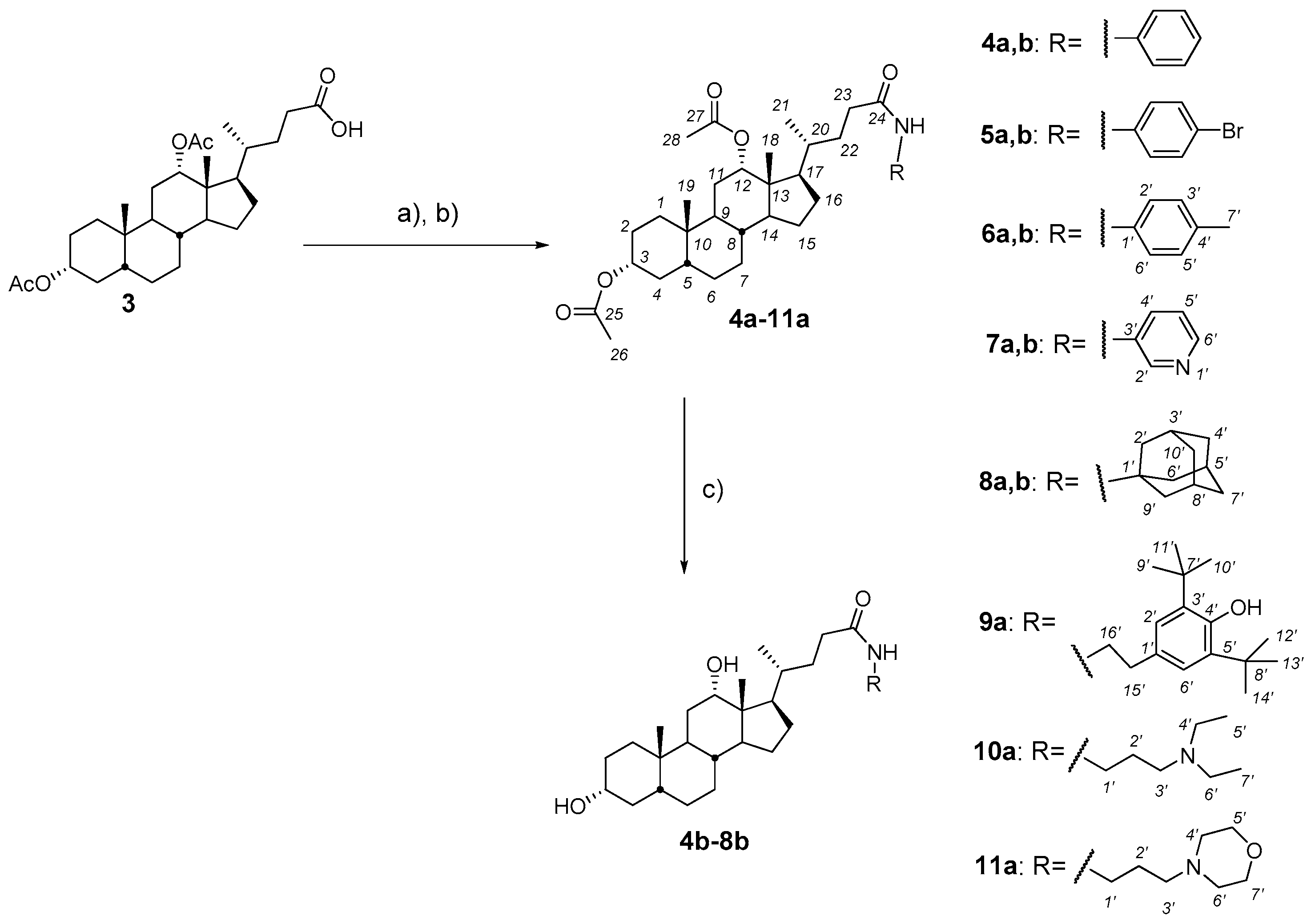
2.3. Synthesis and Testing of Deoxycholic Acid Amides
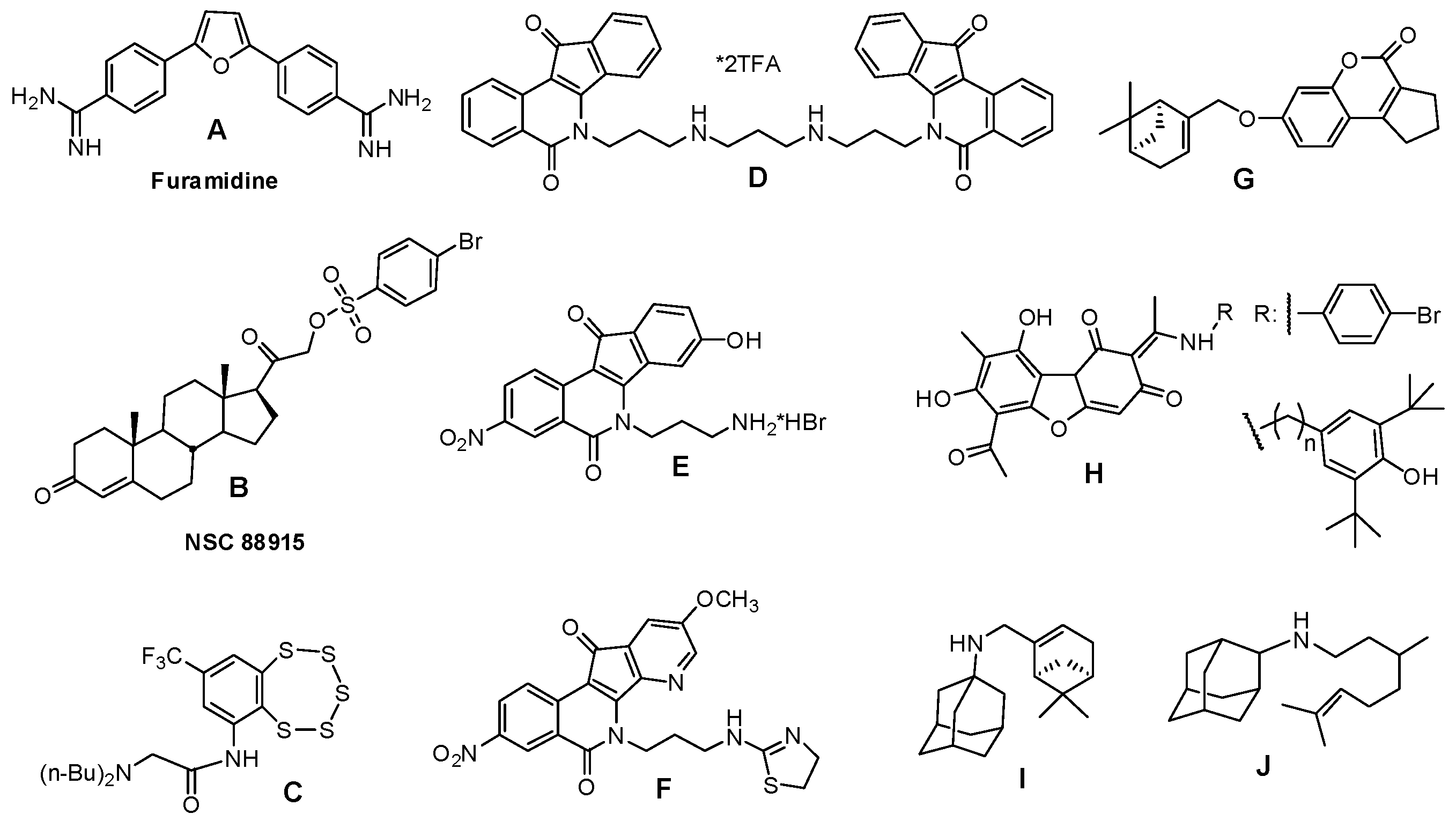
2.4. Molecular Modeling
3. Materials and Methods
3.1. Chemicals and Reagents
- (a)
- General procedures for compound 1a–11a
- (1)
- Oxalyl chloride (6.0 equiv.) and a few drops DMF were added at 0 °C to a solution of diacetoxy bile acid (1–3) (1.0 equiv.), correspondently, in dry CH2Cl2. The reaction mixture was stirred for a further 3 h at 0–5 °C, diluted with benzene and concentrated in vacuum. Then CH2Cl2.was added to the reaction mixture to give a solution of bile acid chloride.
- (2)
- The resulting solution bile acid chloride (1.0 equiv.) was added dropwise at 0 °C to a solution of appropriate amine (1.2 equiv.) and NEt3 (1.5 equiv.) in dry CH2Cl2. The reaction mixture was stirred for a further 18 h at room temperature, diluted with AcOEt (20 mL) and H2O was added. The organic layer was separated and the aqueous layer was extracted with AcOEt (2 × 30 mL). The combined organic layers were washed with brine and dried over calcined MgSO4. The solvent was removed to give an amorphous solid.
- (b)
- General procedures for compound 1b–8bA solution of the appropriate amide derivatives (1a–8a) (1 equiv.)—containing acetoxy groups in the steroid framework—in MeOH was treated with KOH (6 equiv.), refluxed for 2–6 h (monitored by TLC), concentrated under reduced pressure, diluted with H2O (up to ~50–60 mL), acidified with 5% HCl solution to pH 3–4, and extracted with mixture CH2Cl2Et2O (1:3 v/v) (3 × 30 mL). The combined organic layers were washed with a saturated solution of NaHCO3, brine and dried over calcined MgSO4. The solvent was removed to give an amorphous solid.
3.1.1. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,7β-diacetoxy-5β-cholan-24-amide (1a)
3.1.2. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,7β-dihydroxy-5β-cholan-24-amide (1b)
3.1.3. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,7a-diacetoxy-5β-cholan-24-amide (2a)
3.1.4. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,7α-dihydroxy-5β-cholan-24-amide (2b).
3.1.5. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,12a-diacetoxy-5β-cholan-24-amide (3a)
3.1.6. N-(2′-(1H-Indol-2-yl)-ethyl)-3α,12α-dihydroxy-5β-cholan-24-amide (3b)
3.1.7. N-(2′-(1H-Indol-2-yl)-ethyl)-3α-hydroxy-12α-acetoxy-5β-cholan-24-amide (3c)
3.1.8. N-(2′-(1H-Indol-2-yl)-ethyl)-3α-acetoxy-12α-hydroxy-5β-cholan-24-amide (3d)
3.1.9. N-Phenyl-3α,12α-diacetoxy-5β-cholan-24-amide (4a)
3.1.10. N-Phenyl-3α,12α-dihydroxy-5β-cholan-24-amide (4b)
3.1.11. N-(4′-Bromophenyl)-3α,12α-diacetoxy-5β-cholan-24-amide (5a)
3.1.12. N-(4′-Bromophenyl)-3α,12α-dihydroxy-5β-cholan-24-amide (5b)
3.1.13. N-(p-Tolyl)-3α,12α-diacetoxy-5β-cholan-24-amide (6a)
3.1.14. N-(p-Tolyl)-3α,12α-dihydroxy-5β-cholan-24-amide (6b)
3.1.15. N-(Pyridin-3-yl)-3α,12α-diacetoxy-5β-cholan-24-amide (7a)
3.1.16. N-(Pyridin-3-yl)-3α,12α-dihydroxy-5β-cholan-24-amide (7b)
3.1.17. N-(1′-Adamantyl)-3α,12α-diacetoxy-5β-cholan-24-amide (8a)
3.1.18. N-(1′-Adamantyl)-3α,12α-dihidroxy-5β-cholan-24-amide (8b)
3.1.19. N-(2′′-(3′,5′-Di-tret-buthyl-4′-hydroxyphenyl)-ethyl)-3α,12α-diacetoxy-5β-cholan-24-amide (9a)
3.1.20. N-(3′,3′-Diethylaminopropyl)-3α,12α-diacetoxy-5β-cholan-24-amide (10a)
3.1.21. N-(3′-Morpholinopropyl)-3α,12α-diacetoxy-5β-cholan-24-amide (11a).
3.2. Tdp1 Assay
3.3. Analysis of Cytotoxicity
3.4. Virtual Screening and Molecular Modelling
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, J.Y.D.; Su, P.; Barber, J.E.M.; Nash, J.E.; Le, A.D.; Liu, F.; Wong, A.H.C. The neuroprotective effect of nicotine in Parkinson’s disease models is associated with inhibiting PARP-1 and caspase-3 cleavage. PeerJ 2017, 5, e3933. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.S.; Antony, S.; Marchand, C.; Pommier, Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anticancer Agents Med. Chem. 2008, 8, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Laev, S.S.; Salakhutdinov, N.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Jdey, W.; Thierry, S.; Russo, C.; Devun, F.; Al Abo, M.; Noguiez-Hellin, P.; Sun, J.S.; Barillot, E.; Zinovyev, A.; Kuperstein, I.; et al. Drug-Driven Synthetic Lethality: Bypassing Tumor Cell Genetics with a Combination of AsiDNA and PARP Inhibitors. Clin. Cancer Res. 2017, 23, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.N.; Pommier, Y.; Marchand, C. Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin. Ther. Pat. 2011, 21, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Cossa, G.; Gatti, L.; Zunino, F.; Perego, P. Tyrosyl-DNA phosphodiesterase 1 targeting for modulation of camptothecin-based treatment. Curr. Med. Chem. 2010, 17, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Kawale, A.S.; Povirk, L.F. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018, 46, 520–537. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.X.; Morrell, A.; Conda-Sheridan, M.; Marchand, C.; Agama, K.; Bermingham, A.; Stephen, A.G.; Chergui, A.; Naumova, A.; Fisher, R.; et al. Synthesis and biological evaluation of the first dual tyrosyl-DNA phosphodiesterase I (Tdp1)-topoisomerase I (Top1) inhibitors. J. Med. Chem. 2012, 55, 4457–4478. [Google Scholar] [CrossRef] [PubMed]
- Borda, M.A.; Palmitelli, M.; Verón, G.; González-Cid, M.; de Campos Nebel, M. Tyrosyl-DNA-phosphodiesterase I (TDP1) participates in the removal and repair of stabilized-Top2α cleavage complexes in human cells. Mutat. Res. 2015, 781, 37–48. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Katyalm, S.; Patel, P.; Ju, L.; McKinnon, P.J.; Caldecott, K.W. Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair 2009, 8, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Antony, S.; Gupta, S.; Dexheimer, T.S.; Redon, C.E.; Garfield, S.; Shiloh, Y.; Pommier, Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [Google Scholar] [CrossRef] [PubMed]
- Katyal, S.; El-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef] [PubMed]
- Hirano, R.; Interthal, H.; Huang, C.; Nakamura, T.; Deguchi, K.; Choi, K.; Bhattacharjee, M.B.; Arimura, K.; Umehara, F.; Izumo, S.; et al. Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation. EMBO J. 2007, 26, 4732–4743. [Google Scholar] [CrossRef] [PubMed]
- Barthelmes, H.U.; Habermeyer, M.; Christensen, M.O.; Mielke, C.; Interthal, H.; Pouliot, J.J.; Boege, F.; Marko, D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J. Biol. Chem. 2004, 279, 55618–55625. [Google Scholar] [CrossRef] [PubMed]
- Nivens, M.C.; Felder, T.; Galloway, A.H.; Pena, M.M.; Pouliot, J.J.; Spencer, H.T. Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-I and thymidylate synthase. Cancer Chemother. Pharmacol. 2004, 53, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, M.; Wells, O.S.; El-Khamisy, S.F. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2014, 42, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Dexheimer, T.S.; Takeda, S.; Pommier, Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 2012, 287, 12848–12857. [Google Scholar] [CrossRef] [PubMed]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Comeaux, E.; van Waardenburg, R.C. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev. 2014, 46, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Antony, S.; Marchand, C.; Stephen, A.; Thibaut, L.; Agama, K.; Fisher, R.; Pommier, Y. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl–DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007, 35, 4474–4484. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.; Gediya, L.; Stephen, A.; Weidlich, I.; Antony, S.; Marchand, C.; Interthal, H.; Niklaus, M.; Fisher, R.; Njar, V.; et al. 4-Pregnen-21-ol-3,20-dione-21-(4-bromobenzenesufonate) (NSC 88915) and related novel steroid derivatives as tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors. J. Med. Chem. 2009, 52, 7122–7131. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Khomenko, T.; Zhukova, S.; Koval, O.; Zakharova, O.; Anarbaev, R.; Lebedeva, N.; Korchagina, D.; Komarova, N.; Vasiliev, V. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg. Med. Chem. 2015, 9, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.X.; Abdelmalak, M.; Marchand, C.; Agama, K.; Pommier, Y.; Cushman, M. Synthesis and biological evaluation of nitrated 7-, 8-, 9-, and 10-hydroxyindenoisoquinolines as potential dual topoisomerase I (Top1)-tyrosyl-DNA phosphodiesterase I (TDP1) inhibitors. J. Med. Chem. 2015, 58, 3188–3208. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Elsayed, M.; Plescia, C.; Ravji, A.; Redon, C.; Kiselev, E.; Marchand, C.; Zeleznik, O.; Agama, K.; Pommier, Y.; et al. Synthesis and biological evaluation of the first triple inhibitors of human topoisomerase 1, tyrosyl-DNA phosphodiesterase 1 (Tdp1), and tyrosyl-DNA phosphodiesterase 2 (Tdp2). J. Med. Chem. 2017, 60, 3275–3288. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Luzina, O.; Koval, O.; Nilov, D.; Gushchina, I.; Dyrkheeva, N.; ˇSvedas, V.; Salakhutdinov, N.; Lavrik, O. Tyrosyl-DNA phosphodiesterase 1 inhibitors: Usnic acid enamines enhance the cytotoxic effect of camptothecin. J. Nat. Prod. 2016, 79, 2961–2967. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, K.Y.; Suslov, E.V.; Zakharenko, A.L.; Zakharova, O.D.; Rogachev, A.D.; Korchagina, D.V.; Zafar, A.; Reynisson, J.; Nefedov, A.; Volcho, K.P.; et al. Aminoadamantanes Containing Monoterpene-derived Fragments as Potent Tyrosyl-DNA phosphodiesterase 1 Inhibitors. Bioorg. Chem. 2018, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G.J. Crystal Structure of a Transition State Mimic for Tdp1 Assembled from Vanadate, DNA, and a Topoisomerase I-Derived Peptide. Chem. Biol. 2003, 10, 139–147. [Google Scholar] [CrossRef]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G.J. Insights into Substrate Binding and Catalytic Mechanism of Human Tyrosyl-DNA Phosphodiesterase (Tdp1) from Vanadate and Tungstate-inhibited Structures. J. Mol. Biol. 2002, 324, 917–932. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational Design of Nucleoside–Bile Acid Conjugates Incorporating a Triazole Moiety for Anticancer Evaluation and SAR Exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, T.; Cheng, D.; Chu, C.; Tong, S.; Yan, J.; Li, Q.-Y. Synthesis and Biological Activity of Some Bile Acid-Based Camptothecin Analogues. Molecules 2014, 19, 3761–3776. [Google Scholar] [CrossRef] [PubMed]
- Popadyuk, I.I.; Salomatina, O.V.; Salakhutdinov, N.F. Modern approaches to modification of bile acids for the synthesis of compounds possessing valuable physicochemical and biological properties. Russ. Chem. Rev. 2017, 86, 388–443. [Google Scholar] [CrossRef]
- Sharma, R.; Long, A.; Gilmer, J.F. Advances in bile acid medicinal chemistry. Curr. Med. Chem. 2011, 18, 4029–4052. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P. Bile Acid Scaffolds in Supramolecular Chemistry: The Interplay of Design and Synthesis. Molecules 2007, 12, 2106–2122. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Logan, G.; Reynisson, J. Wine Compounds as a Source for HTS Screening Collections. A Feasibility Study. Mol. Inf. 2012, 31, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Brossard, D.; Lechevrel, M.; El Kihel, L.; Quesnelle, C.; Khalid, M.; Moslemi, S.; Reimund, J.M. Synthesis and biological evaluation of bile carboxamide derivatives with pro-apoptotic effect on human colon adenocarcinoma cell lines. Eur. J. Med. Chem. 2014, 86, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Interthal, H.; Pouliott, J.J.; Champoux, J.J. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc. Natl. Acad. Sci. USA 2001, 98, 12009–12014. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; Lavrik, O.I. AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett. 2011, 585, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Fijitsu Limited. SciGress Ultra v. F.J 2.6; Fijitsu Limited: Tokyo, Japan, 2000. [Google Scholar]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible Docking Using Tabu Search and an Empirical Estimate of Binding Affinity. Proteins Struct. Funct. Bioinform. 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins Struct. Funct. Bioinform. 2005, 61, 272–287. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon-Marvin. Version 15.7.20.0. 2015. Available online: http://www.chemaxon.com (accessed on 16 March 2018).
Sample Availability: Samples of the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 1 μM | Compound | IC50 1 μM |
|---|---|---|---|
| 1a | 0.32 ± 0.11 | 1b | 2.65 ± 0.3 |
| 2a | 0.38 ± 0.12 | 2b | 2.6 ± 0.4 |
| 3a | 0.65 ± 0.16 | 3b | 2.7 ± 0.2 |
| 3c | 0.95 ± 0.05 | 3d | 0.48 ± 0.04 |
| Fur 2 | 1.2 ± 0.3 |
| Compound | IC50 1 μM | Compound | IC50 1 μM |
|---|---|---|---|
| 4a | 0.43 ± 0.13 | 4b | 6.7 ± 0.7 |
| 5a | 0.42 ± 0.01 | 5b | 1.3 ± 0.2 |
| 6a | 1.00 ± 0.05 | 6b | 7.6 ± 3.9 |
| 7a | 4.08 ± 0.08 | 7b | >15 |
| 8a | 0.47 ± 0.08 | 8b | 2.3 ± 0.4 |
| 9a | 0.29 ± 0.12 | ||
| 10a | >15 | ||
| 11a | >15 | ||
| Fur 2 | 1.2 ± 0.3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomatina, O.V.; Popadyuk, I.I.; Zakharenko, A.L.; Zakharova, O.D.; Fadeev, D.S.; Komarova, N.I.; Reynisson, J.; Arabshahi, H.J.; Chand, R.; Volcho, K.P.; et al. Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Molecules 2018, 23, 679. https://doi.org/10.3390/molecules23030679
Salomatina OV, Popadyuk II, Zakharenko AL, Zakharova OD, Fadeev DS, Komarova NI, Reynisson J, Arabshahi HJ, Chand R, Volcho KP, et al. Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Molecules. 2018; 23(3):679. https://doi.org/10.3390/molecules23030679
Chicago/Turabian StyleSalomatina, Oksana V., Irina I. Popadyuk, Alexandra L. Zakharenko, Olga D. Zakharova, Dmitriy S. Fadeev, Nina I. Komarova, Jóhannes Reynisson, H. John Arabshahi, Raina Chand, Konstantin P. Volcho, and et al. 2018. "Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors" Molecules 23, no. 3: 679. https://doi.org/10.3390/molecules23030679
APA StyleSalomatina, O. V., Popadyuk, I. I., Zakharenko, A. L., Zakharova, O. D., Fadeev, D. S., Komarova, N. I., Reynisson, J., Arabshahi, H. J., Chand, R., Volcho, K. P., Salakhutdinov, N. F., & Lavrik, O. I. (2018). Novel Semisynthetic Derivatives of Bile Acids as Effective Tyrosyl-DNA Phosphodiesterase 1 Inhibitors. Molecules, 23(3), 679. https://doi.org/10.3390/molecules23030679