Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT



Abstract

1. Introduction
2. Theoretical Background
| Electronegativity | |
| Global hardness | |
| Electrophilicity | |
| Electron-donating power | |
| Electron-accepting power | |
| Net electrophilicity |
| Nucleophilic Fukui function | |
| Electrophilic Fukui function | |
| Dual descriptor | |
| Nucleophilic Parr function | |
| Electrophilic Parr function |
3. Settings and Computational Methods
4. Results and Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Argirova, M.D. Photosensitizer Activity of Model Melanoidins. J. Agric. Food Chem. 2005, 53, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. A Comparison of the Chemical Reactivity of Naringenin Calculated with M06 Family of Density Functionals. Chem. Cent. J. 2013, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Morán, G.; Ruiz-Nieto, S.; Gerli-Candia, L.; Flores-Holguín, N.; Favila-Pérez, A.; Glossman-Mitnik, D. Computational Nanochemistry Study of the Molecular Structure and Properties of Ethambutol. J. Mol. Model. 2013, 19, 3507–3515. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nanochemistry Report on the Oxicams—Conceptual DFT and Chemical Reactivity. J. Phys. Chem. B 2013, 117, 6639–6651. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. Computational Nanochemistry Study of the Chemical Reactivity Properties of the Rhodamine B Molecule. Procedia Comput. Sci. 2013, 18, 816–825. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I.; Salgado-Morán, G.; Glossman-Mitnik, D. Computational Nutraceutics: Chemical Reactivity Properties of the Flavonoid Naringin by Means of Conceptual DFT. J. Chem. 2013, 2013, 850297. [Google Scholar] [CrossRef]
- Cervantes-Navarro, F.; Glossman-Mitnik, D. Density Functional Study of the Effects of Substituents on the Chemical Reactivity of the Indigo Molecule. J. Theor. Comput. Chem. 2013, 12, 1350013. [Google Scholar] [CrossRef]
- Alvarado-González, M.; Flores-Holguín, N.; Glossman-Mitnik, D. Computational Nanochemistry Study of the Molecular Structure and Properties of the Chlorophyll a Molecule. Int. J. Photoenergy 2013, 2013, 424620. [Google Scholar] [CrossRef]
- Glossman-Mitnik, D. Chemical Reactivity Theory within DFT Applied to the Study of the Prunin Flavonoid. Eur. Int. J. Sci. Technol. 2014, 3, 195–207. [Google Scholar]
- Glossman-Mitnik, D. Computational Chemistry of Natural Products: A Comparison of the Chemical Reactivity of Isonaringin Calculated with the M06 Family of Density Functionals. J. Mol. Model. 2014, 20, 2316. [Google Scholar] [CrossRef] [PubMed]
- Glossman-Mitnik, D. Computational Nanochemistry Study of the Molecular Structure, Spectra and Chemical Reactivity Properties of the BFPF Green Fluorescent Protein Chromophore. In Biosensors Nanotechnology; Tiwari, A., Turner, A.P., Eds.; John Wiley & Sons: Hoboken, NY, USA, 2014; pp. 201–238. [Google Scholar]
- Glossman-Mitnik, D. Computational Nanochemistry Report of the Molecular Structure, Spectra and Chemical Reactivity Properties of Pheophorbide A. In Design and Applications of Nanomaterials for Sensors; Seminario, J.M., Ed.; Springer Science + Business Media: Dordrecht, The Netherlands, 2014; pp. 359–402. [Google Scholar]
- Martínez-Araya, J.I.; Grand, A.; Glossman-Mitnik, D. Towards the Rationalization of Catalytic Activity Values by Means of Local Hyper-Softness on the Catalytic Site: A Criticism About the Use of Net Electric Charges. Phys. Chem. Chem. Phys. 2015, 17, 29764–29775. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rojo, R.; Baldenebro-López, J.; Glossman-Mitnik, D. Study of Chemical Reactivity in Relation to Experimental Parameters of Efficiency in Coumarin Derivatives for Dye Sensitized Solar Cells Using DFT. Phys. Chem. Chem. Phys. 2015, 17, 14122–14129. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I.; Glossman-Mitnik, D. The Substituent Effect from the Perspective of Local Hyper-Softness. An Example Applied on Normeloxicam, Meloxicam and 4-Meloxicam: Non-Steroidal Anti-Inflammatory Drugs. Chem. Phys. Lett. 2015, 618, 162–167. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. Validation of the Koopmans’ Theorem by Means of the Calculation of the Conceptual DFT Descriptors of Three Fluorescent DNA Staining Dyes. Chem. Inform. 2016, 2, 1–7. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Molecular Electron Density Theory Study of the Chemical Reactivity of cis- and trans-Resveratrol. Molecules 2016, 21, 1650. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Theoretical Study of the Chemical Reactivity of Neohesperidin Dihydrochalcone Through Conceptual DFT Descriptors. SDRP J. Comput. Chem. Mol. Model. 2016, 1. [Google Scholar] [CrossRef][Green Version]
- Mendoza-Huízar, L.H.; Salgado-Morán, G.; Ramírez-Tagle, R.; Glossman-Mitnik, D. A Theoretical Quantum Study of the Intramolecular Interactions and Chemical Reactivity of Polymorphs A and B of Famotidine in the Gas, DMSO, and Aqueous Phases. Comput. Theor. Chem. 2016, 1075, 54–62. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Comparison of the Minnesota Family of Density Functionals for the Calculation of Conceptual DFT Descriptors: Citrus Flavonoids as a Test Case. Res. J. Chem. Sci. 2017, 7, 46–58. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. A Comparative Study of the Glycating Power of Simple Carbohydrates in the Maillard Reaction by Means of Conceptual DFT Descriptors. Bri. J. Appl. Sci. Technol. 2017, 21, 32795. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. A Conceptual DFT Study of the Chemical Reactivity of Magnesium Octaethylprphyrin (MgOEP) as Predicted by the Minnesota Family of Density Functionals. Quím. Nova 2017, 40, 402–406. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Pyridoxamine Derivatives as Non Enzymatic Glycation Inhibitors: The Conceptual DFT Viewpoint. Res. J. Life Sci. Bioinform. Pharm. Chem. Sci. 2017, 2, 103–122. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Molecular Modeling Study of the Structures, Properties and Glycating Power of Some Reducing Disacharides. MOJ Drug Des. Dev. Ther. 2017, 1, 00003. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Computational Prediction of the Reactivity sites of Alzheimer Amyloid β-Peptides Aβ40 and Aβ42. ChemXpress 2017, 10, 120. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Descriptors of Amino Acids with Potential Corrosion Inhibition Properties Calculated with the Latest Minnesota Density Functionals. Front. Chem. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Sastre, S.; Frau, J.; Glossman-Mitnik, D. Computational Prediction of the Protonation Sites of Ac-Lys-(Ala)n-Lys-NH2 Peptides through Conceptual DFT and MEDT Descriptors. Molecules 2017, 22, 458. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Ramis, R.; Glossman-Mitnik, D. Computational Prediction of the Preferred Glycation Sites of Model Helical Peptides Derived from Human Serum Albumin (HSA) and Lysozyme Helix 4 (LH4). Theor. Chem. Acc. 2017, 136, 39. [Google Scholar] [CrossRef]
- Frau, J.; Muñoz, F.; Glossman-Mitnik, D. Application of DFT Concepts to the Study of the Chemical Reactivity of Some Resveratrol Derivatives Through the Assessment of the Validity of the “Koopmans in DFT” (KID) Procedure. J. Theor. Comput. Chem. 2017, 16, 1750006. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Study of Advanced Glycation Endproduct Inhibitors. Molecules 2017, 22, 226. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Glossman-Mitnik, D. A Conceptual DFT Study of the Molecular Properties of Glycating Carbonyl Compounds. Chem. Cent. J. 2017, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Hernández-Haro, N.; Glossman-Mitnik, D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. [Google Scholar] [CrossRef]
- Nursten, H. (Ed.) The Maillard Reaction—Chemistry, Biochemistry and Implications; The Royal Society of Chemistry: Cambridge, UK, 2005. [Google Scholar]
- Ono, Y.; Watanabe, H.; Hayase, F. Identification of the Blue Pigment Formed in a D-Glucose-Glycine Reaction System. Bioscience, Biotechnology and Biochemistry 2010, 74, 2526–2528. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Toro-Labbé, A. (Ed.) Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Chattaraj, P. (Ed.) Chemical Reactivity Theory—A Density Functional View; CRC Press. Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Domingo, L.R. Molecular Electron Density Theory: A Modern View of Reactivity in Organic Chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, R. Searching for Zwitterionic Intermediates in Hetero Diels–Alder Reactions between Methyl α,p-dinitrocinnamate and Vinyl-Alkyl Ethers. Comput. Theor. Chem. 2014, 1046, 93–98. [Google Scholar] [CrossRef]
- Jasinski, R.; Mroz, K.; Kacka, A. Experimental and Theoretical DFT Study on Synthesis of Sterically Crowded 2,3,3,(4)5-Tetrasubstituted-4-nitroisoxazolidines via 1,3-Dipolar Cycloaddition Reactions Between Ketonitrones and Conjugated Nitroalkenes. J. Heterocycl. Chem. 2016, 53, 1424–1429. [Google Scholar] [CrossRef]
- Jasinski, R.; Jasinska, E.; Dresler, E. A DFT Computational Study of the Molecular Mechanism of [3 + 2] Cycloaddition Reactions between Nitroethene and Benzonitrile N-oxides. J. Mol. Model. 2017, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Huzinaga, S.; Andzelm, J.; Klobukowski, M.; Radzio-Audzelm, E.; Sakai, Y.; Tatewaki, H. Gaussian Basis Sets for Molecular Calculations; Elsevier: Amsterdam, The Netherlands, 1984. [Google Scholar]
- Easton, R.; Giesen, D.; Welch, A.; Cramer, C.; Truhlar, D. The MIDI! Basis Set for Quantum Mechanical Calculations of Molecular Geometries and Partial Charges. Theor. Chem. Acc. 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Gledhill, J.D.; De Proft, F.; Tozer, D.J. Range-Separation Parameter in Tuned Exchange-Correlation Functionals: Successive Ionizations and the Fukui Function. J. Chem. Theory Comput. 2016, 12, 4879–4884. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Moore, B.; Planchat, A.; Adamo, C.; Autschbach, J. Performance of an Optimally Tuned Range-Separated Hybrid Functional for 0-0 Electronic Excitation Energies. J. Chem. Theory Comput. 2014, 10, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.; Yang, W. Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Parr, R.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Cárdenas, C.; Rabi, N.; Ayers, P.; Morell, C.; Jaramillo, P.; Fuentealba, P. Chemical Reactivity Descriptors for Ambiphilic Reagents: Dual Descriptor, Local Hypersoftness, and Electrostatic Potential. J. Phys. Chem. A 2009, 113, 8660–8667. [Google Scholar] [CrossRef] [PubMed]
- Ayers, P.; Morell, C.; De Proft, F.; Geerlings, P. Understanding the Woodward-Hoffmann Rules by Using Changes in Electron Density. Chem. A Eur. J. 2007, 13, 8240–8247. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Ayers, P.; Grand, A.; Gutiérrez-Oliva, S.; Toro-Labbé, A. Rationalization of the Diels-Alder Reactions through the Use of the Dual Reactivity Descriptor Δf(r). Phys. Chem. Chem. Phys. 2008, 10, 7239–7246. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.; Hocquet, A.; Grand, A.; Jamart-Grégoire, B. A Conceptual DFT Study of Hydrazino Peptides: Assessment of the Nucleophilicity of the Nitrogen Atoms by Means of the Dual Descriptor Δf(r). J. Mol. Struct. THEOCHEM 2008, 849, 46–51. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J. Understanding the Local Reactivity in Polar Organic Reactions through Electrophilic and Nucleophilic Parr Functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Chamorro, E.; Pérez, P.; Domingo, L.R. On the Nature of Parr Functions to Predict the Most Reactive Sites along Organic Polar Reactions. Chem. Phys. Lett. 2013, 582, 141–143. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scalmani, G.; Scuseria, G.E. Can Short-Range Hybrids Describe Long-Range-Dependent Properties? J. Chem. Phys. 2009, 131, 044108. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. An Improved and Broadly Accurate Local Approximation to the Exchange-Correlation Density Functional: the MN12-L Functional for Electronic Structure Calculations in Chemistry and Physics. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Exchange-Correlation Functional with Good Accuracy for Both Structural and Energetic Properties while Depending Only on the Density and Its Gradient. J. Chem. Theory Comput. 2012, 8, 2310–2319. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Head-Gordon, M. Systematic Optimization of Long-Range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Egger, D.A.; Weissman, S.; Refaely-Abramson, S.; Sharifzadeh, S.; Dauth, M.; Baer, R.; Kümmel, S.; Neaton, J.B.; Zojer, E.; Kronik, L. Outer-Valence Electron Spectra of Prototypical Aromatic Heterocycles From an Optimally Tuned Range-Separated Hybrid Functional. J. Chem. Theory Comput. 2014, 10, 1934–1952. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.E.; Wong, B.M. Nonempirically Tuned Range-Separated DFT Accurately Predicts Both Fundamental and Excitation Gaps in DNA and RNA Nucleobases. J. Chem. Theory Comput. 2012, 8, 2682–2687. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.E.; Azoulay, J.D.; Wong, B.M.; Allendorf, M.D. Novel Metal–Organic Framework Linkers for Light Harvesting Applications. Chem. Sci. 2014, 5, 2081–2090. [Google Scholar] [CrossRef]
- Karolewski, A.; Stein, T.; Baer, R.; Kümmel, S. Communication: Tailoring the Optical Gap in Light-Harvesting Molecules. J. Chem. Phys. 2011, 134, 151101. [Google Scholar] [CrossRef] [PubMed]
- Karolewski, A.; Kronik, L.; Kümmel, S. Using Optimally Tuned Range Separated Hybrid Functionals in Ground-State Calculations: Consequences and Caveats. J. Chem. Phys. 2013, 138, 204115. [Google Scholar] [CrossRef] [PubMed]
- Koppen, J.V.; Hapka, M.; Szczeniak, M.M.; Chalasinski, G. Optical Absorption Spectra of Gold Clusters Au(n) (n = 4, 6, 8,12, 20) From Long-Range Corrected Functionals with Optimal Tuning. J. Chem. Phys. 2012, 137, 114302. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Kuritz, N.; Stein, T.; Baer, R.; Kronik, L. Charge-Transfer-Like π→π* Excitations in Time-Dependent Density Functional Theory: A Conundrum and Its Solution. J. Chem. Theory Comput. 2011, 7, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.T.; Prado, A.D.S.; Martins, J.B.L.; de Oliveira Neto, P.H.; Ceschin, A.M.; da Cunha, W.F.; da Silva Filho, D.A. Improving the Description of the Optical Properties of Carotenoids by Tuning the Long-Range Corrected Functionals. J. Phys. Chem. A 2016, 120, 4944–4950. [Google Scholar] [CrossRef] [PubMed]
- Manna, A.K.; Lee, M.H.; McMahon, K.L.; Dunietz, B.D. Calculating High Energy Charge Transfer States Using Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2015, 11, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Moore, B., II; Autschbach, J. Longest-Wavelength Electronic Excitations of Linear Cyanines: The Role of Electron Delocalization and of Approximations in Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2013, 9, 4991–5003. [Google Scholar]
- Niskanen, M.; Hukka, T.I. Modeling of Photoactive Conjugated Donor-Acceptor Copolymers: The Effect of the Exact HF Exchange in DFT Functionals on Geometries and Gap Energies of Oligomer and Periodic Models. Phys. Chem. Chem. Phys. 2014, 16, 13294–13305. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.L.; Leal, L.A.; da Cunha, W.F.; Timóteo de Sousa Júnior, R.; Ribeiro Junior, L.A.; Antonio da Silva Filho, D. Optimally Tuned Functionals Improving the Description of Optical and Electronic Properties of the Phthalocyanine Molecule. J. Mol. Model. 2017, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.; Zheng, S.; Hyla, A.; Laine, R.; Goodson, T., III; Geva, E.; Dunietz, B.D. Ab Initio Calculation of the Electronic Absorption of Functionalized Octahedral Silsesquioxanes via Time-Dependent Density Functional Theory with Range-Separated Hybrid Functionals. J. Phys. Chem. A 2012, 116, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.; Geva, E.; Dunietz, B.D. Calculating Off-Site Excitations in Symmetric Donor-Acceptor Systems via Time-Dependent Density Functional Theory with Range-Separated Density Functionals. J. Chem. Theory Comput. 2012, 8, 2661–2668. [Google Scholar] [CrossRef] [PubMed]
- Refaely-Abramson, S.; Baer, R.; Kronik, L. Fundamental and Excitation Gaps in Molecules of Relevance for Organic Photovoltaics from an Optimally Tuned Range-Separated Hybrid Functional. Phys. Rev. B 2011, 84, 075144. [Google Scholar] [CrossRef]
- Stein, T.; Kronik, L.; Baer, R. Prediction of Charge-Transfer Excitations in Coumarin-Based Dyes Using a Range-Separated Functional Tuned from First Principles. J. Chem. Phys. 2009, 131, 244119. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.; Kronik, L.; Baer, R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Autschbach, J. Electronic Energy Gaps for π-Conjugated Oligomers and Polymers Calculated with Density Functional Theory. J. Chem. Theory Comput. 2014, 10, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Vertical Excitation Energies From the Adiabatic Connection. J. Chem. Phys. 2016, 145, 194107. [Google Scholar] [CrossRef] [PubMed]
- Baerends, E.J.; Gritsenko, O.V.; van Meer, R. The Kohn-Sham Gap, the Fundamental Gap and the Optical Gap: The Physical Meaning of Occupied and Virtual Kohn-Sham Orbital Energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, R.; Gritsenko, O.V.; Baerends, E.J. Physical Meaning of Virtual Kohn-Sham Orbitals and Orbital Energies: An Ideal Basis for the Description of Molecular Excitations. J. Chem. Theory Comput. 2014, 10, 4432–4441. [Google Scholar] [CrossRef] [PubMed]
- Zhurko, G.; Zhurko, D. Chemcraft program Revision 1.6; Zhurko, G.A., Ed.; Akzo Nobel Coatings Inc.: High Point, NC, USA, 2012. [Google Scholar]
- Poater, A.; Duran, M.; Jaque, P.; Toro-Labbé, A.; Solà, M. Molecular Structure and Bonding of Copper Cluster Monocarbonyls CunCO (N = 1−9). J. Phys. Chem. B 2006, 110, 6526–6536. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
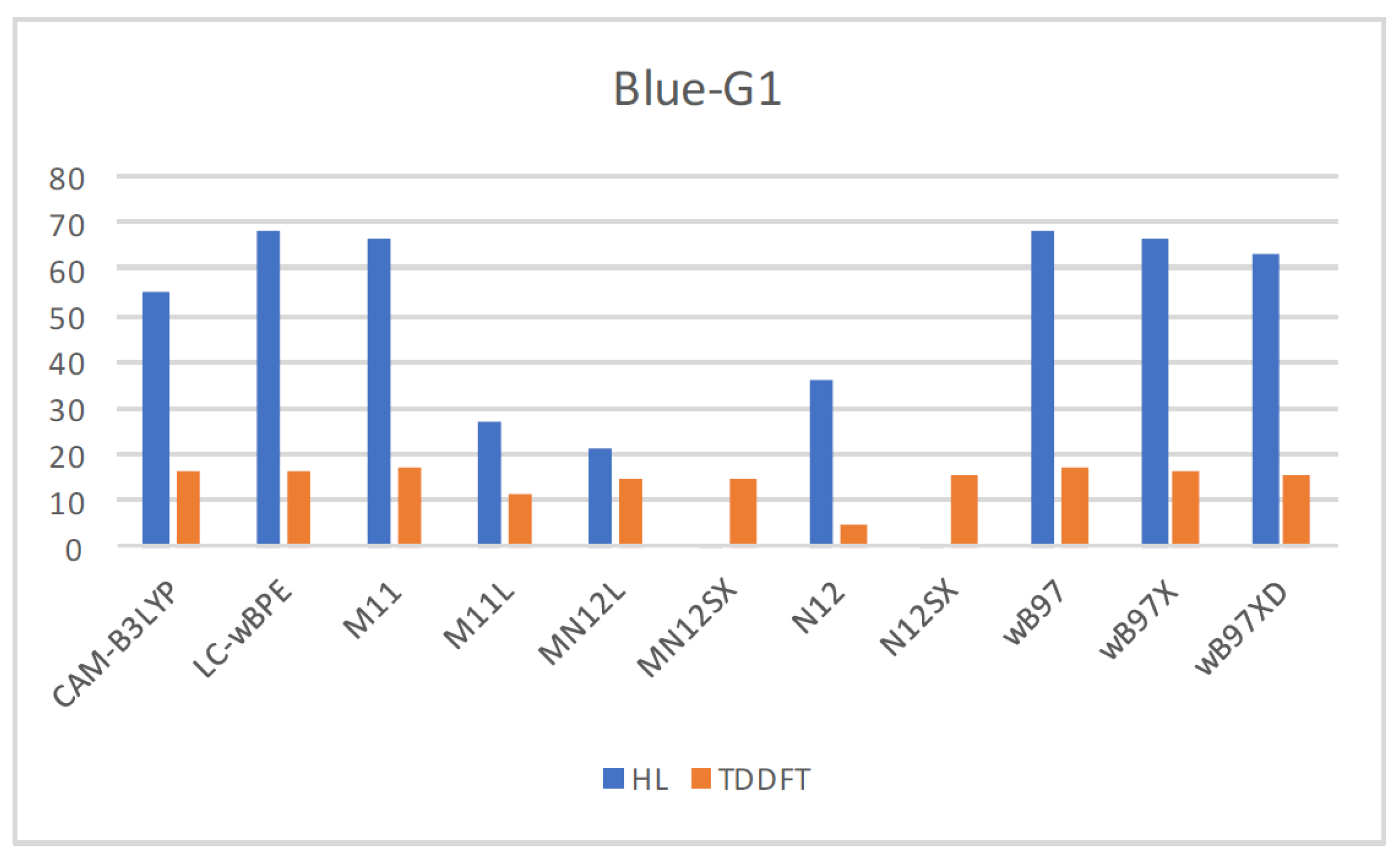
| Eo | E+ | E- | HOMO | LUMO | SOMO | J(I) | J(A) | J(HL) | HL | |
|---|---|---|---|---|---|---|---|---|---|---|
| CAM-B3LYP | −2395.4460 | −2395.2561 | −2395.5734 | −0.2380 | −0.0783 | −0.1748 | 0.0480 | 0.0490 | 0.0686 | 0.0965 |
| LC-wBPE | −2395.0237 | −2394.8290 | −2395.1654 | −0.2812 | −0.0560 | −0.2234 | 0.0865 | 0.0857 | 0.1217 | 0.1674 |
| M11 | −2395.2791 | −2395.0804 | −2395.4158 | −0.2761 | −0.0603 | −0.2104 | 0.0774 | 0.0764 | 0.1087 | 0.1501 |
| M11L | −2395.2507 | −2395.0467 | −2395.3823 | −0.1974 | −0.1406 | −0.1235 | 0.0066 | 0.0090 | 0.0112 | 0.0171 |
| MN12L | −2394.3267 | −2394.1354 | −2394.4448 | −0.1850 | −0.1254 | −0.1122 | 0.0063 | 0.0073 | 0.0097 | 0.0132 |
| MN12SX | −2394.4578 | −2394.2578 | −2394.5853 | −0.1995 | −0.1271 | −0.1277 | 0.0004 | 0.0004 | 0.0006 | 0.0006 |
| N12 | −2396.1437 | −2395.9620 | −2396.2534 | −0.1732 | −0.1199 | −0.1013 | 0.0086 | 0.0102 | 0.0133 | 0.0186 |
| N12SX | −2395.4309 | −2395.2397 | −2395.5531 | −0.1926 | −0.1199 | −0.1239 | 0.0014 | 0.0023 | 0.0027 | 0.0040 |
| wB97 | −2396.1313 | −2395.9395 | −2396.2668 | −0.2774 | −0.0513 | −0.2165 | 0.0856 | 0.0842 | 0.1201 | 0.1652 |
| wB97X | −2395.9266 | −2395.7344 | −2396.0603 | −0.2715 | −0.0546 | −0.2102 | 0.0793 | 0.0791 | 0.1120 | 0.1556 |
| wB97XD | −2395.7877 | −2395.5925 | −2395.9205 | −0.2619 | −0.0643 | −0.1995 | 0.0668 | 0.0685 | 0.0957 | 0.1353 |
| J | J | J | J | |
|---|---|---|---|---|
| CAM-B3LYP | 0.0005 | 0.0970 | 0.1226 | 0.1564 |
| LC-wBPE | 0.0004 | 0.1722 | 0.2035 | 0.2666 |
| M11 | 0.0005 | 0.1538 | 0.1610 | 0.2226 |
| M11L | 0.0012 | 0.0156 | 0.0568 | 0.0590 |
| MN12L | 0.0005 | 0.0136 | 0.0387 | 0.0410 |
| MN12SX | 0.0004 | 0.0000 | 0.0009 | 0.0010 |
| N12 | 0.0008 | 0.0188 | 0.0544 | 0.0575 |
| N12SX | 0.0004 | 0.0037 | 0.0101 | 0.0108 |
| wB97 | 0.0007 | 0.1698 | 0.1783 | 0.2462 |
| wB97X | 0.0001 | 0.1583 | 0.1654 | 0.2290 |
| wB97XD | 0.0009 | 0.1353 | 0.1483 | 0.2008 |
| Electronegativity () | Chemical Hardness () | Electrophilicity () |
| 4.4428 | 1.9956 | 4.9457 |
| Electron-Donating Power () | Electron-Accepting Power () | Net Electrophilicity () |
| 7.1959 | 5.7429 | 12.9388 |
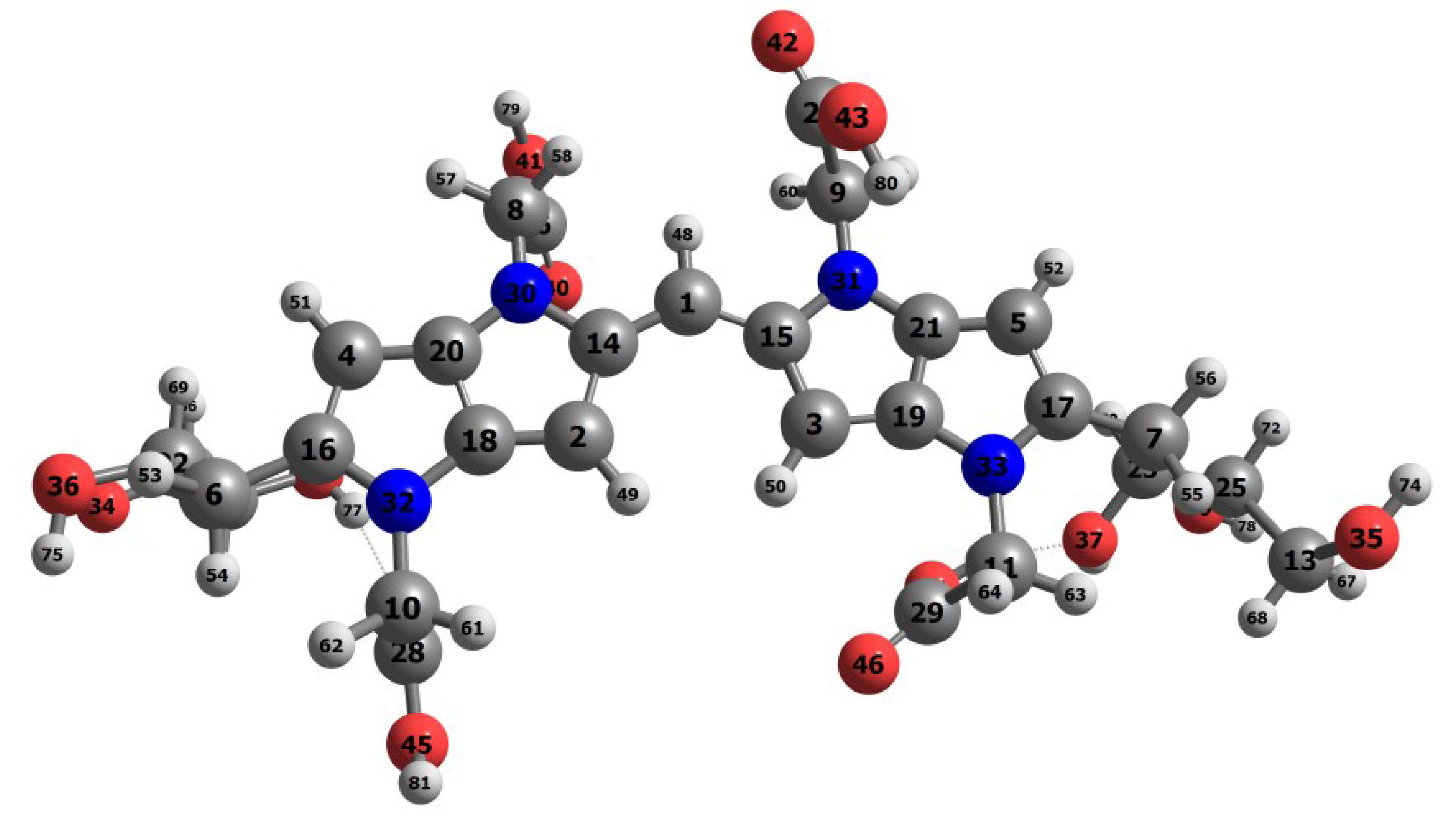
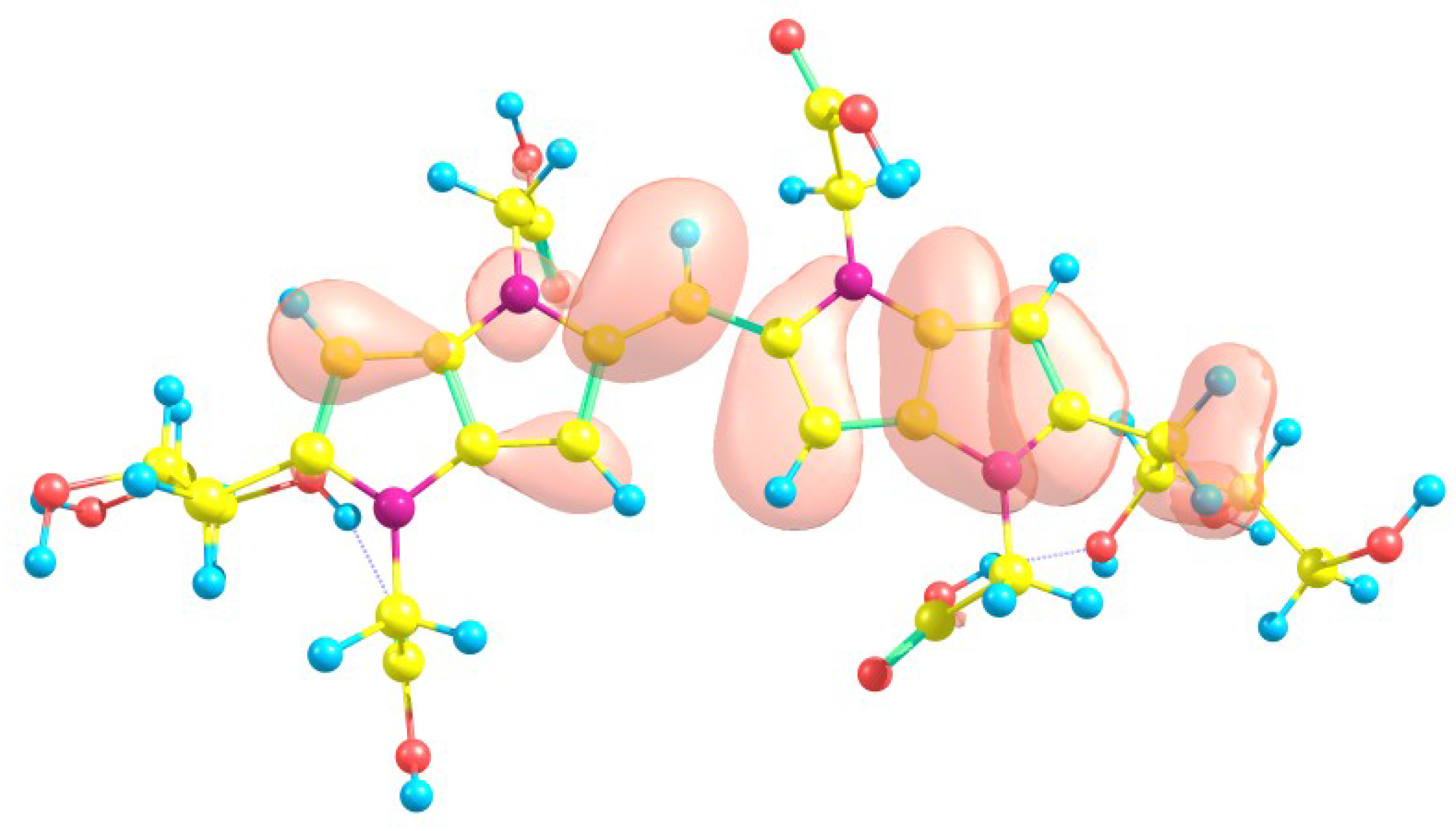
| Atom | f (M) | f (N) | P (M) | P (M) | P (H) | P (H) |
|---|---|---|---|---|---|---|
| 1C | 21.86 | 15.00 | 0.4112 | −0.1674 | 0.2228 | −0.0445 |
| 2C | 9.23 | 8.64 | 0.1947 | −0.0404 | 0.1207 | 0.0106 |
| 3C | 8.05 | 7.72 | 0.1928 | −0.0218 | 0.1169 | 0.0226 |
| 4C | −6.14 | −4.69 | −0.0644 | 0.0567 | −0.0054 | 0.0611 |
| 5C | −5.06 | −3.96 | −0.0548 | 0.0339 | −0.0043 | 0.0489 |
| 6C | −0.26 | 0.01 | −0.0244 | −0.0181 | 0.0096 | 0.0130 |
| 7C | −0.04 | −0.23 | −0.0173 | −0.0168 | 0.0079 | 0.0107 |
| 8C | −0.02 | 0.08 | −0.0039 | 0.0003 | 0.0013 | 0.0003 |
| 9C | −0.02 | 0.13 | −0.0029 | 0.0006 | 0.0017 | −0.0001 |
| 10C | −0.02 | −0.05 | 0.0002 | 0.0001 | 0.0002 | 0.0002 |
| 11C | −0.02 | −0.06 | −0.0003 | 0.0004 | 0.0000 | 0.0004 |
| 12C | −0.10 | −0.04 | 0.0003 | 0.0016 | 0.0003 | 0.0014 |
| 13C | 0.00 | −0.01 | −0.0001 | 0.0000 | 0.0001 | 0.0001 |
| 14C | −12.52 | −9.92 | −0.1063 | 0.2541 | 0.0003 | 0.1435 |
| 15C | −12.95 | −9.52 | −0.1114 | 0.2506 | −0.0026 | 0.1419 |
| 16C | −0.62 | −0.35 | 0.1831 | 0.1843 | 0.1033 | 0.1175 |
| 17C | −2.11 | −1.73 | 0.1521 | 0.1854 | 0.0895 | 0.1199 |
| 18C | −2.66 | −3.17 | −0.0041 | 0.1051 | 0.0277 | 0.0730 |
| 19C | −3.40 | −3.58 | −0.0113 | 0.1010 | 0.0224 | 0.0744 |
| 20C | 3.27 | 3.56 | 0.1269 | 0.0461 | 0.0792 | 0.0502 |
| 21C | 1.70 | 2.17 | 0.1183 | 0.0710 | 0.0727 | 0.0615 |
| 22C | −0.27 | −0.03 | 0.0048 | 0.0067 | 0.0043 | 0.0074 |
| 23C | −0.23 | −0.01 | 0.0042 | 0.0073 | 0.0074 | 0.0108 |
| 24C | −0.17 | 0.03 | 0.0000 | 0.0025 | 0.0004 | 0.0025 |
| 25C | −0.02 | −0.02 | 0.0012 | 0.0015 | 0.0009 | 0.0014 |
| 26C | 0.16 | 0.06 | 0.0000 | −0.0009 | 0.0012 | 0.0000 |
| 27C | 0.26 | −0.02 | 0.0000 | −0.0004 | 0.0015 | −0.0004 |
| 28C | −0.01 | −0.01 | −0.0011 | −0.0012 | −0.0001 | −0.0002 |
| 29C | −0.02 | −0.02 | −0.0006 | −0.0004 | 0.0000 | 0.0000 |
| 30N | 1.35 | 1.87 | 0.0288 | −0.0186 | 0.0239 | 0.0086 |
| 31N | 1.91 | 1.93 | 0.0313 | −0.0272 | 0.0250 | 0.0041 |
| 32N | −0.07 | 0.14 | −0.0118 | −0.0209 | 0.0061 | 0.0069 |
| 33N | −0.14 | 0.22 | −0.0071 | −0.0184 | 0.0057 | 0.0079 |
| 34O | −0.07 | −0.11 | 0.0000 | 0.0009 | 0.0000 | 0.0009 |
| 35O | 0.00 | −0.01 | 0.0001 | 0.0002 | 0.0002 | 0.0002 |
| 36O | −0.11 | −0.18 | 0.0004 | 0.0017 | 0.0005 | 0.0017 |
| 37O | −0.01 | −0.03 | 0.0008 | 0.0012 | 0.0007 | 0.0009 |
| 38O | −0.71 | −0.66 | 0.0017 | 0.0116 | 0.0026 | 0.0119 |
| 39O | −0.02 | −0.03 | 0.0001 | 0.0003 | 0.0003 | 0.0005 |
| 40O | −0.07 | 0.02 | 0.0020 | 0.0013 | 0.0018 | 0.0014 |
| 41O | 0.00 | 0.04 | 0.0001 | 0.0001 | 0.0001 | 0.0001 |
| 42O | 0.01 | 0.13 | 0.0005 | −0.0002 | 0.0005 | −0.0002 |
| 43O | −0.02 | 0.04 | 0.0019 | 0.0009 | 0.0013 | 0.0005 |
| 44O | −0.02 | −0.03 | 0.0008 | 0.0011 | 0.0006 | 0.0008 |
| 45O | −0.01 | −0.03 | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
| 46O | −0.02 | −0.12 | 0.0002 | −0.0002 | 0.0001 | −0.0001 |
| 47O | −0.02 | −0.06 | 0.0003 | 0.0007 | 0.0001 | 0.0005 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frau, J.; Glossman-Mitnik, D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559. https://doi.org/10.3390/molecules23030559
Frau J, Glossman-Mitnik D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules. 2018; 23(3):559. https://doi.org/10.3390/molecules23030559
Chicago/Turabian StyleFrau, Juan, and Daniel Glossman-Mitnik. 2018. "Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT" Molecules 23, no. 3: 559. https://doi.org/10.3390/molecules23030559
APA StyleFrau, J., & Glossman-Mitnik, D. (2018). Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules, 23(3), 559. https://doi.org/10.3390/molecules23030559