Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition

 ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
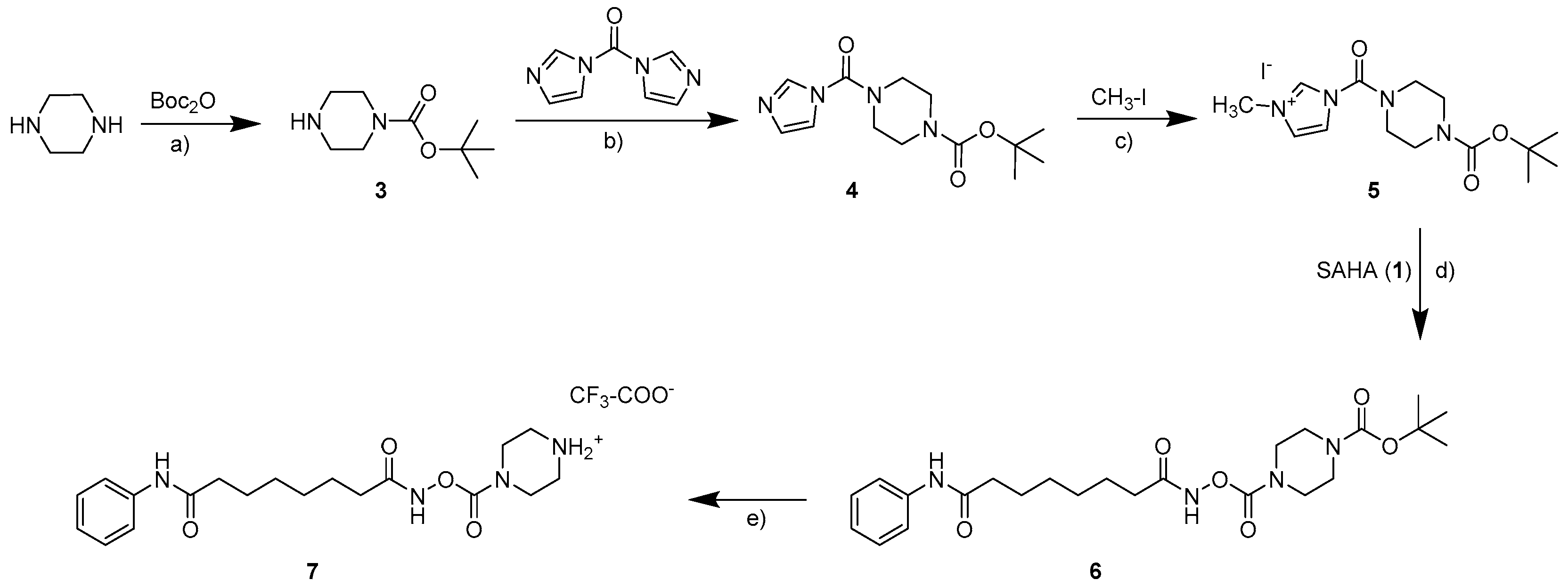
2.1. Synthesis
2.2. In Vitro Testing
2.3. Stability Analysis
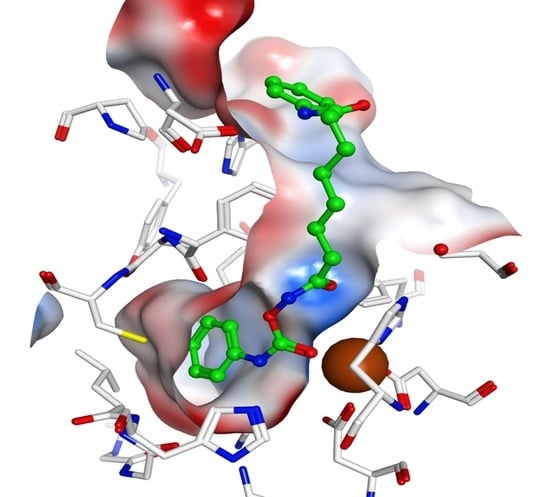
2.4. Docking Studies
2.5. Cellular Testing
2.6. Carbamate of Bufexamac
3. Materials and Methods
3.1. Synthesis
3.1.1. General
3.1.2. General Procedure for the Preparation of the Carbamates 2a–2d and 9
3.2. Enzymes and In Vitro Testing
3.3. Docking Studies
3.4. Cellular Testing
3.4.1. Cell Proliferation Assay
3.4.2. Acetylation Levels
3.4.3. Cellular HDAC Inhibition Assay
3.5. Stability Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keller, K.; Jung, M. Histone Deacteylase (HDAC) Inhibitors in Recent Clinical Trials for Cancer Therapy; Springer: Berlin, Germany, 2014. [Google Scholar]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Ren, S.; Li, W.; Wang, X.; He, M.; Guo, Y.; Sun, L.; He, Y.; Ge, Y.; Yu, Q. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Barbarotta, L.; Hurley, K. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [PubMed]
- Libby, E.N.; Becker, P.S.; Burwick, N.; Green, D.J.; Holmberg, L.; Bensinger, W.I. Panobinostat: A review of trial results and future prospects in multiple myeloma. Expert Rev. Hematol. 2015, 8, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Schlimme, S.; Hauser, A.T.; Carafa, V.; Heinke, R.; Kannan, S.; Stolfa, D.A.; Cellamare, S.; Carotti, A.; Altucci, L.; Jung, M.; et al. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem 2011, 6, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Seki, H.; Pellett, S.; Silhar, P.; Stowe, G.N.; Blanco, B.; Lardy, M.A.; Johnson, E.A.; Janda, K.D. Synthesis/biological evaluation of hydroxamic acids and their prodrugs as inhibitors for Botulinum neurotoxin A light chain. Bioorg. Med. Chem. 2014, 22, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Stowell, J.C.; Huot, R.I.; Van Voast, L. The synthesis of N-hydroxy-N′-phenyloctanediamide and its inhibitory effect on proliferation of AXC rat prostate cancer cells. J. Med. Chem. 1995, 38, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Esposito, M.; Sbardella, G.; Massa, S. A new facile and expeditious synthesis of N-hydroxy-N′-phenyloctanediamide, a potent inducer of terminal cytodifferentiation. Org. Prep. Proced. Int. 2001, 33, 391–394. [Google Scholar] [CrossRef]
- Batey, R.A.; Santhakumar, V.; Yoshina-Ishii, C.; Taylor, S.D. An efficient new protocol for the formation of unsymmetrical tri- and tetrasubstituted ureas. Tetrahedron Lett. 1998, 39, 6267–6270. [Google Scholar] [CrossRef]
- Batey, R.A.; Yoshina-Ishii, C.; Taylor, S.D.; Santhakumar, V. A new protocol for the formation of carbamates and thiocarbamates using carbamoyl imidazolium salts. Tetrahedron Lett. 1999, 40, 2669–2672. [Google Scholar] [CrossRef]
- Grzyb, J.A.; Batey, R.A. Carbamoylimidazolium salts as diversification reagents: An application to the synthesis of tertiary amides from carboxylic acids. Tetrahedron Lett. 2003, 44, 7485–7488. [Google Scholar] [CrossRef]
- Grzyb, J.A.; Shen, M.; Yoshina-Ishii, C.; Chi, W.; Brown, R.S.; Batey, R.A. Carbamoylimidazolium and thiocarbamoylimidazolium salts: Novel reagents for the synthesis of ureas, thioureas, carbamates, thiocarbamates and amides. Tetrahedron 2005, 61, 7153–7175. [Google Scholar] [CrossRef]
- Heltweg, B.; Trapp, J.; Jung, M. In vitro assays for the determination of histone deacetylase activity. Methods 2005, 36, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.L.; Schwabe, J.W.R. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.L.; Saito, M.; Hess, D.; Wang, X.N.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Bressi, J.C.; Jennings, A.J.; Skene, R.; Wu, Y.Q.; Melkus, R.; De Jong, R.; O’Connell, S.; Grimshaw, C.E.; Navre, M.; Gangloff, A.R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Brosch, G.; Loidl, P.; Jung, M. A non-isotopic assay for histone deacetylase activity. Nucleic Acids Res. 1999, 27, 2057–2058. [Google Scholar] [CrossRef] [PubMed]
- Bonfils, C.; Kalita, A.; Dubay, M.; Siu, L.L.; Carducci, M.A.; Reid, G.; Martell, R.E.; Besterman, J.M.; Li, Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin. Cancer Res. 2008, 14, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Kannan, S.; Hauser, A.T.; Moraes Mourao, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke Schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Suite 2012 Protein Preparation Wizard, Epik Version 2.3; Impact Version 5.8; Prime Version 3.1; Schrödinger, LLC: New York, NY, USA, 2012.
- Molecular Operating Environment (MOE), 2012.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2012.
- Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A.T.; Shaik, T.B.; Duclaud, S.; Robaa, D.; Erdmann, F.; et al. Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 2016, 59, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J. Med. Chem. 2016, 59, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, L.; Dobler, M.R.; Radetich, B.; Zhu, Y.; Atadja, P.W.; Claiborne, T.; Grob, J.E.; McRiner, A.; Pancost, M.R.; Patnaik, A.; et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011, 19, 4626–4634. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) ± SE or Inhibition (%) @ Conc. (µM) | Release of SAHA (%) | ||
|---|---|---|---|---|
| HDAC1 | HDAC6 | HDAC8 | ||
| SAHA (1) | 0.065 ± 0.002 | 0.061 ± 0.002 | 5.74 ± 0.08 | - |
| 2a | 1.52 ± 0.02 | 2.11 ± 0.02 | 35 @ 100 | 2.2 |
| 2b | 0.74 ± 0.01 | 0.637 ± 0.002 | 52 @ 100 | 0.5 |
| 2c | 0.79 ± 0.01 | 0.58 ± 0.01 | 68 @ 100 | 14.5 |
| 2d | 0.132 ± 0.001 | 0.187 ± 0.007 | 6.49 ± 0.12 | 4.6 |
| 7 | 5.09 ± 0.26 | 3.71 ± 0.01 | not tested | 3.7 |
| Compound | GI50 (µM) ± SE | IC50 (µM) ± SE | ||
|---|---|---|---|---|
| HL60 | U937 | HL60 | U937 | |
| SAHA | 0.54 ± 0.03 | 0.43 ± 0.09 | 0.65 ± 0.05 | 0.73 ± 0.005 |
| 2a | 1.02 ± 0.09 | 0.49 ± 0.09 | 3.25 ± 0.02 | 5.30 ± 0.59 |
| 2b | 1.06 ± 0.11 | 0.68 ± 0.09 | 1.89 ± 0.04 | 3.02 ± 0.03 |
| 2c | 0.99 ± 0.17 | 0.88 ± 0.13 | 1.42 ± 0.53 | 2.07 ± 0.04 |
| 2d | 0.58 ± 0.05 | 0.42 ± 0.13 | 0.69 ± 0.004 | 0.71 ± 0.002 |
| 7 | 25.92 ± 1.96 | not tested | 22.35 ± 0.28 | not tested |
| Compound | IC50 (µM) ± SE or Inhibition (%) @ Concentration (µM) | GI50 [µM] ± SE | |
|---|---|---|---|
| HDAC1 | HDAC6 | ||
| Bufexamac (8) | 31.66 ± 4.75 | 2.24 ± 0.05 | 31.78 ± 0.57 |
| 9 | 49 @ 300 | 13.72 ± 0.16 | 33.99 ± 0.06 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, K.; Hauser, A.-T.; Melesina, J.; Sippl, W.; Jung, M. Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition. Molecules 2018, 23, 321. https://doi.org/10.3390/molecules23020321
King K, Hauser A-T, Melesina J, Sippl W, Jung M. Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition. Molecules. 2018; 23(2):321. https://doi.org/10.3390/molecules23020321
Chicago/Turabian StyleKing, Kristina, Alexander-Thomas Hauser, Jelena Melesina, Wolfgang Sippl, and Manfred Jung. 2018. "Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition" Molecules 23, no. 2: 321. https://doi.org/10.3390/molecules23020321
APA StyleKing, K., Hauser, A.-T., Melesina, J., Sippl, W., & Jung, M. (2018). Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition. Molecules, 23(2), 321. https://doi.org/10.3390/molecules23020321