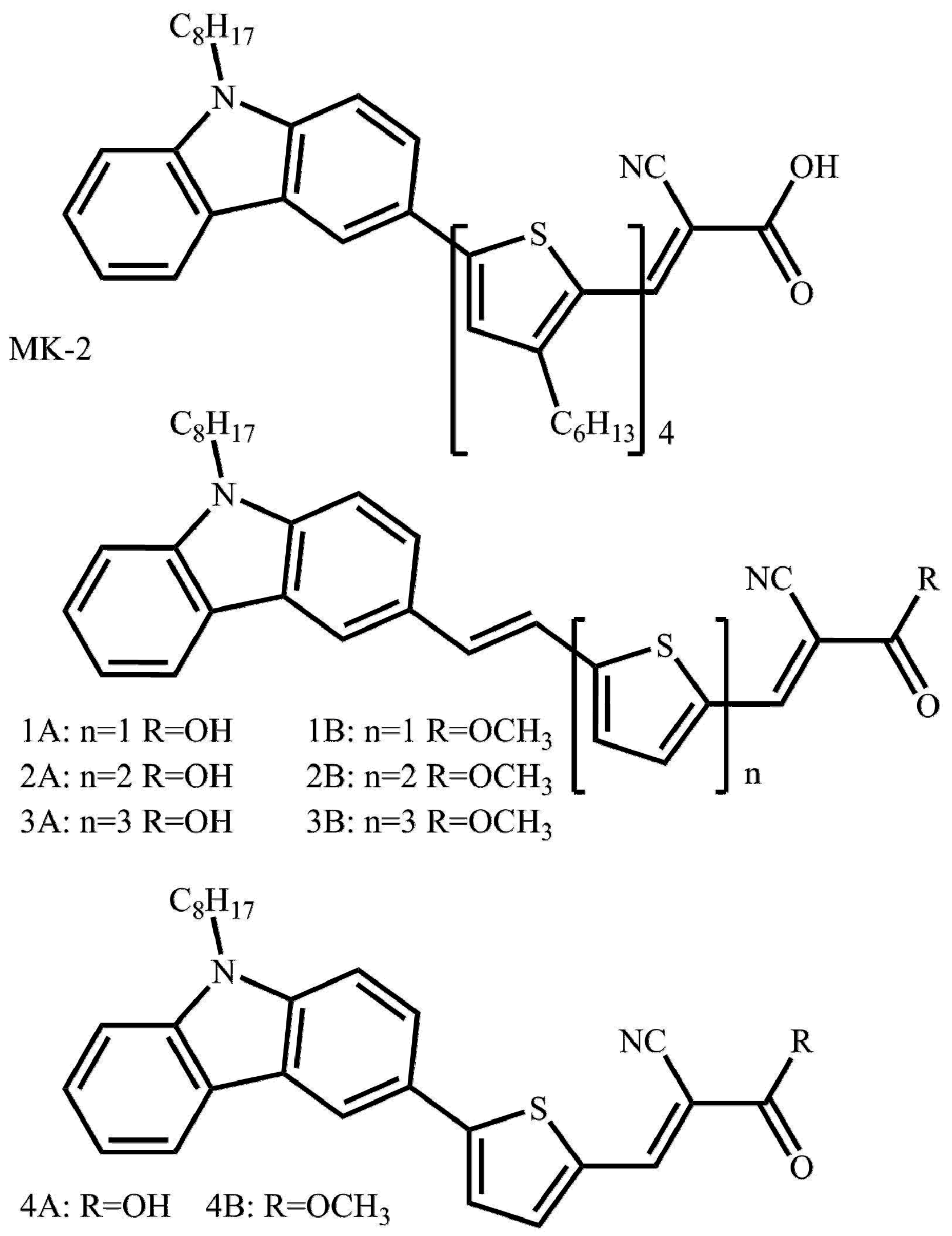
Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study



Abstract
:1. Introduction
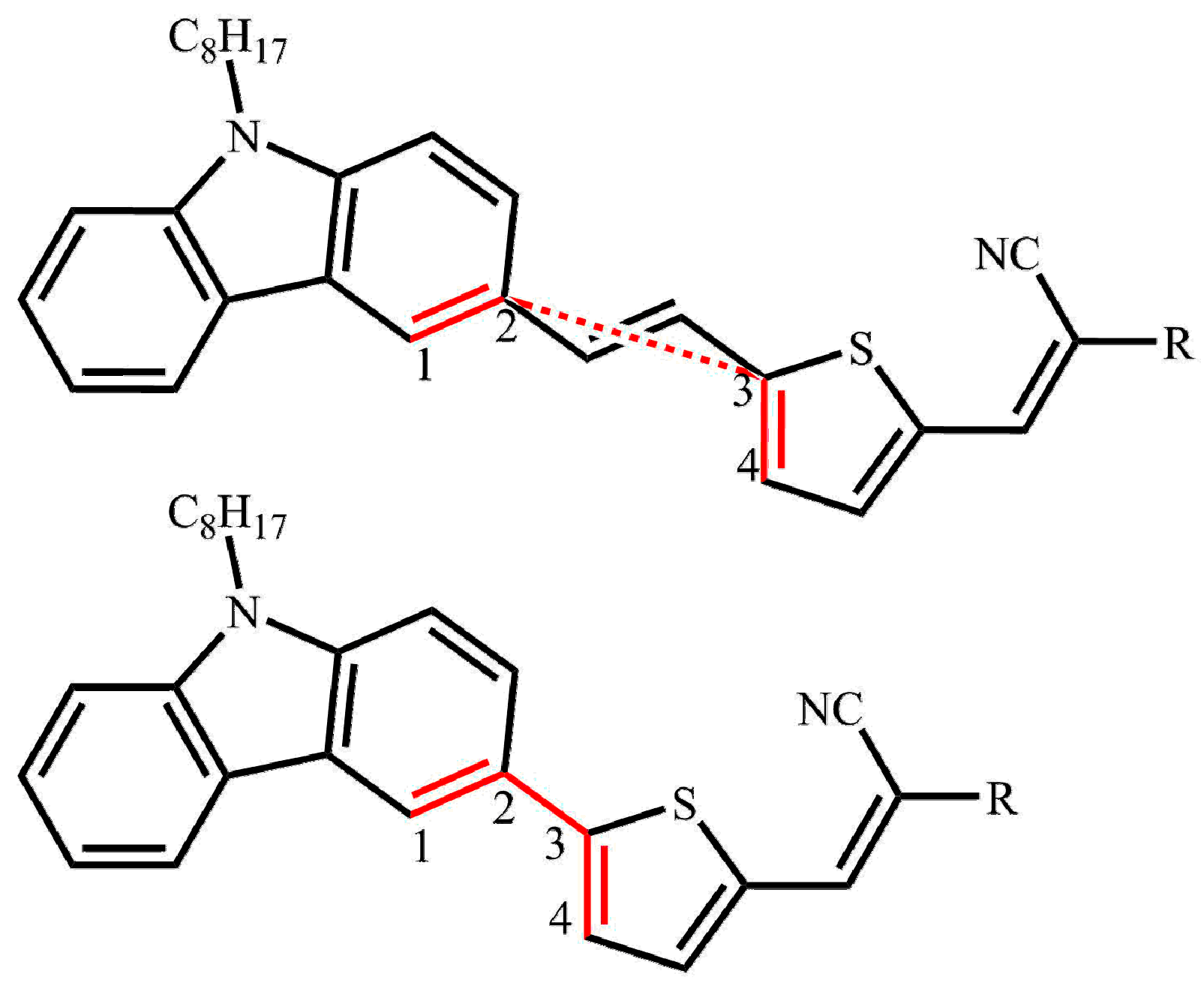
2. Results and Discussion
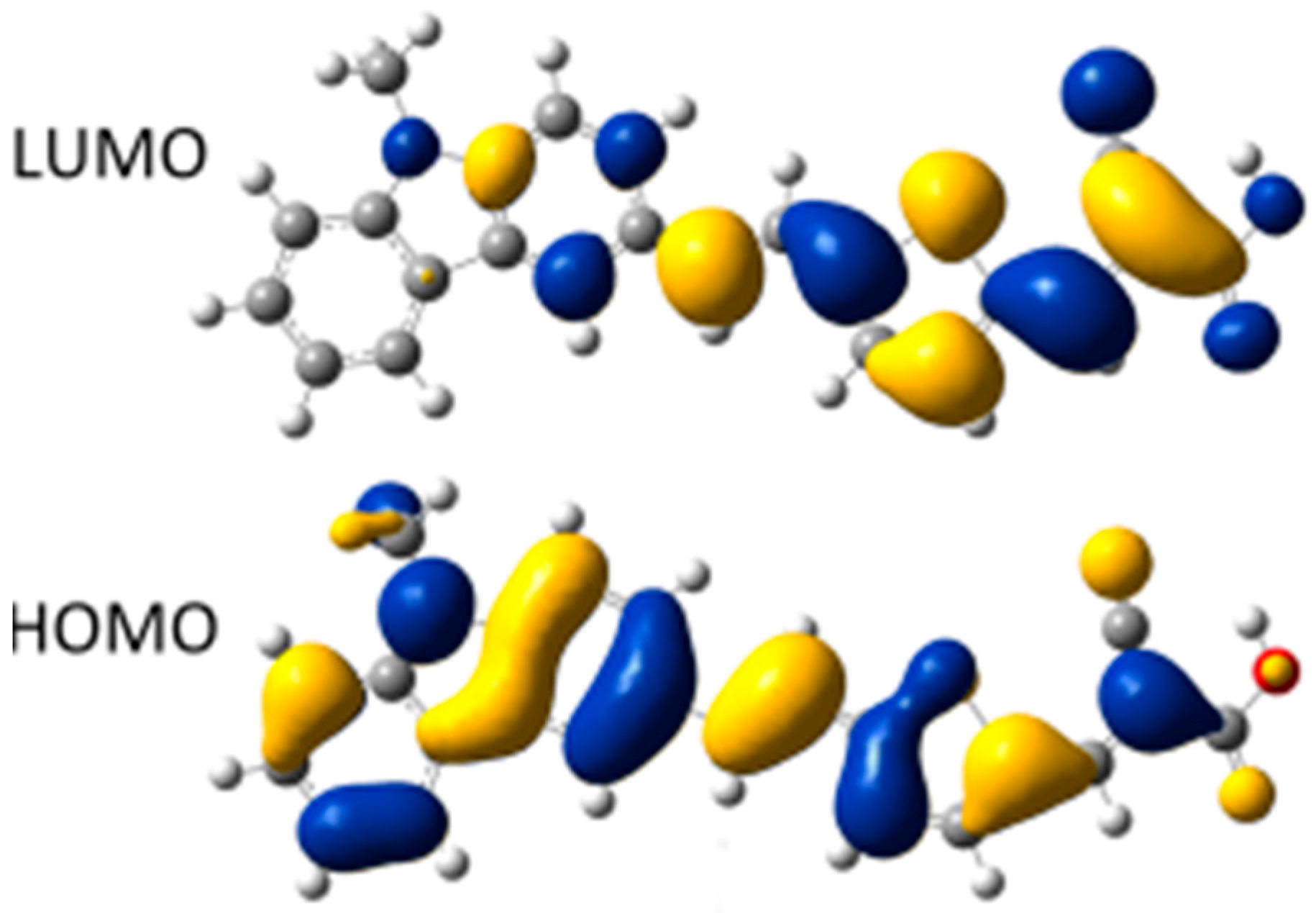
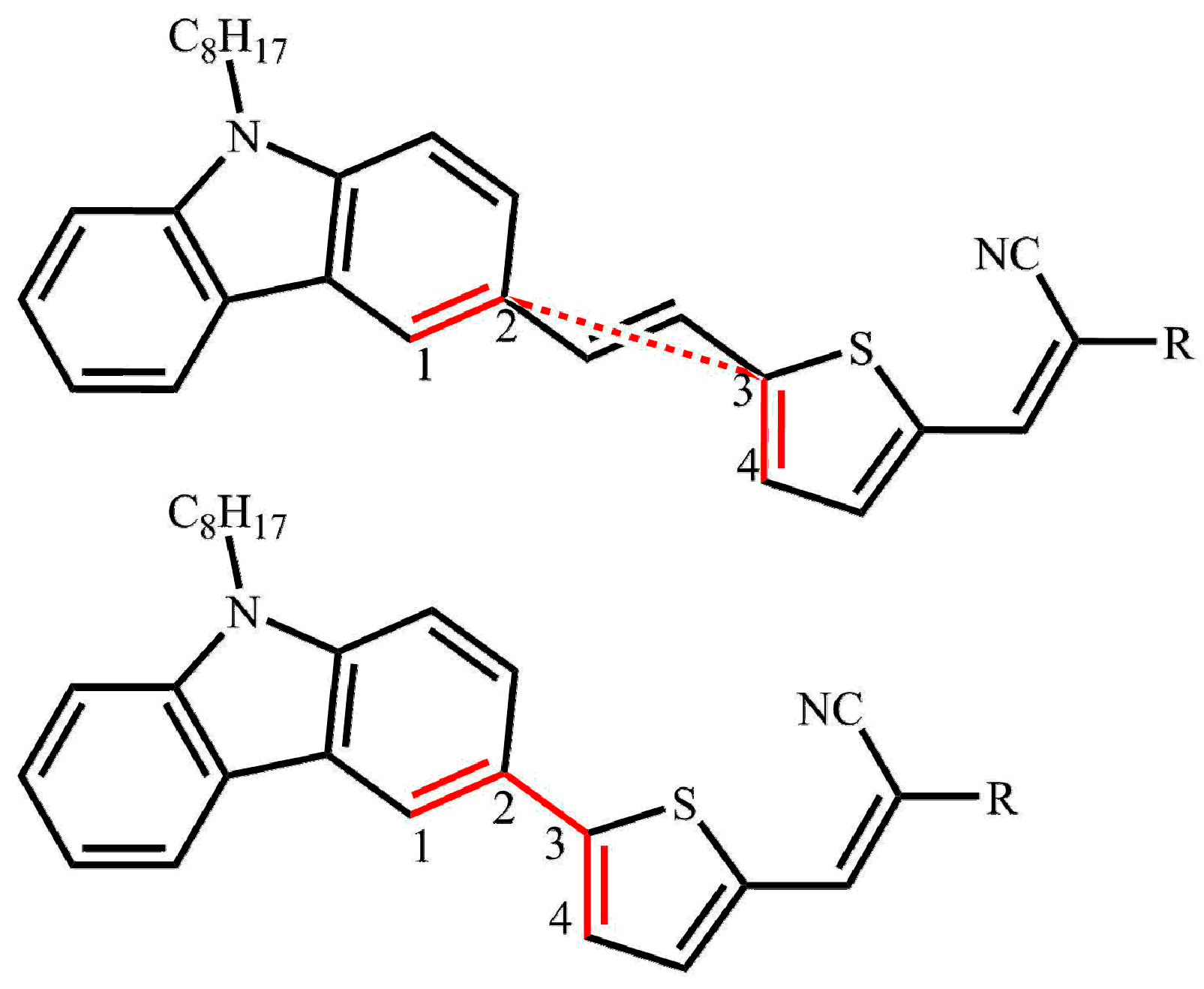
2.1. Calculated Ground-State Geometries
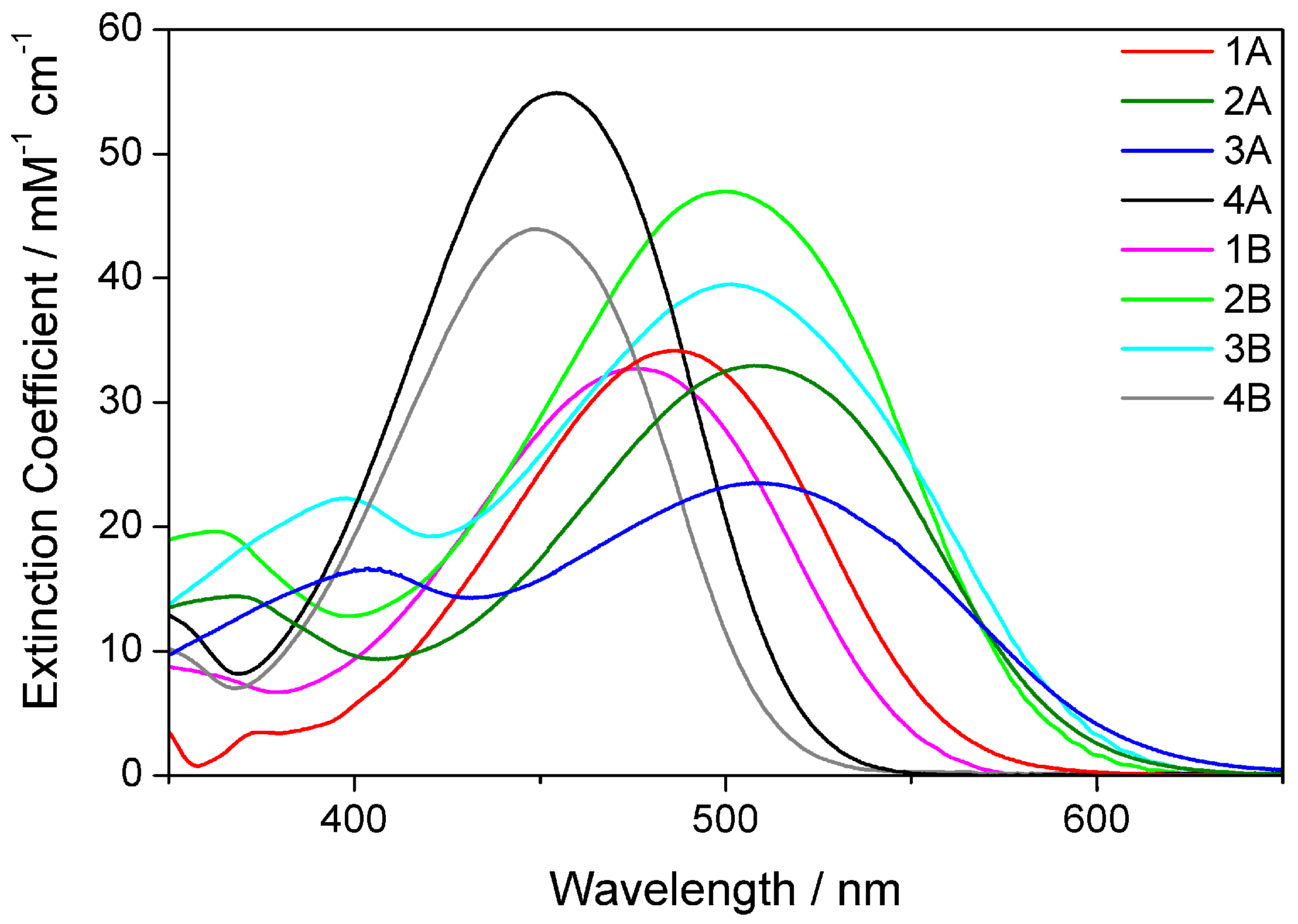
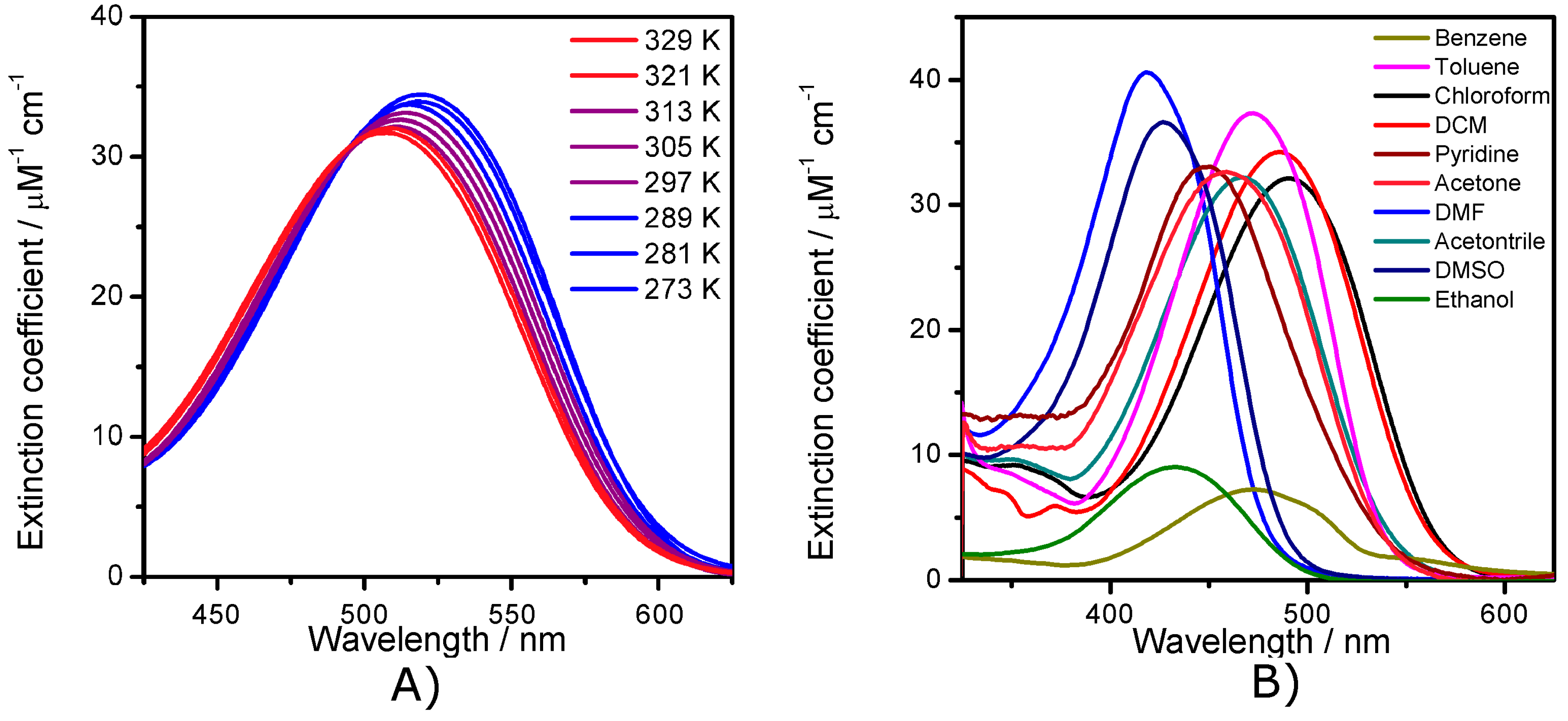
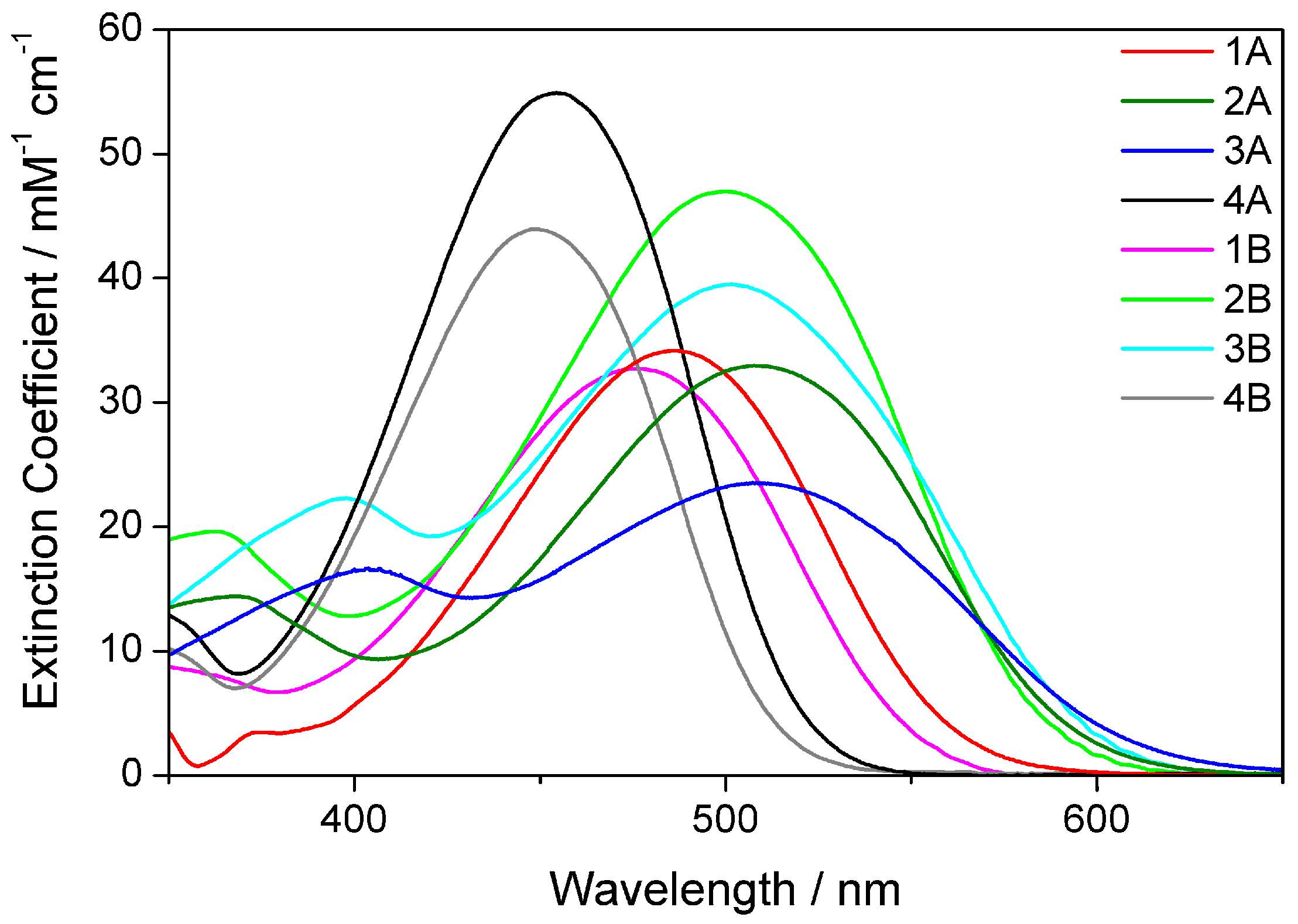
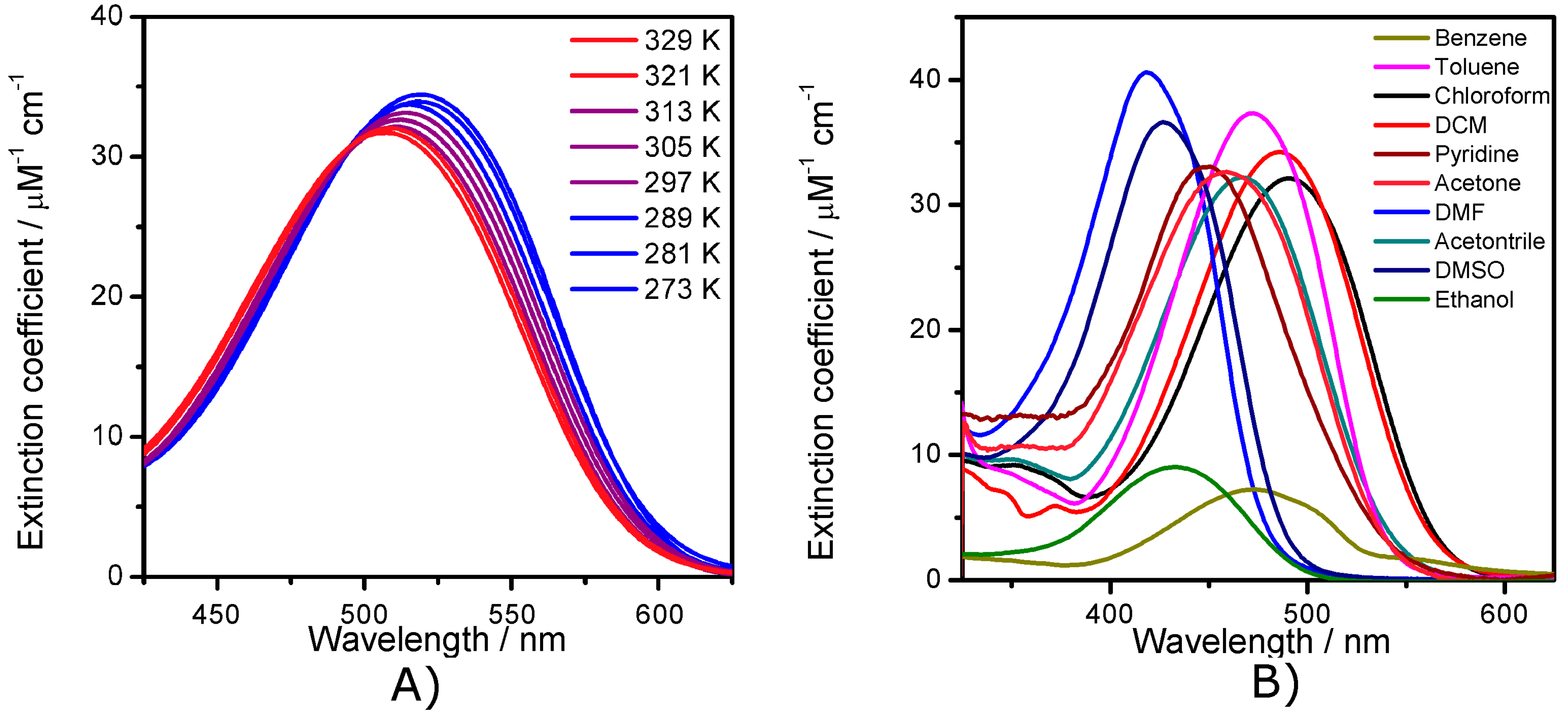
2.2. Electronic Absorption Spectroscopy
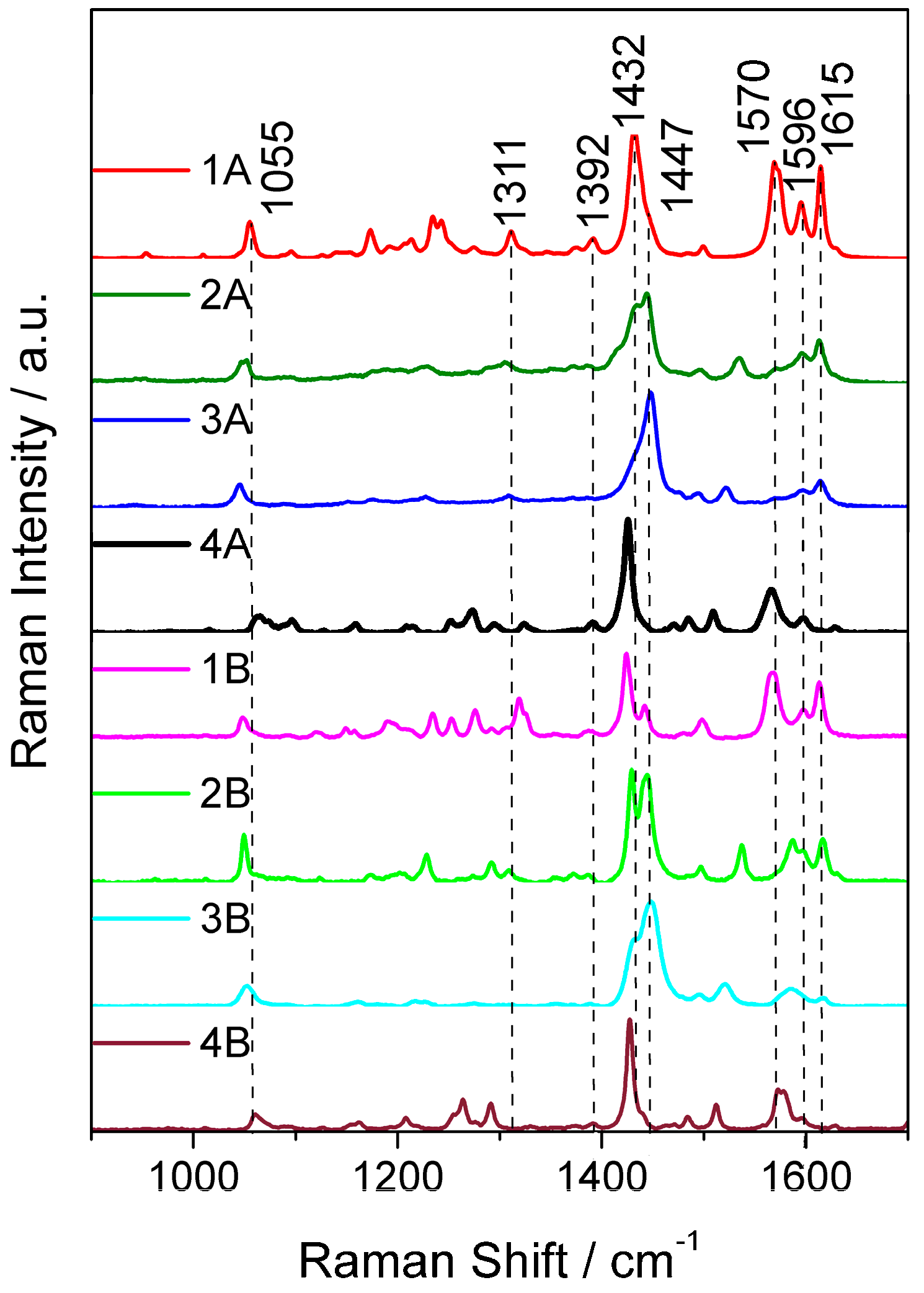
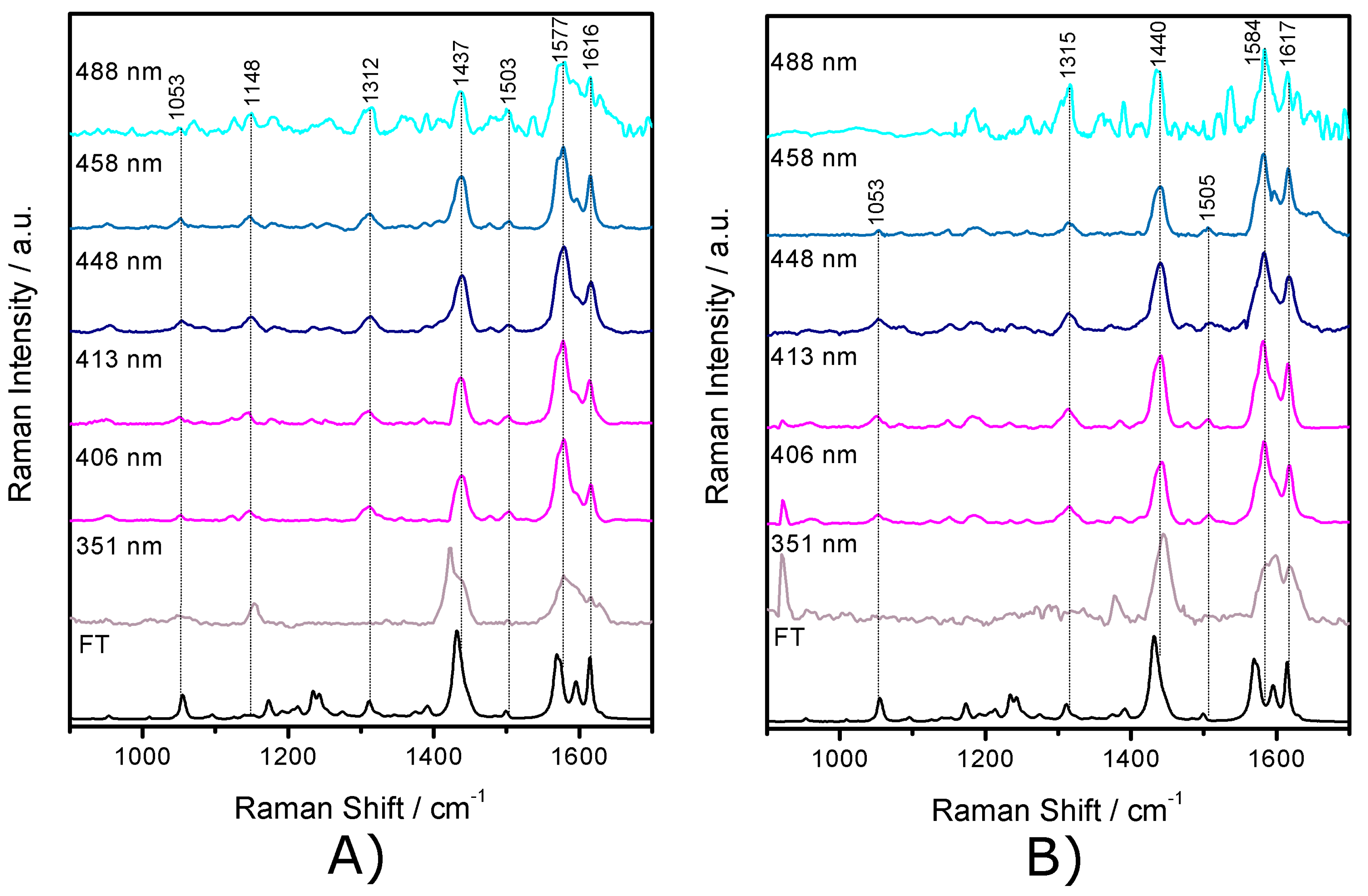
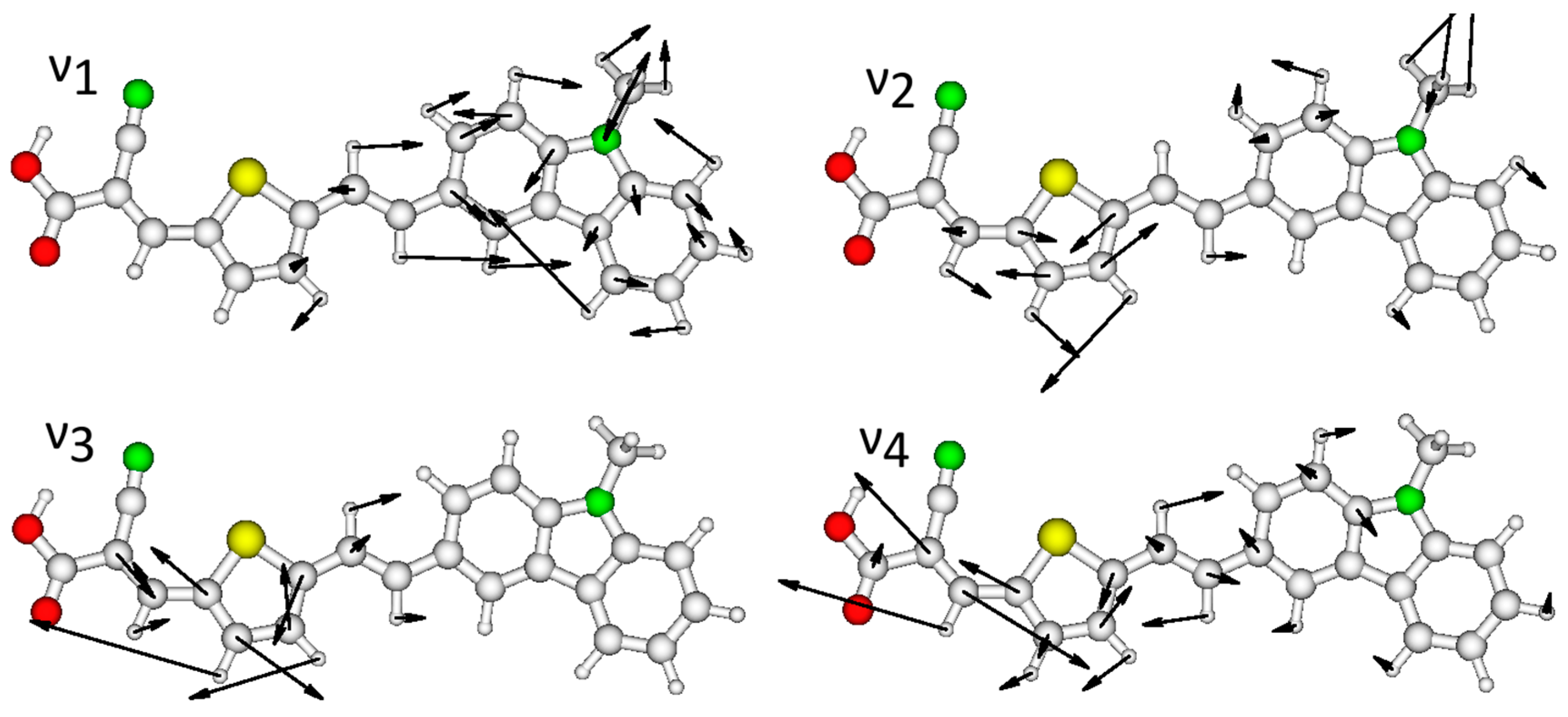
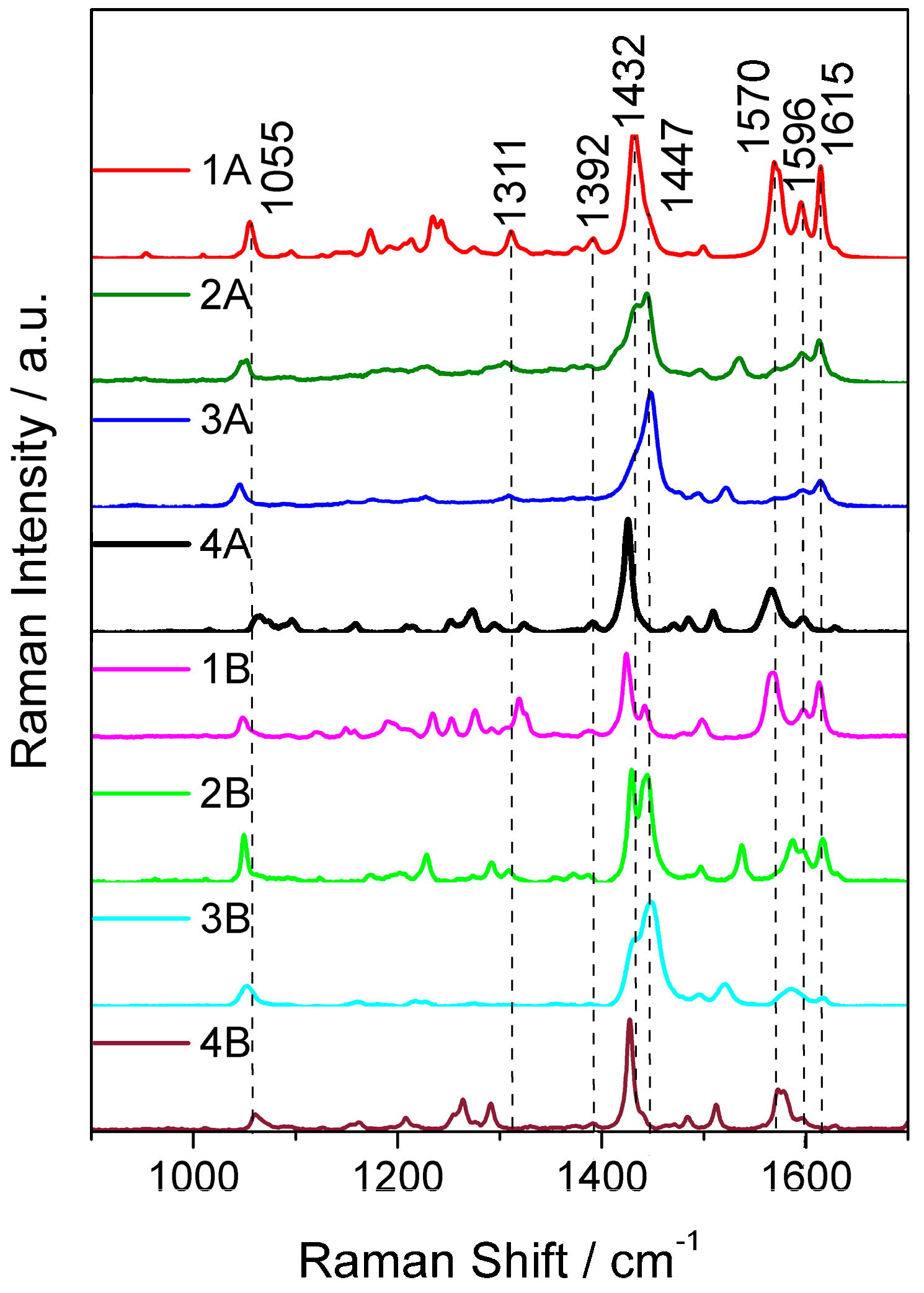
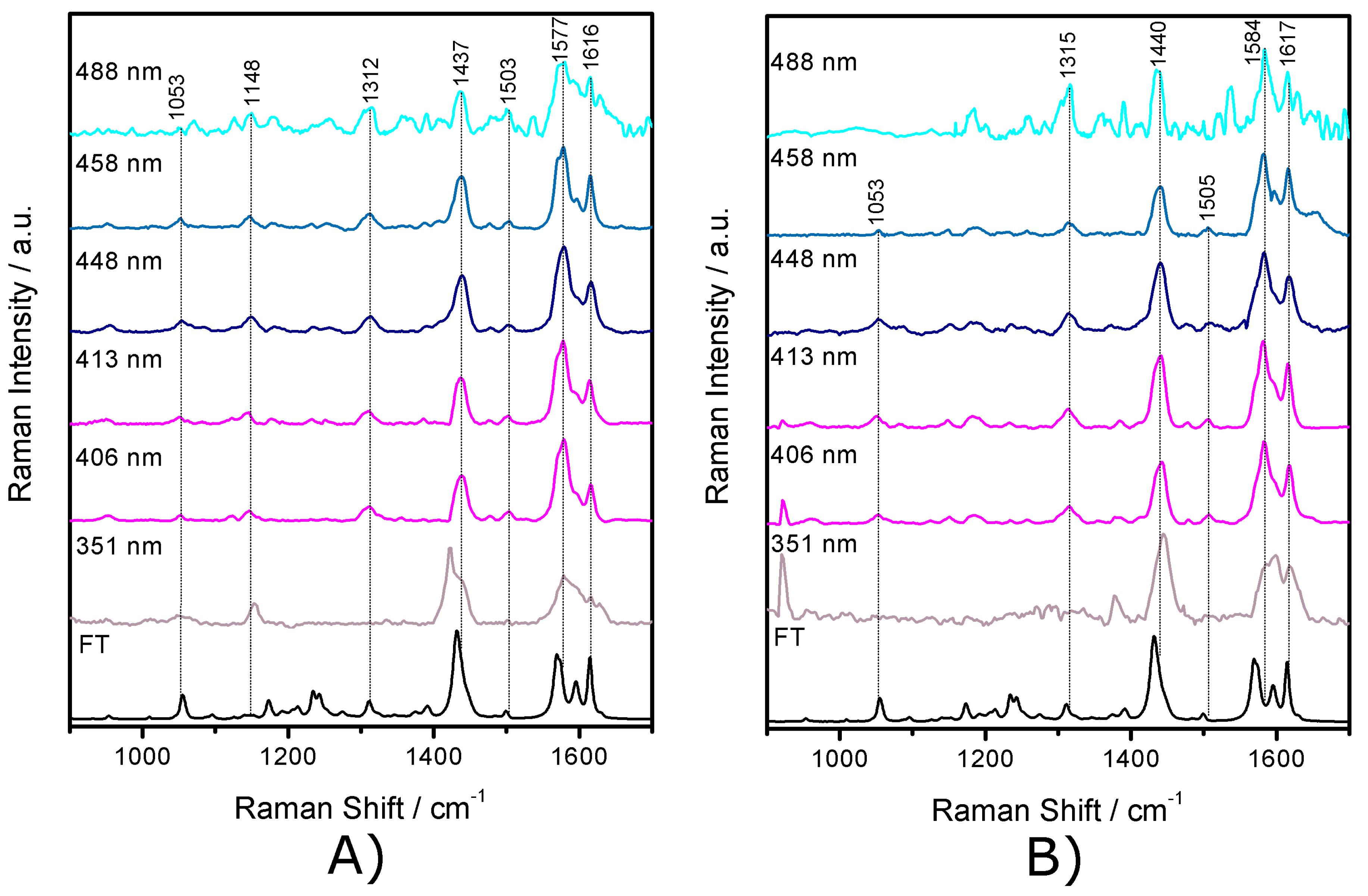
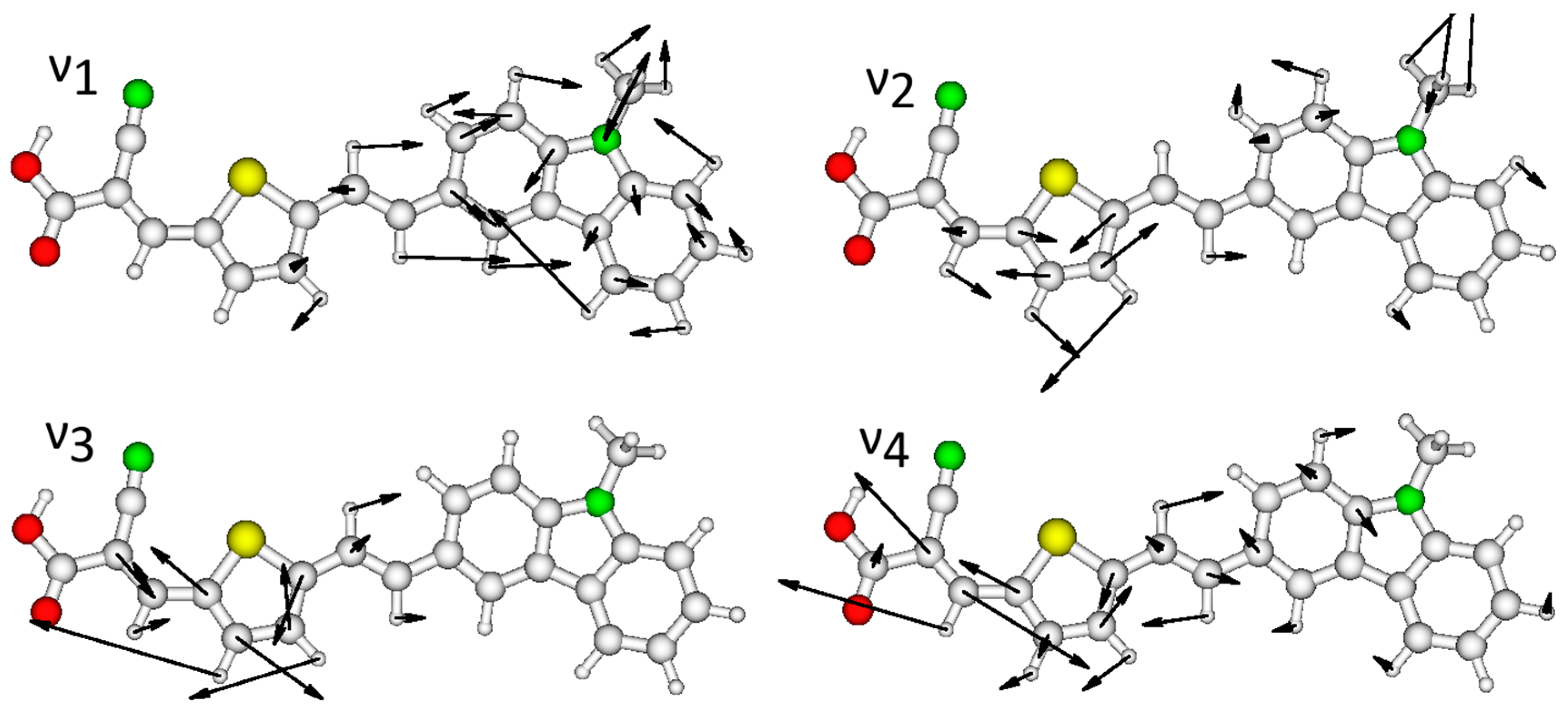
2.3. Resonance Raman Spectroscopy
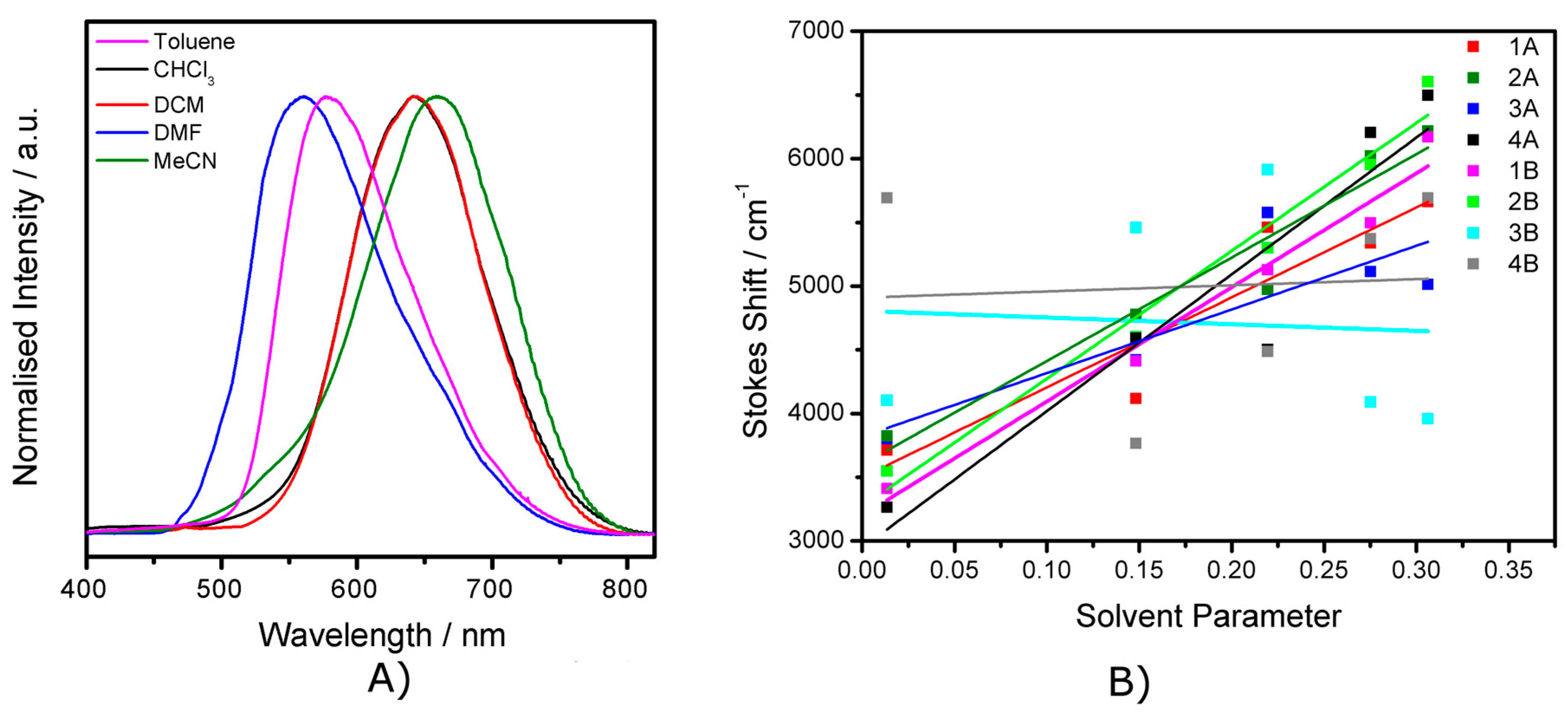
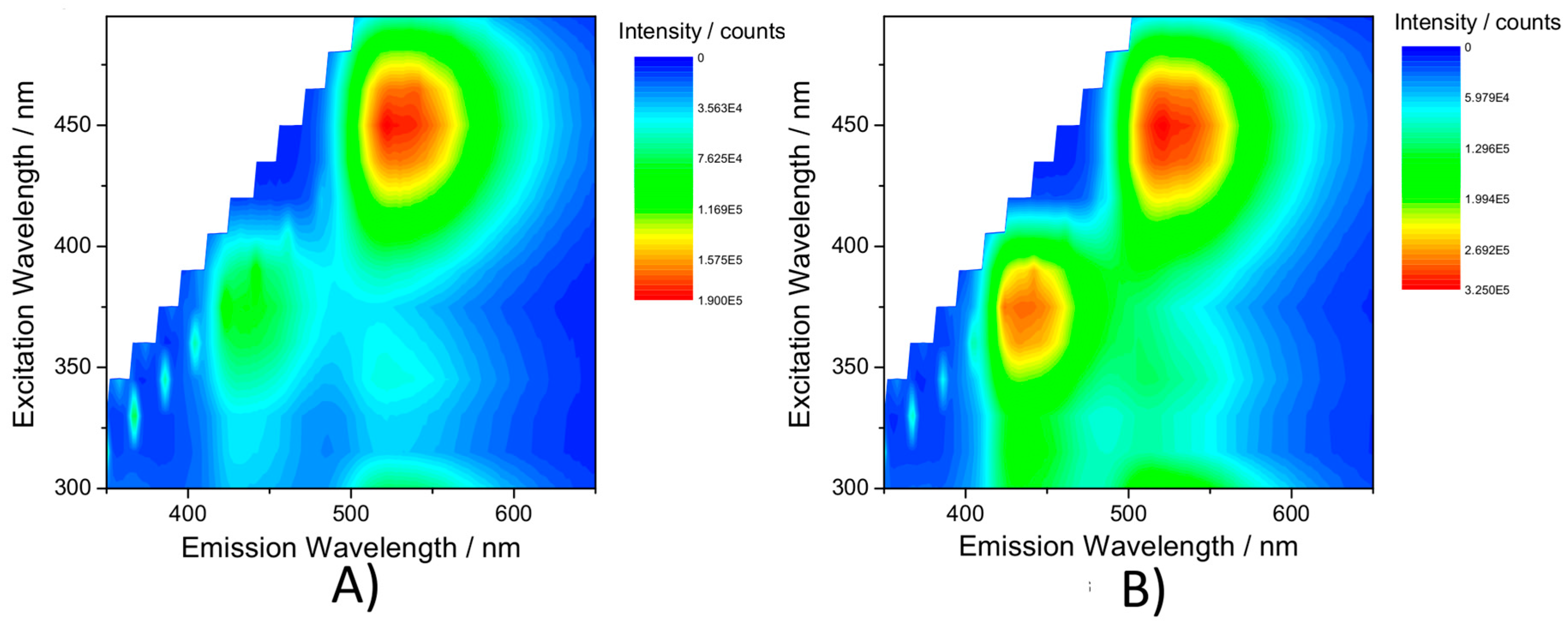
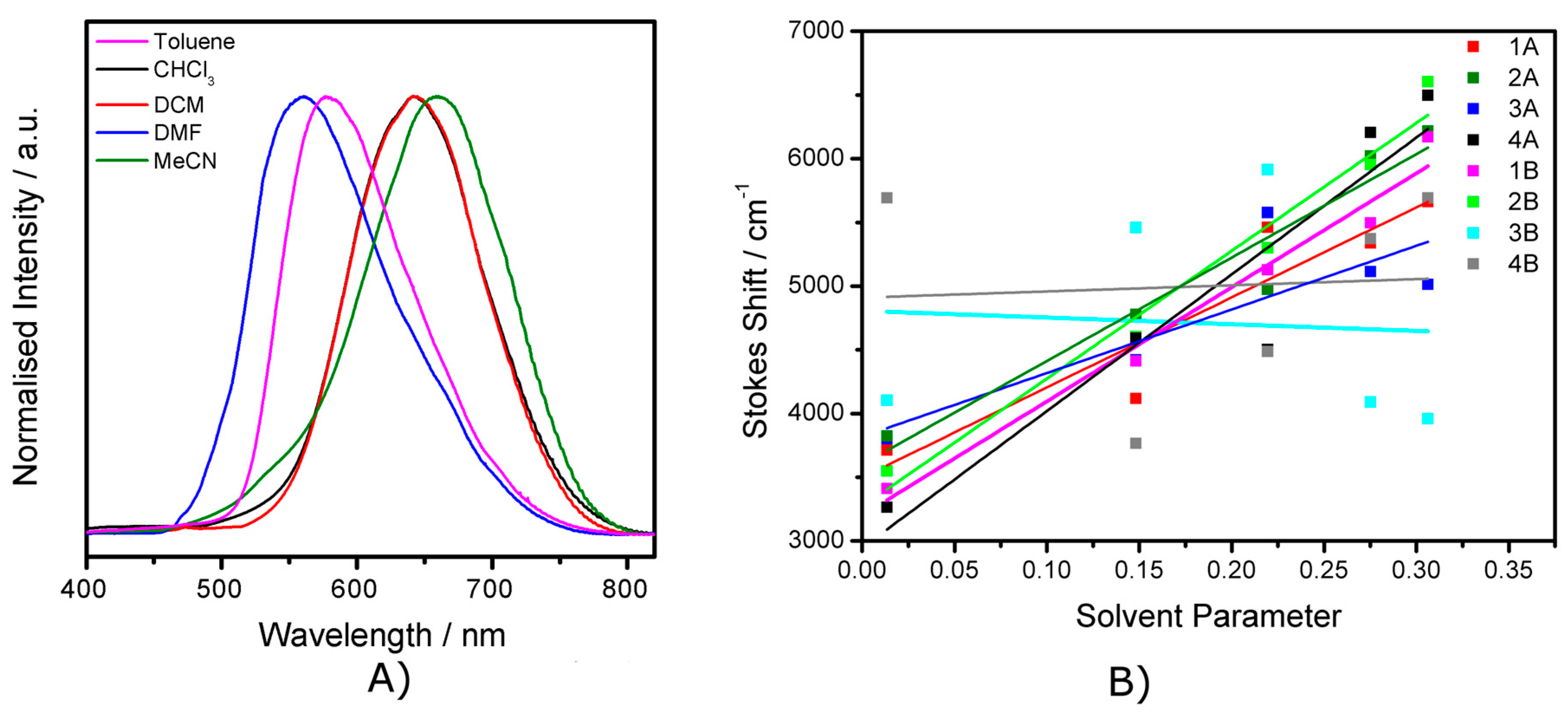
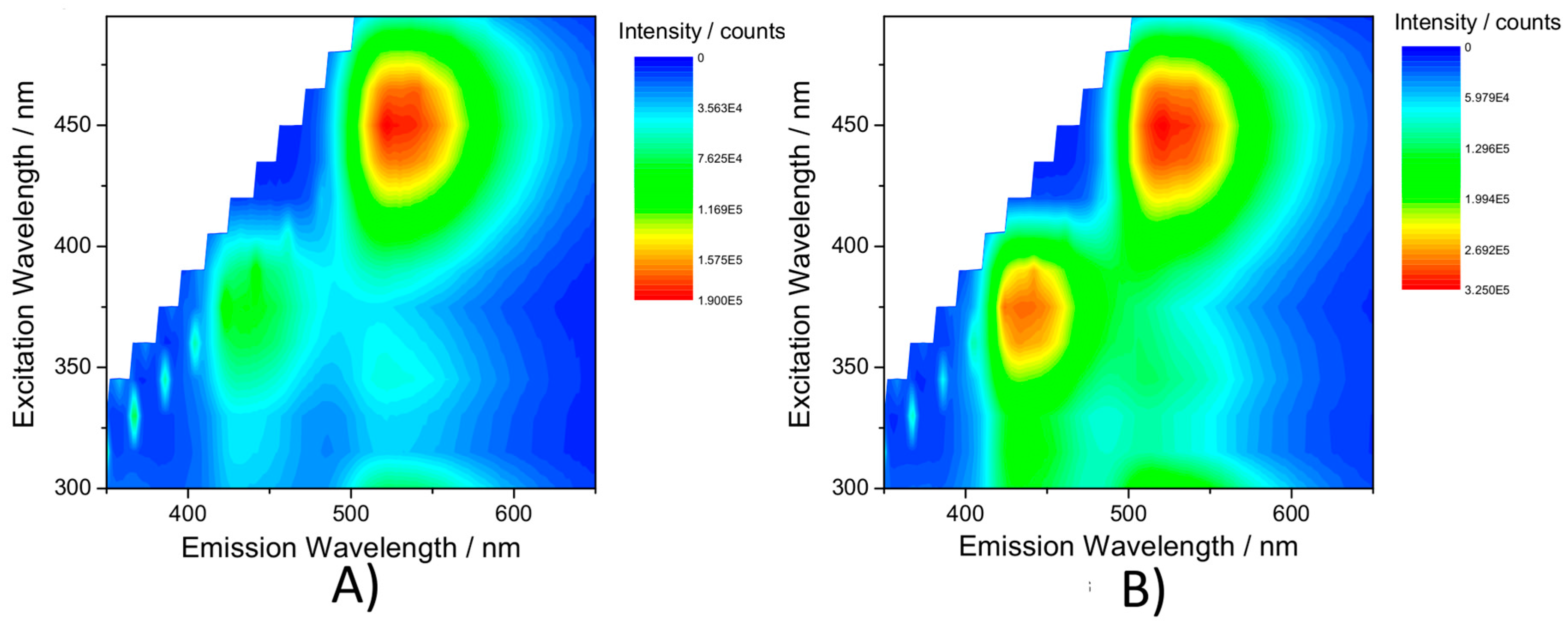
2.4. Emission Spectroscopy
2.5. The Effect of the Bridge on Properties
3. Materials and Methods
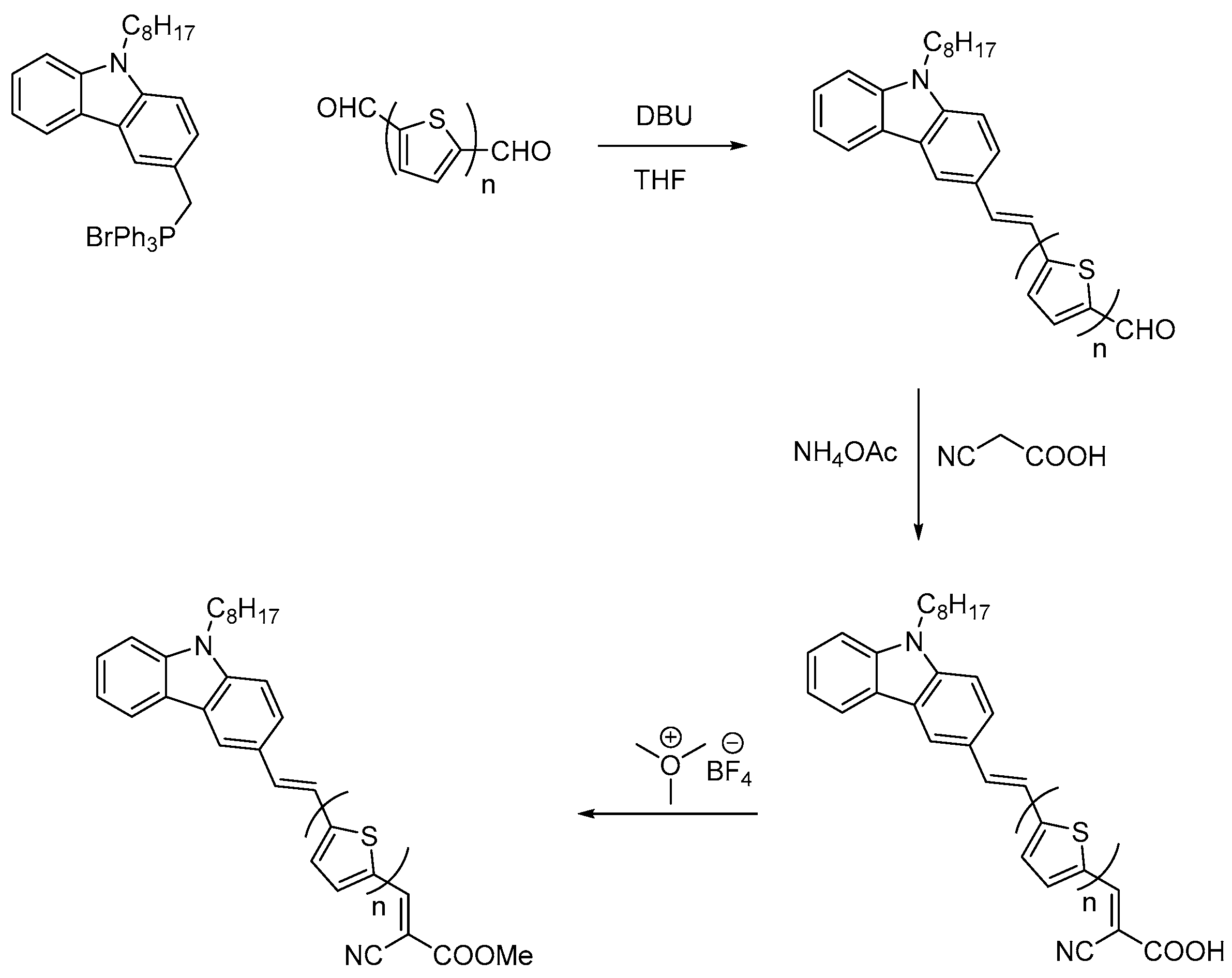
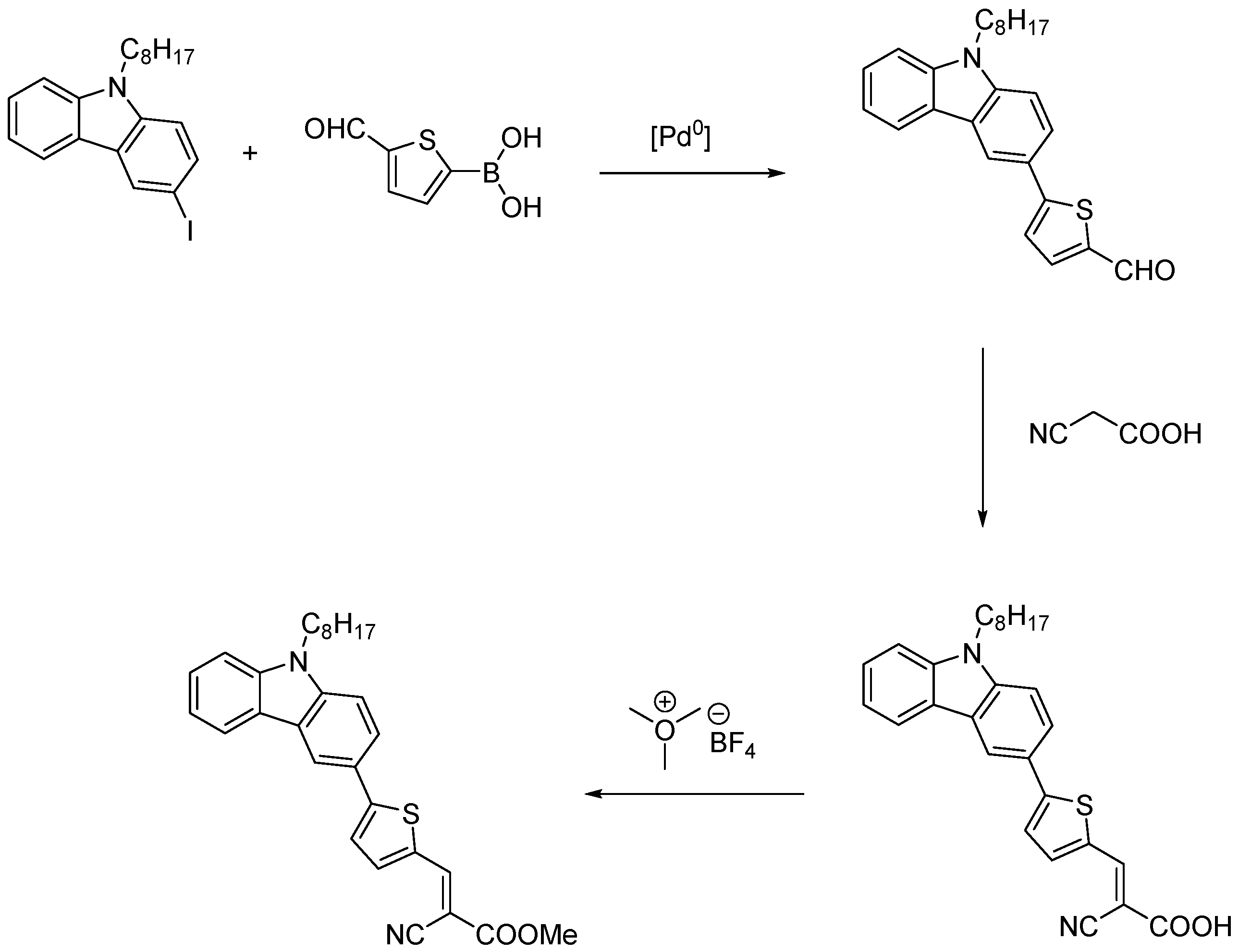
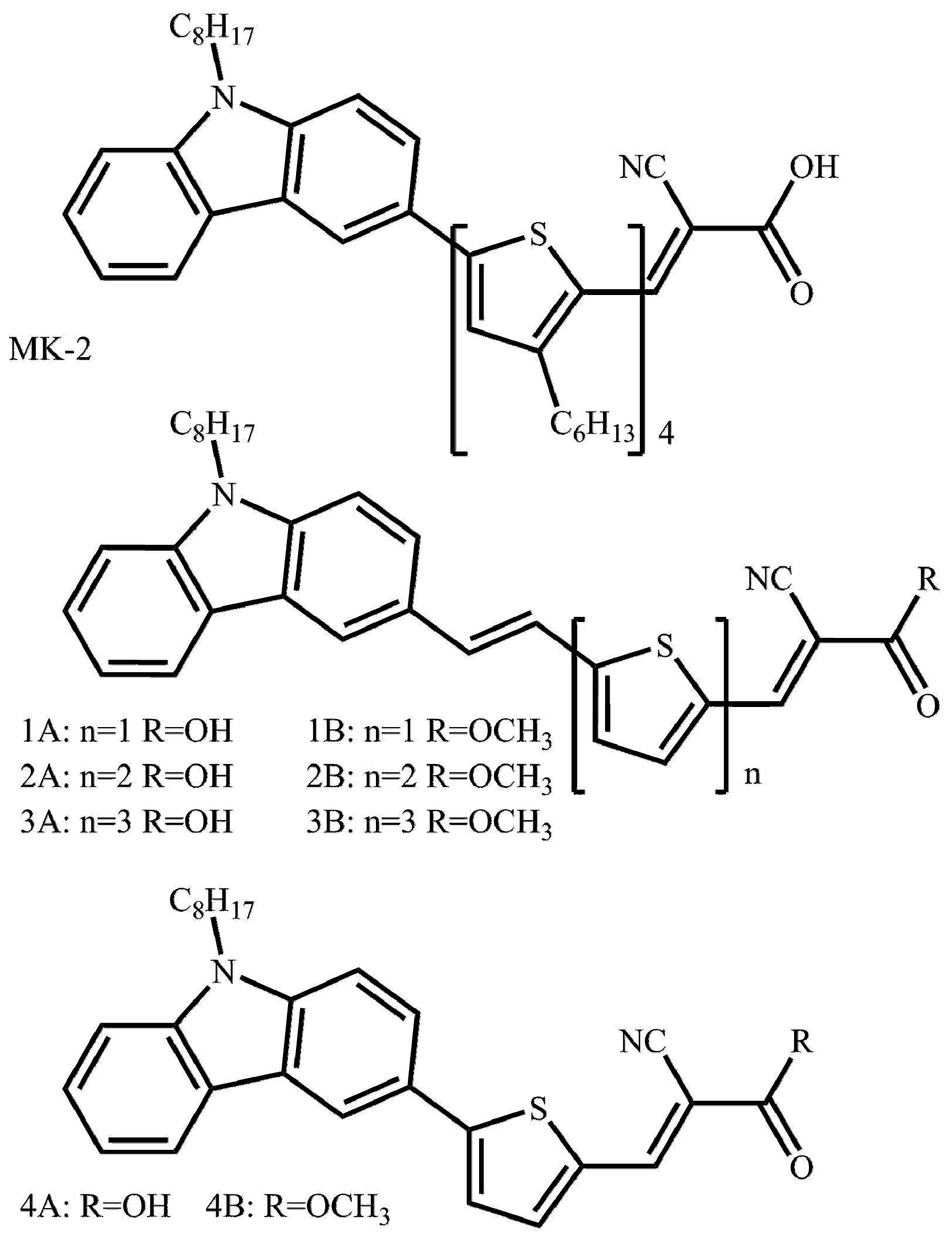
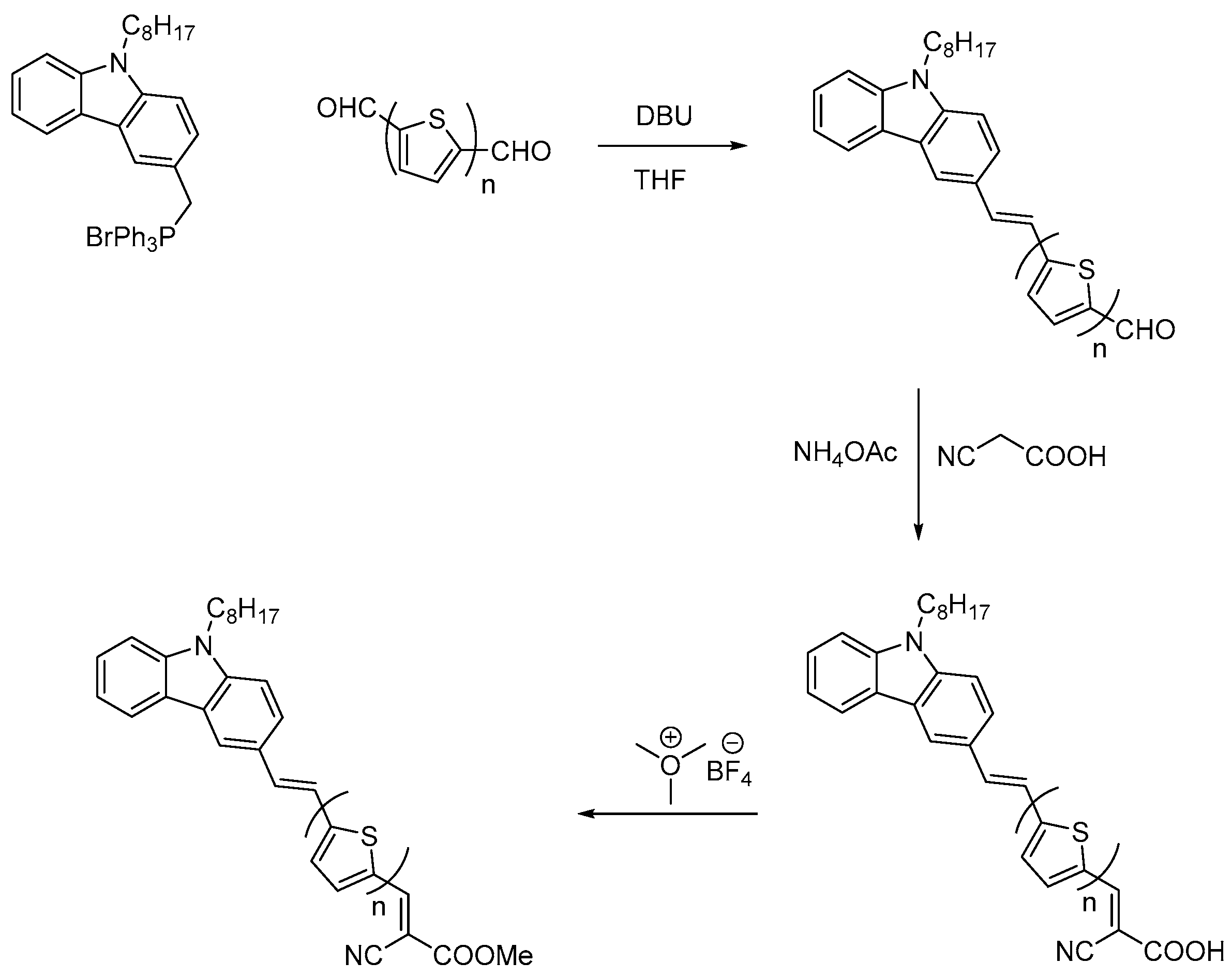
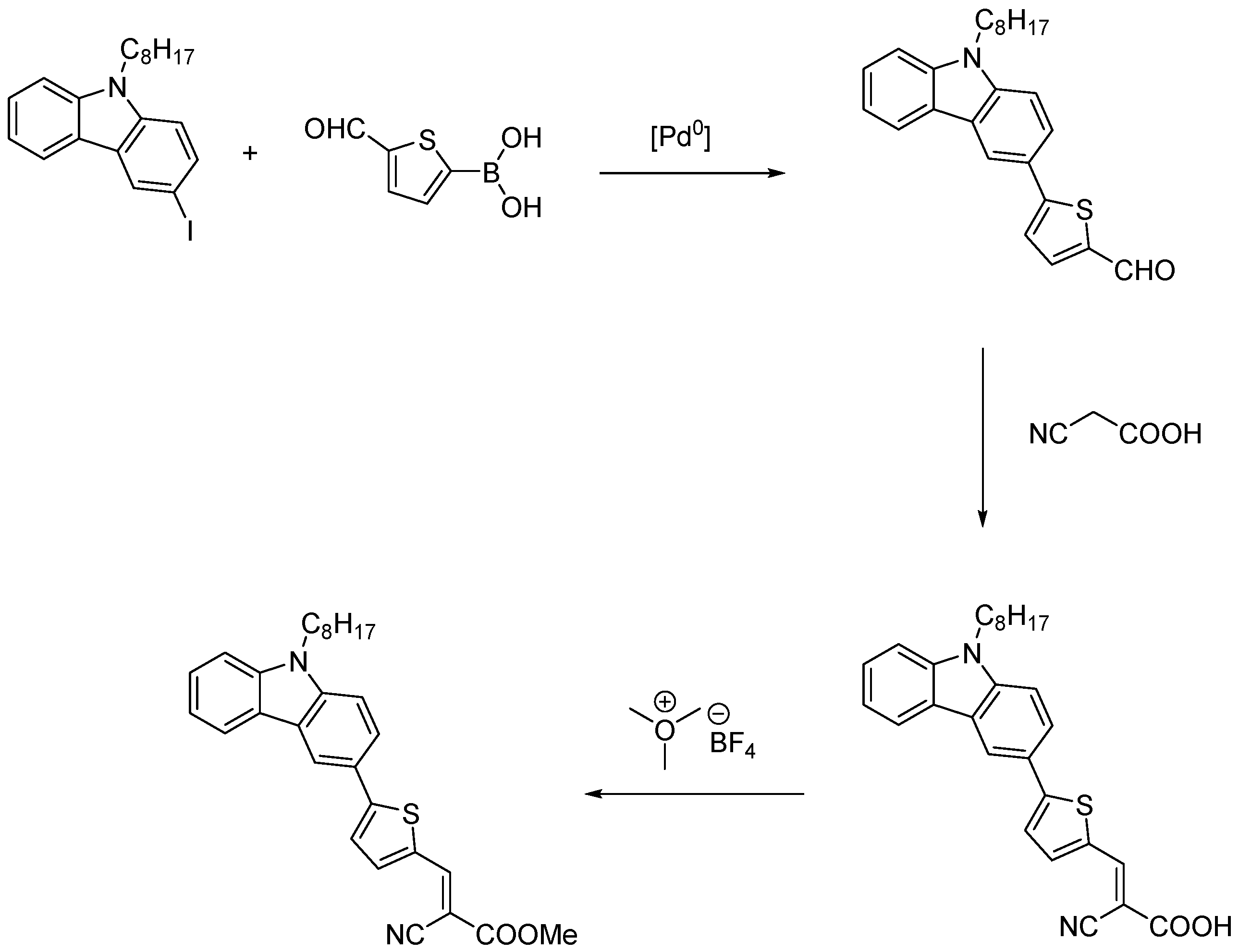
3.1. Synthesis
3.1.1. General Procedure for Wittig Condensation
3.1.2. General Procedure for the Knoevenagel Condensation
3.1.3. General Procedure for Esterification of Cyanoacrylic Acids:
3.2. Computation Methods
3.3. Physical Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martin, C.; Borreguero, C.; Kennes, K.; van der Auweraer, M.; Hofkens, J.; de Miguel, G.; García-Frutos, E.M. Simple donor–acceptor luminogen based on an azaindole derivative as solid-state emitter for organic light-emitting devices. ACS Energy Lett. 2017, 2, 2653–2658. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, D.; Zhang, S.; Tao, Y.; Huang, W. CN-containing donor-acceptor-type small-molecule materials for thermally activated delayed fluorescence OLEDs. J. Mater. Chem. C 2017, 5, 7699–7714. [Google Scholar] [CrossRef]
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913–915. [Google Scholar] [CrossRef]
- Arbačiauskienė, E.; Kazlauskas, K.; Miasojedovas, A.; Juršėnas, S.; Jankauskas, V.; Holzer, W.; Getautis, V.; Šačkus, A. Multifunctional polyconjugated molecules with carbazolyl and pyrazolyl moieties for optoelectronic applications. Synth. Met. 2010, 160, 490–498. [Google Scholar] [CrossRef]
- Xie, B.; Bi, S.; Wu, R.; Yin, L.; Ji, C.; Cai, Z.; Li, Y. Efficient small molecule photovoltaic donor based on 2,3-diphenyl-substituted quinoxaline core for solution-processed organic solar cells. RSC Adv. 2017, 7, 23779–23786. [Google Scholar] [CrossRef]
- Deng, D.; Zhang, Y.; Zhang, J.; Wang, Z.; Zhu, L.; Fang, J.; Xia, B.; Wang, Z.; Lu, K.; Ma, W.; et al. Fluorination-enabled optimal morphology leads to over 11% efficiency for inverted small-molecule organic solar cells. Nat. Commun. 2016, 7, 13740–13749. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.; Kim, C.; Nguyen, T.-Q. Small molecule solution-processed bulk heterojunction solar cells. Chem. Mater. 2011, 23, 470–482. [Google Scholar] [CrossRef]
- Gupta, A.; Ali, A.; Bilic, A.; Gao, M.; Hegedus, K.; Singh, B.; Watkins, S.E.; Wilson, G.J.; Bach, U.; Evans, R.A. Absorption enhancement of oligothiophene dyes through the use of a cyanopyridone acceptor group in solution-processed organic solar cells. Chem. Commun. 2012, 48, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Ishi-i, T.; Amemori, S.; Okamura, C.; Yanaga, K.; Kuwahara, R.; Mataka, S.; Kamada, K. Self-assembled triphenylamine-hexaazatriphenylene two-photon absorption dyes. Tetrahedron 2013, 69, 29–37. [Google Scholar] [CrossRef]
- Andreu, R.; Galán, E.; Garín, J.; Herrero, V.; Lacarra, E.; Orduna, J.; Alicante, R.; Villacampa, B. Linear and v-shaped nonlinear optical chromophores with multiple 4H-pyran-4-ylidene moieties. J. Org. Chem. 2010, 75, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.C.; Bourdakos, K.N.; Bhalla, V.; Kamtekar, K.T.; Bryce, M.R.; Fox, M.A.; Vaughan, H.L.; Dias, F.B.; Monkman, A.P. Tuning the intramolecular charge transfer emission from deep blue to green in ambipolar systems based on dibenzothiophene S,S-dioxide by manipulation of conjugation and strength of the electron donor units. J. Org. Chem. 2010, 75, 6771–6781. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hagiwara, H.; Ogata, Y.; Staykov, A.; Bishop, S.R.; Perry, N.H.; Chang, Y.J.; Ida, S.; Tanaka, K.; Ishihara, T. Impact of alkoxy chain length on carbazole-based, visible light-driven, dye sensitized photocatalytic hydrogen production. J. Mater. Chem. A 2015, 3, 21713–21721. [Google Scholar] [CrossRef]
- Karpicz, R.; Puzinas, S.; Krotkus, S.; Kazlauskas, K.; Jursenas, S.; Grazulevicius, J.V.; Grigalevicius, S.; Gulbinas, V. Impact of intramolecular twisting and exciton migration on emission efficiency of multifunctional fluorene-benzothiadiazole-carbazole compounds. J. Chem. Phys. 2011, 134, 204508–204517. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Jiang, K.-J.; Li, Y.-F.; Li, S.-L.; Yang, L.-M. Efficient structural modification of triphenylamine-based organic dyes for dye-sensitized solar cells. J. Phys. Chem. C 2008, 112, 11591–11599. [Google Scholar] [CrossRef]
- Barnsley, J.E.; Shillito, G.E.; Larsen, C.B.; van der Salm, H.; Wang, L.E.; Lucas, N.T.; Gordon, K.C. Benzo[c][1,2,5]thiadiazole donor–acceptor dyes: A synthetic, spectroscopic, and computational study. J. Phys. Chem. A 2016, 120, 1853–1866. [Google Scholar] [CrossRef] [PubMed]
- Barnsley, J.E.; Lomax, B.A.; McLay, J.R.W.; Larsen, C.B.; Lucas, N.T.; Gordon, K.C. Flicking the switch on donor–acceptor interactions in hexaazatrinaphthalene dyes: A spectroscopic and computational study. ChemPhotoChem 2017, 1, 432–441. [Google Scholar] [CrossRef]
- Hara, K.; Sato, T.; Katoh, R.; Furube, A.; Ohga, Y.; Shinpo, A.; Suga, S.; Sayama, K.; Sugihara, H.; Arakawa, H. Molecular design of coumarin dyes for efficient dye-sensitized solar cells. J. Phys. Chem. B 2003, 107, 597–606. [Google Scholar] [CrossRef]
- Zhang, L.; Cole, J.M.; Dai, C. Variation in optoelectronic properties of azo dye-sensitized TiO2 semiconductor interfaces with different adsorption anchors: Carboxylate, sulfonate, hydroxyl and pyridyl groups. ACS Appl. Mater. Interfaces 2014, 6, 7535–7546. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hong, J.; Oh, J.H.; Yang, C. Naphthalene diimide incorporated thiophene-free copolymers with acene and heteroacene units: Comparison of geometric features and electron-donating strength of co-units. Chem. Mater. 2013, 25, 3251–3259. [Google Scholar] [CrossRef]
- Shillito, G.E.; Larsen, C.B.; McLay, J.R.W.; Lucas, N.T.; Gordon, K.C. Effect of bridge alteration on ground-and excited-state properties of Ruthenium(II) complexes with electron-donor-substituted dipyrido[3,2-a:2′,3′-c]phenazine ligands. Inorg. Chem. 2016, 55, 11170–11184. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.D.; Mikroyannidis, J.A.; Roy, M.S.; Thomas, K.R.J.; Ball, R.J.; Kurchania, R. Dithienylthienothiadiazole-based organic dye containing two cyanoacrylic acid anchoring units for dye-sensitized solar cells. RSC Adv. 2012, 2, 11457–11464. [Google Scholar] [CrossRef]
- Wang, Z.-S.; Koumura, N.; Cui, Y.; Takahashi, M.; Sekiguchi, H.; Mori, A.; Kubo, T.; Furube, A.; Hara, K. Hexylthiophene-functionalized carbazole dyes for efficient molecular photovoltaics: Tuning of solar-cell performance by structural modification. Chem. Mater. 2008, 20, 3993–4003. [Google Scholar] [CrossRef]
- Soni, S.S.; Fadadu, K.B.; Vaghasiya, J.V.; Solanki, B.G.; Sonigara, K.K.; Singh, A.; Das, D.; Iyer, P.K. Improved molecular architecture of D-π-A carbazole dyes: 9% PCE with a cobalt redox shuttle in dye sensitized solar cells. J. Mater. Chem. A 2015, 3, 21664–21671. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Afroz, M.A.; Sonigara, K.K.; Raju, T.B.; Soni, S.S.; Iyer, P.K. Effect of fluorine substitution and position on phenylene spacer in carbazole based organic sensitizers for dye sensitized solar cells. Phys. Chem. Chem. Phys. 2017, 19, 28579–28587. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hagiwara, H.; Iribe, A.; Ogata, Y.; Shiomi, K.; Staykov, A.; Ida, S.; Tanaka, K.; Ishihara, T. Spacer effects in metal-free organic dyes for visible-light-driven dye-sensitized photocatalytic hydrogen production. J. Mater. Chem. A 2014, 2, 12952–12961. [Google Scholar] [CrossRef]
- Clifford, J.N.; Palomares, E.; Nazeeruddin, M.K.; Grätzel, M.; Nelson, J.; Li, X.; Long, N.J.; Durrant, J.R. Molecular control of recombination dynamics in dye-sensitized nanocrystalline TiO2 films: Free energy vs distance dependence. J. Am. Chem. Soc. 2004, 126, 5225–5233. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.M.; Gordon, K.C.; Officer, D.L.; Hall, S.B.; Collis, G.E.; Burrell, A.K. Theoretical and spectroscopic study of a series of styryl-substituted terthiophenes. J. Phys. Chem. A 2003, 107, 11505–11516. [Google Scholar] [CrossRef]
- Diaz-Quijada, G.A.; Weinberg, N.; Holdcroft, S.; Pinto, B.M. Investigation of barriers to conformational interchange in oligothiophenes and oligo(thienyl)furans. J. Phys. Chem. A 2002, 106, 1266–1276. [Google Scholar] [CrossRef]
- DiCésare, N.; Belletête, M.; Marrano, C.; Leclerc, M.; Durocher, G. Conformational analysis (ab initio HF/3-21g*) and optical properties of symmetrically disubstituted terthiophenes. J. Phys. Chem. A 1998, 102, 5142–5149. [Google Scholar] [CrossRef]
- McGoverin, C.M.; Walsh, T.J.; Gordon, K.C.; Kay, A.J.; Woolhouse, A.D. Predicting nonlinear optical properties in push–pull molecules based on methyl pyridinium donor and 3-cyano-5,5-dimethyl-2(5H)-furanylidene-propanedinitrile acceptor units using vibrational spectroscopy and density functional theory. Chem. Phys. Lett. 2007, 443, 298–303. [Google Scholar] [CrossRef]
- Earles, J.C.; Gordon, K.C.; Stephenson, A.W.I.; Partridge, A.C.; Officer, D.L. Spectroscopic and computational study of β-ethynylphenylene substituted zinc and free-base porphyrins. Phys. Chem. Chem. Phys. 2011, 13, 1597–1605. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Møller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. A 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Lind, S.J.; Gordon, K.C.; Gambhir, S.; Officer, D.L. A spectroscopic and DFT study of thiophene-substituted metalloporphyrins as dye-sensitized solar cell dyes. Phys. Chem. Chem. Phys. 2009, 11, 5598–5607. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.; Hernández, V.; Kanemitsu, Y.; Navarrete, J.T.L. Infrared and raman spectra of a new radical cation charged defect created on a well–barrier–well thiophene-based oligomer. J. Raman Spectrosc. 2000, 31, 565–570. [Google Scholar] [CrossRef]
- Hernández, V.; Casado, J.; Kanemitsu, Y.; López Navarrete, J.T. Vibrational study of a well–barrier–well thiophene–based oligomer in relation to the effective π-conjugation length. J. Chem. Phys. 1999, 110, 690–6915. [Google Scholar] [CrossRef]
- Hernandez, V.; Castiglioni, C.; Del Zoppo, M.; Zerbi, G. Confinement potential and π-electron delocalization in polyconjugated organic materials. Phys. Rev. B 1994, 50, 9815–9823. [Google Scholar] [CrossRef]
- Furukawa, Y. Electronic absorption and vibrational spectroscopies of conjugated conducting polymers. J. Phys. Chem. A 1996, 100, 15644–15653. [Google Scholar] [CrossRef]
- Svedberg, F.; Alaverdyan, Y.; Johansson, P.; Käll, M. Raman spectroscopic studies of terthiophenes for molecular electronics. J. Phys. Chem. B 2006, 110, 25671–25677. [Google Scholar] [CrossRef] [PubMed]
- Huff, G.S.; Gallaher, J.K.; Hodgkiss, J.M.; Gordon, K.C. No single DFT method can predict Raman cross-sections, frequencies and electronic absorption maxima of oligothiophenes. Synth. Met. 2017, 231, 1–6. [Google Scholar] [CrossRef]
- Mitchell, R.; Wagner, K.; Barnsley, J.E.; van der Salm, H.; Gordon, K.C.; Officer, D.L.; Wagner, P. Synthesis and light-harvesting potential of cyanovinyl β-substituted porphyrins and dyads. Eur. J. Org. Chem. 2017, 2017, 5750–5762. [Google Scholar] [CrossRef]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. of Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Reish, M.E.; Kay, A.J.; Teshome, A.; Asselberghs, I.; Clays, K.; Gordon, K.C. Testing computational models of hyperpolarizability in a merocyanine dye using spectroscopic and DFT methods. J. Phys. Chem. A 2012, 116, 5453–5463. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; pp. 185–197. ISBN 0-306-46093-9. [Google Scholar]
- Suppan, P. Chemistry and Light; The Royal Society of Chemistry: Cambridge, UK, 1994; pp. 1–295. [Google Scholar]
- Akerlof, G. Dielectric constants of some organic solvent-water mixtures at various temperatures. J. Am. Chem. Soc. 1932, 54, 4125–4139. [Google Scholar] [CrossRef]
- Elliott, A.B.S.; Horvath, R.; Sun, X.-Z.; Gardiner, M.G.; Müllen, K.; Lucas, N.T.; George, M.W.; Gordon, K.C. Long-lived charge transfer excited states in HBC-polypyridyl complex hybrids. Inorg. Chem. 2016, 55, 4710–4719. [Google Scholar] [CrossRef] [PubMed]
- Reish, M.E.; Huff, G.S.; Lee, W.; Uddin, M.A.; Barker, A.J.; Gallaher, J.K.; Hodgkiss, J.M.; Woo, H.Y.; Gordon, K.C. Thermochromism, Franck–Condon analysis and interfacial dynamics of a donor–acceptor copolymer with a low band gap. Chem. Mater. 2015, 27, 2770–2779. [Google Scholar] [CrossRef]
- Pastore, M.; Mosconi, E.; De Angelis, F.; Grätzel, M. A computational investigation of organic dyes for dye-sensitized solar cells: Benchmark, strategies, and open issues. J. Phys. Chem. C 2010, 114, 7205–7212. [Google Scholar] [CrossRef]
- Clark, R.J.H.; Dines, T.J. Resonance Raman spectroscopy, and its application to inorganic chemistry. Angewandte Chemie Int. Ed. 1986, 25, 131–158. [Google Scholar] [CrossRef]
- Tsuboi, M.; Hirakawa, A.Y. A correlation between vibronic coupling, adiabatic potential, and Raman scattering: A theoretical background of a proposed rule. J. Raman Spectrosc. 1976, 5, 75–86. [Google Scholar] [CrossRef]
- Horvath, R.; Huff, G.S.; Gordon, K.C.; George, M.W. Probing the excited state nature of coordination complexes with blended organic and inorganic chromophores using vibrational spectroscopy. Coord. Chem. Rev. 2016, 325, 41–58. [Google Scholar] [CrossRef]
- Horvath, R.; Gordon, K.C. Understanding excited-state structure in metal polypyridyl complexes using resonance Raman excitation profiles, time-resolved resonance Raman spectroscopy and density functional theory. Coord. Chem. Rev. 2010, 254, 2505–2518. [Google Scholar] [CrossRef]
- Albrecht, A.C. On the theory of raman intensities. J. Chem. Phys. 1961, 34, 1476–1484. [Google Scholar] [CrossRef]
- Hirakawa, A.Y.; Tsuboi, M. Molecular geometry in an excited electronic state and a preresonance Raman effect. Science 1975, 188, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.M.; Gordon, K.C.; Kwok, W.M.; Phillips, D.L.; Officer, D.L. Tuning from π,π* to charge-transfer excited states in styryl-substituted terthiophenes: An ultrafast and steady-state emission study. J. Phys. Chem. A 2006, 110, 7696–7702. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Crowe, L.L.; Wagner, P.; Gambhir, S.; Partridge, A.C.; Earles, J.C.; Clarke, T.M.; Gordon, K.C.; Officer, D.L. Indanedione-substituted poly(terthiophene)s: Processable conducting polymers with intramolecular charge transfer interactions. Macromolecules 2010, 43, 3817–3827. [Google Scholar] [CrossRef]
- Larsen, C.B.; Barnsley, J.E.; van der Salm, H.; Fraser, M.G.; Lucas, N.T.; Gordon, K.C. Synthesis and optical properties of unsymmetrically substituted triarylamine hexaazatrinaphthalenes. Eur. J. Org. Chem. 2017, 2017, 2432–2440. [Google Scholar] [CrossRef]
- Leriche, P.; Raimundo, J.-M.; Turbiez, M.; Monroche, V.; Allain, M.; Sauvage, F.-X.; Roncali, J.; Frere, P.; Skabara, P.J. Linearly extended tetrathiafulvalene analogues with fused thiophene units as π-conjugated spacers. J. Mater. Chem. 2003, 13, 1324–1332. [Google Scholar] [CrossRef]
- Brzeczek, A.; Ledwon, P.; Data, P.; Zassowski, P.; Golba, S.; Walczak, K.; Lapkowski, M. Synthesis and properties of 1,3,5-tricarbazolylbenzenes with star-shaped architecture. Dyes Pigment. 2015, 113, 640–648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. {III}. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Pascual-ahuir, J.L.; Silla, E.; Tuñon, I. Gepol: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. Gaussview Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Arslan, H.; Algül, Ö. Synthesis and ab initio/DFT studies on 2-(4-methoxyphenyl)benzo[d]thiazole. Int. J. Mol. Sci. 2007, 8, 760–776. [Google Scholar] [CrossRef]
- Walsh, P.J.; Gordon, K.C.; Lundin, N.J.; Blackman, A.G. Photoexcitation in Cu(I) and Re(I) complexes containing substituted dipyrido[3,2-a:2′,3′-c]phenazine: A spectroscopic and density functional theoretical study. J. Phys. Chem. A 2005, 109, 5933–5942. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Experimental | Calculated | ||||||
|---|---|---|---|---|---|---|---|---|
| B3LYP | CAM-B3LYP | |||||||
| nm | eV | nm | eV | eVexpt–eVb3lyp | nm | eV | eVexpt–eVcam-b3lyp | |
| 1A | 487 | 2.55 | 533 | 2.33 | 0.22 | 452 | 2.74 | −0.20 |
| 2A | 509 | 2.44 | 607 | 2.04 | 0.39 | 487 | 2.55 | −0.11 |
| 3A | 508 | 2.44 | 664 | 1.87 | 0.57 | 508 | 2.44 | 0.00 |
| 4A | 467 | 2.66 | 480 | 2.58 | 0.07 | 385 | 3.22 | −0.57 |
| 1B | 494 | 2.51 | 522 | 2.38 | 0.13 | 422 | 2.94 | −0.43 |
| 2B | 517 | 2.40 | 596 | 2.08 | 0.32 | 452 | 2.74 | −0.34 |
| 3B | 519 | 2.39 | 656 | 1.89 | 0.50 | 508 | 2.44 | −0.05 |
| 4B | 473 | 2.62 | 491 | 2.53 | 0.10 | 392 | 3.16 | −0.54 |
| Compound | Dihedral Angle/° |
|---|---|
| 1A | 0.4 |
| 1B | 0.1 |
| 4A | 23.8 |
| 4B | 24.3 |
| Compound | λem/nm | ||||
|---|---|---|---|---|---|
| Toluene | CHCl3 | DCM | DMF | MeCN | |
| 1A | 577 | 641 | 642 | 561 | 659 |
| 2A | 605 | 652 | 703 | 586 | 654 |
| 3A | 622 | 672 | 715 | 602 | 649 |
| 4A | 527 | 588 | 577 | 518 | 517 |
| 1B | 590 | 617 | 635 | 665 | 662 |
| 2B | 597 | 663 | 684 | 716 | 731 |
| 3B | 620 | 701 | 718 | 627 | 611 |
| 4B | 522 | 552 | 567 | 597 | 596 |
| Compound | Lifetime/ns | Quantum Yield | Decay Rates in DCM/s−1 | |||
|---|---|---|---|---|---|---|
| Toluene | DCM | Toluene | DCM | kr | knr | |
| 1B | * | 5.1 ± 0.4 | 0.07 | 0.35 | 6.9 × 107 | 1.3 × 108 |
| 2B | 5.5 ± 0.1 | 17.9 ± 0.4 | 0.46 | 0.70 | 3.9 × 107 | 1.7 × 107 |
| 3B | * | 6.1 ± 0.2 | 0.23 | 0.23 | 3.8 × 107 | 1.3 × 108 |
| 4B (450 nm) | 5.8 ± 0.1 | 2.5 ± 0.1 | 0.02 | 0.05 | 1.4 × 107 # | 2.7 × 108 # |
| 4B (550 nm) | 6.4 ± 0.1 | 3.5 ± 0.2 | ||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutton, J.J.; Barnsley, J.E.; Mapley, J.I.; Wagner, P.; Officer, D.L.; Gordon, K.C. Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study. Molecules 2018, 23, 421. https://doi.org/10.3390/molecules23020421
Sutton JJ, Barnsley JE, Mapley JI, Wagner P, Officer DL, Gordon KC. Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study. Molecules. 2018; 23(2):421. https://doi.org/10.3390/molecules23020421
Chicago/Turabian StyleSutton, Joshua J., Jonathan E. Barnsley, Joseph I. Mapley, Pawel Wagner, David L. Officer, and Keith C. Gordon. 2018. "Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study" Molecules 23, no. 2: 421. https://doi.org/10.3390/molecules23020421
APA StyleSutton, J. J., Barnsley, J. E., Mapley, J. I., Wagner, P., Officer, D. L., & Gordon, K. C. (2018). Modulation of Donor-Acceptor Distance in a Series of Carbazole Push-Pull Dyes; A Spectroscopic and Computational Study. Molecules, 23(2), 421. https://doi.org/10.3390/molecules23020421