Synthesis and Biological Evaluations of NO-Donating Oxa- and Aza-Pentacycloundecane Derivatives as Potential Neuroprotective Candidates



Abstract
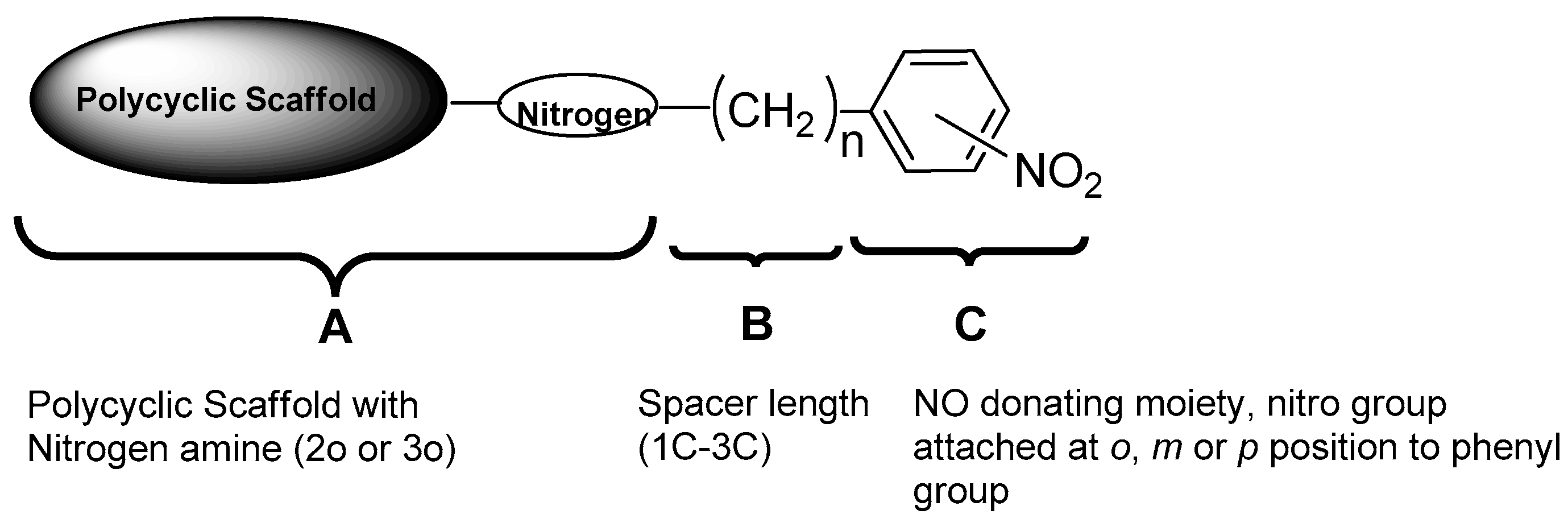
: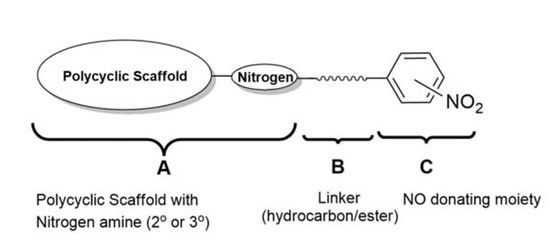
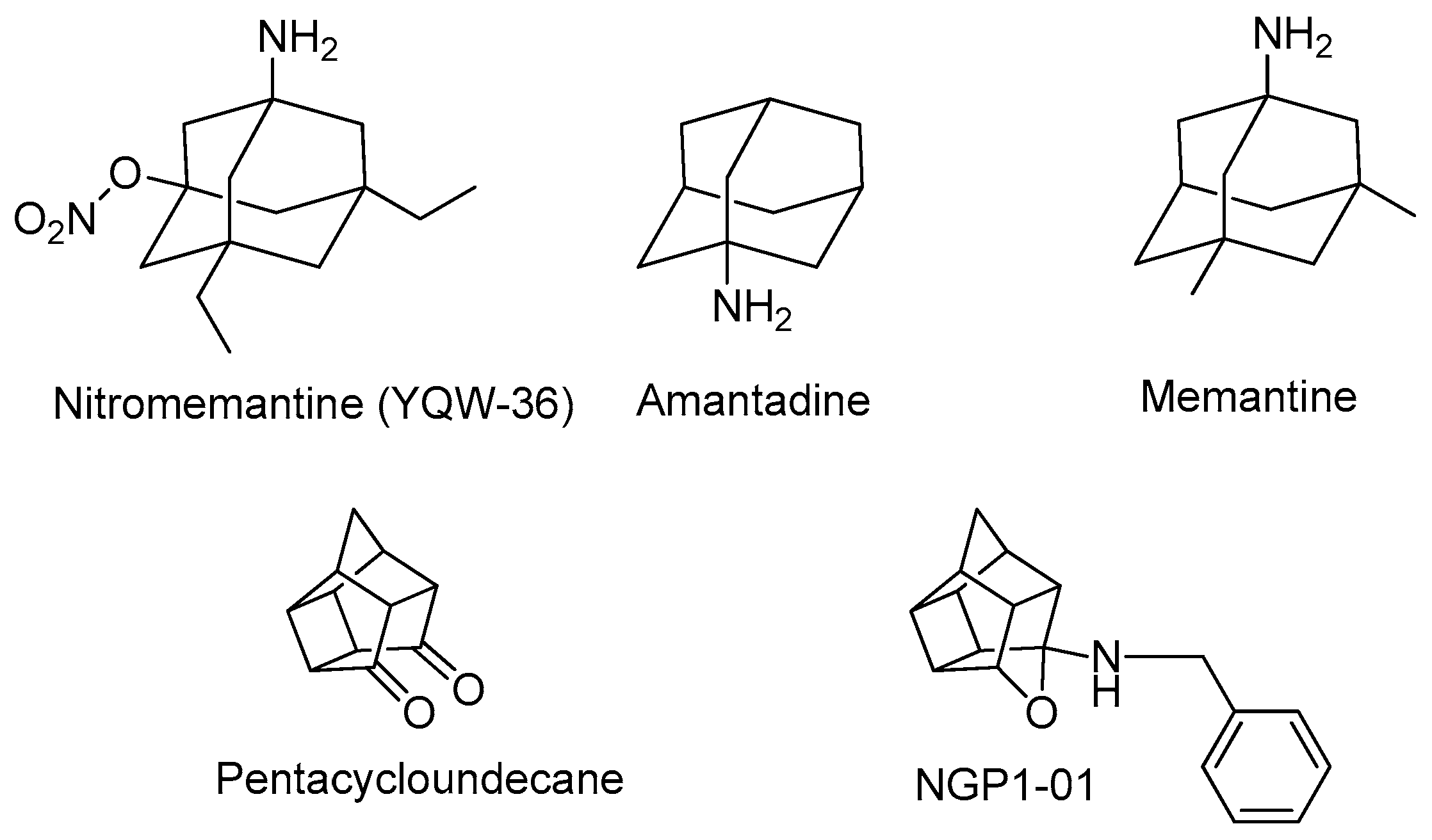
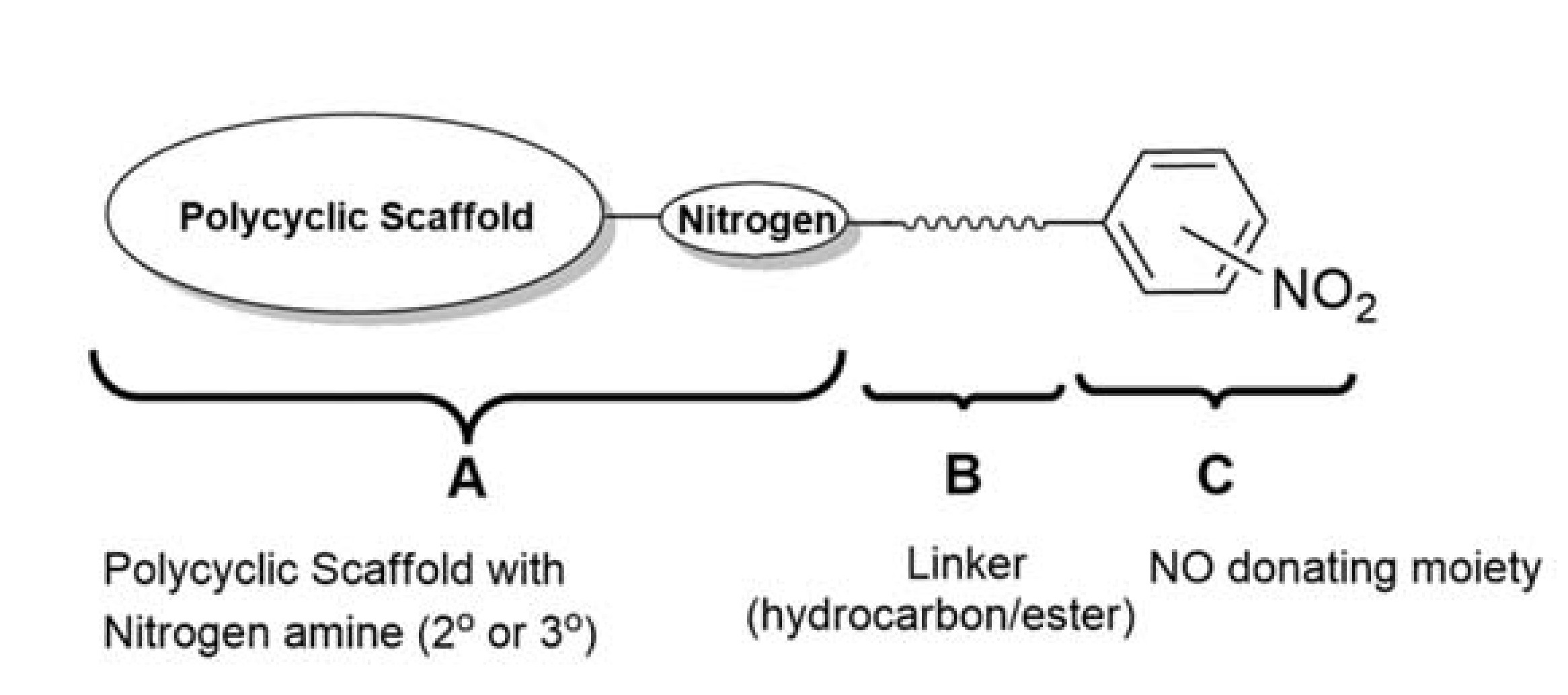
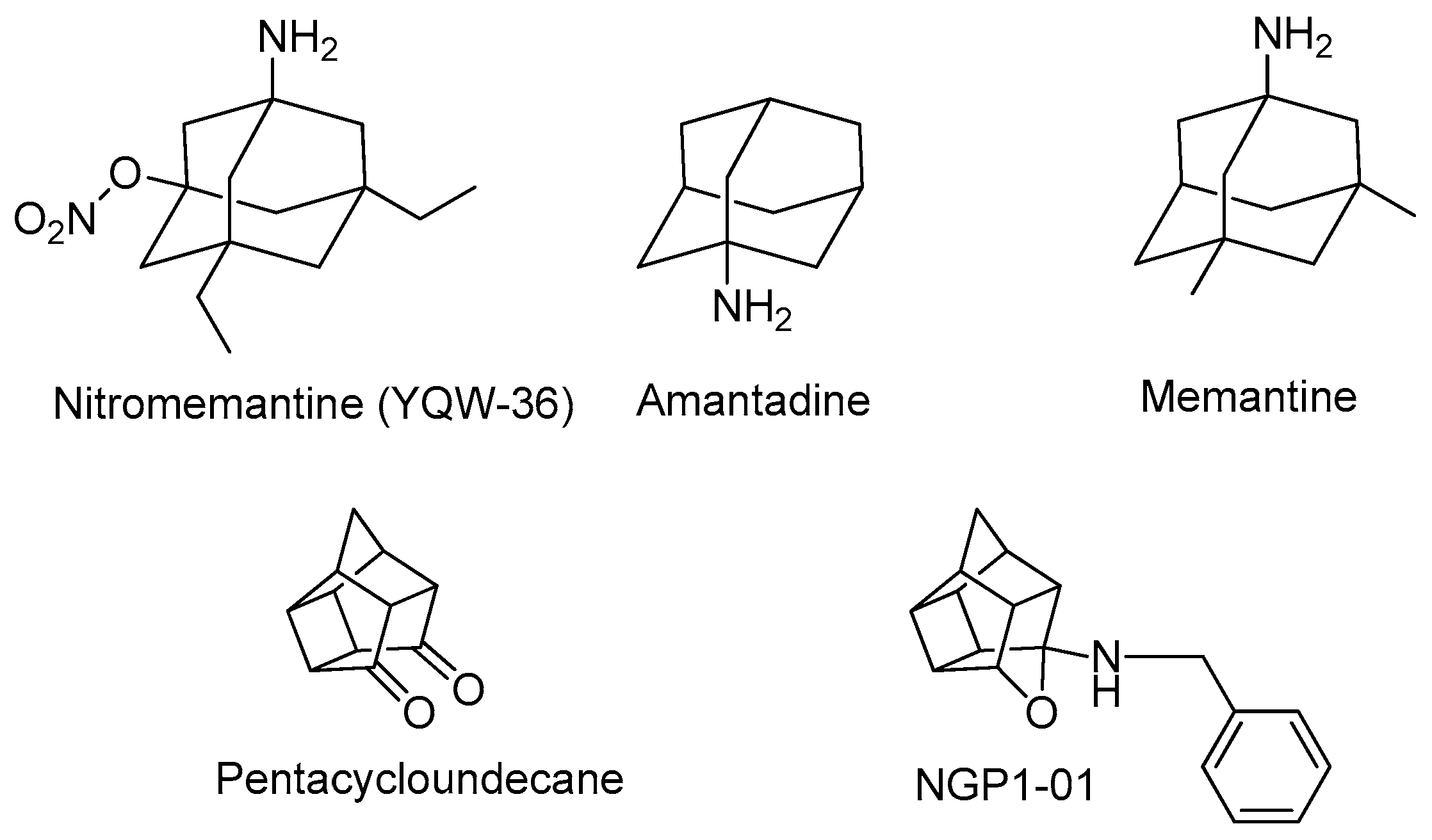
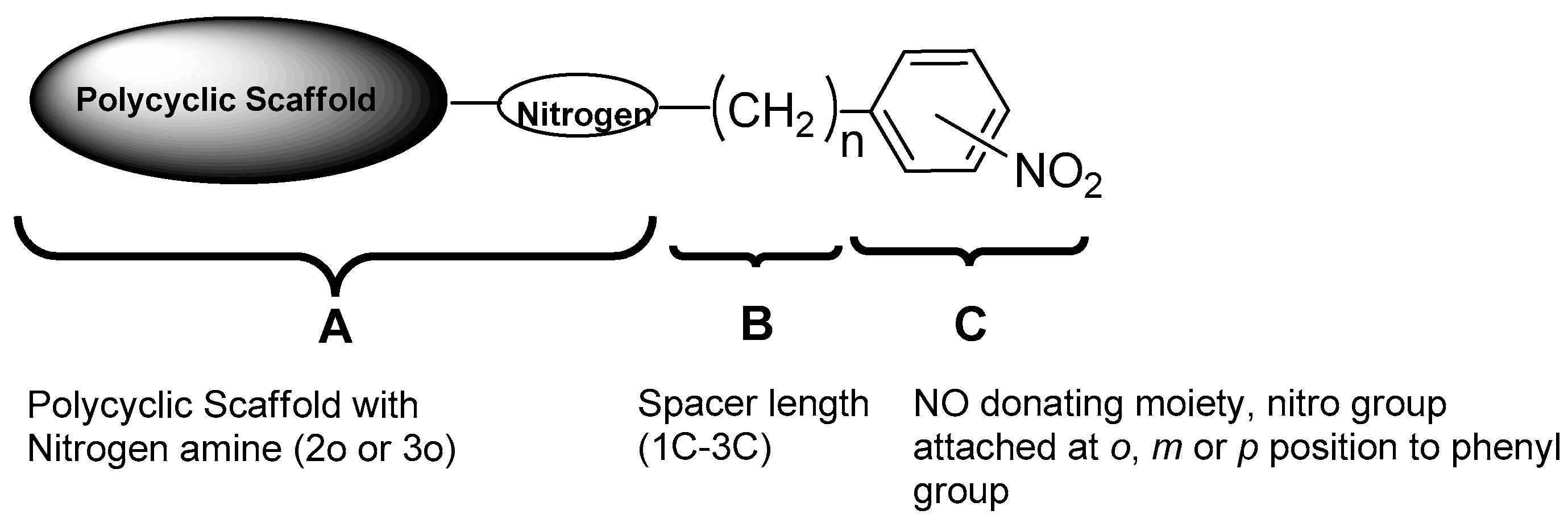
1. Introduction
2. Results and Discussion
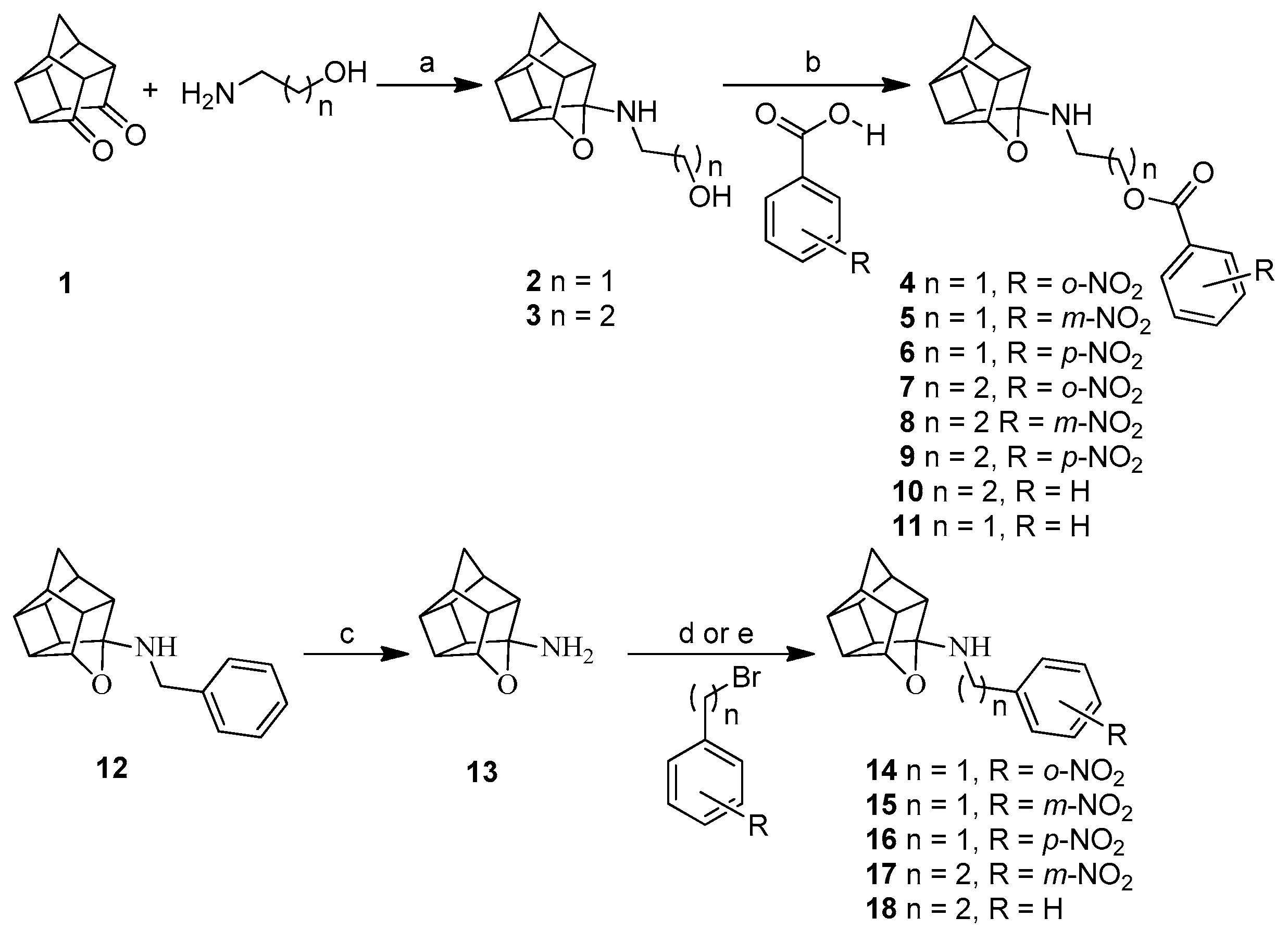
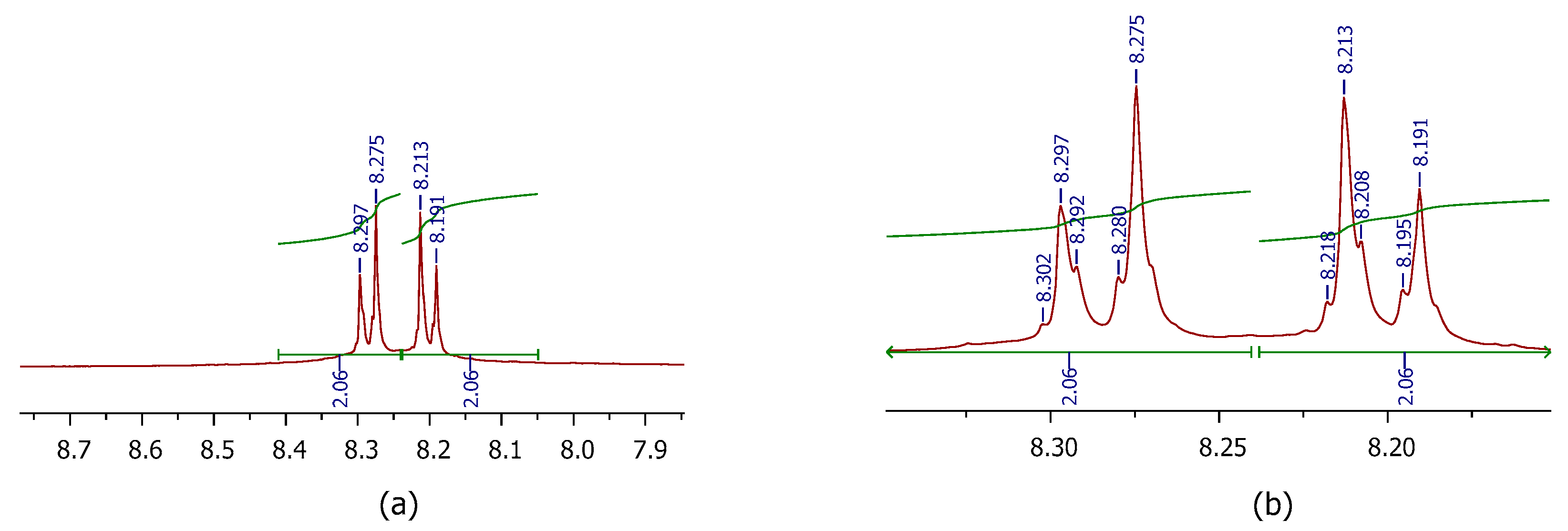
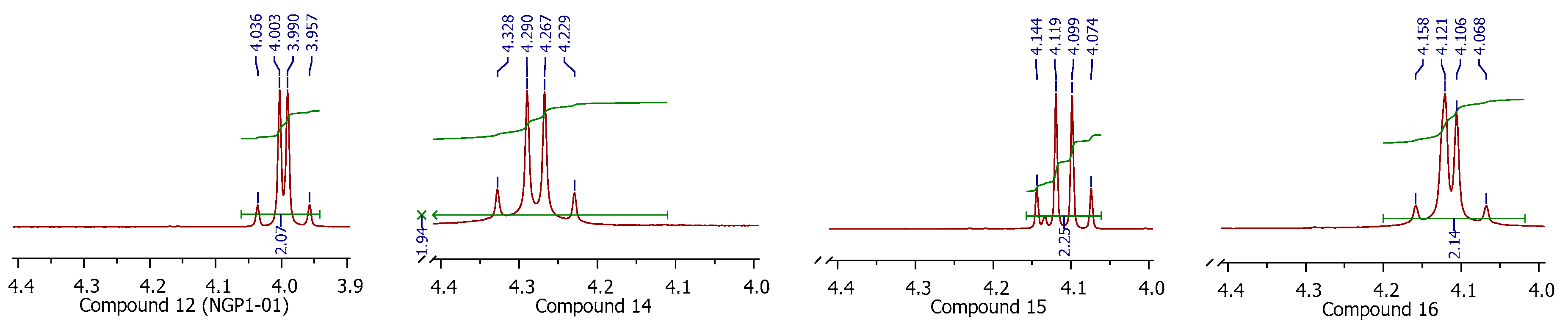
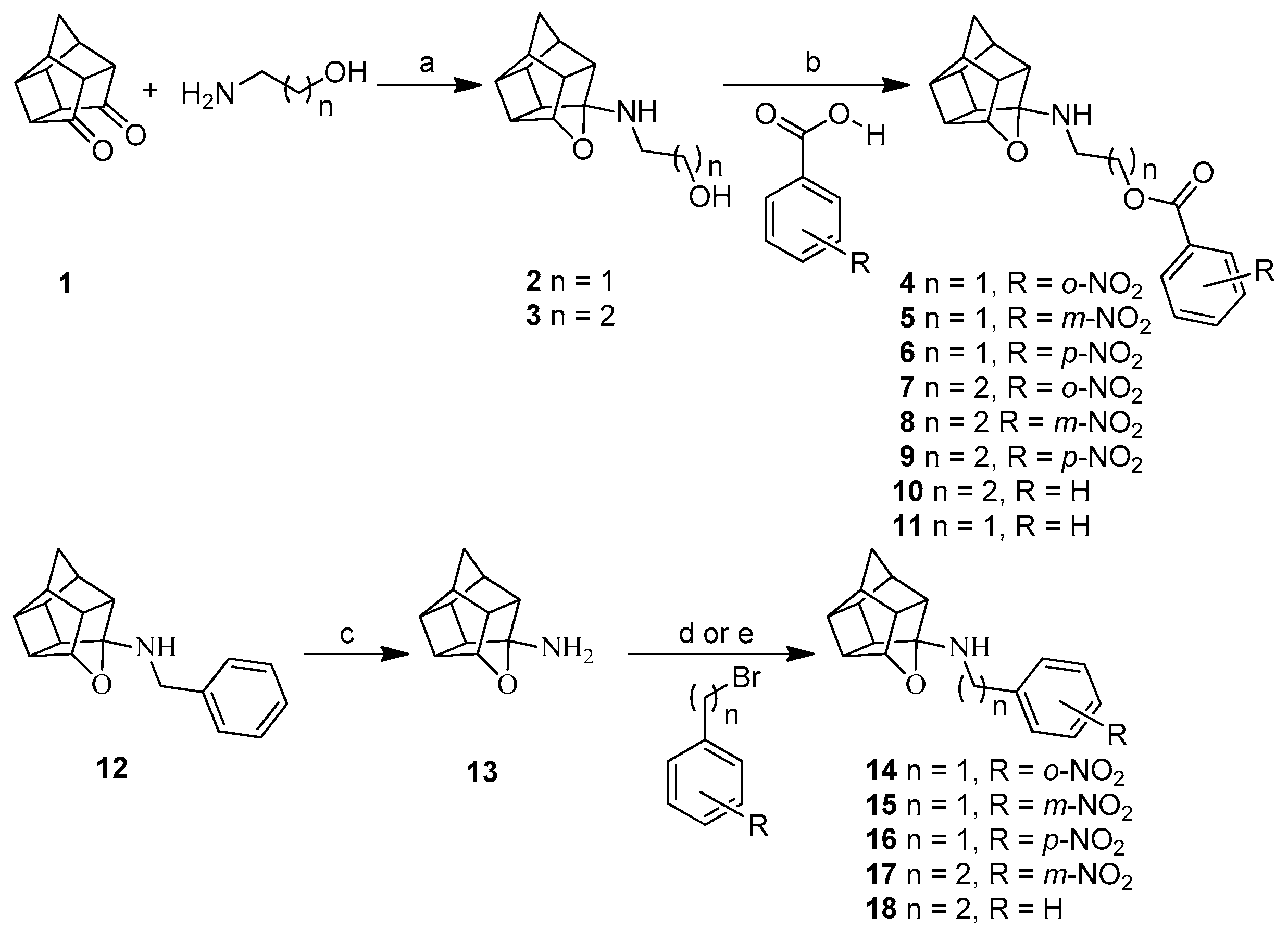
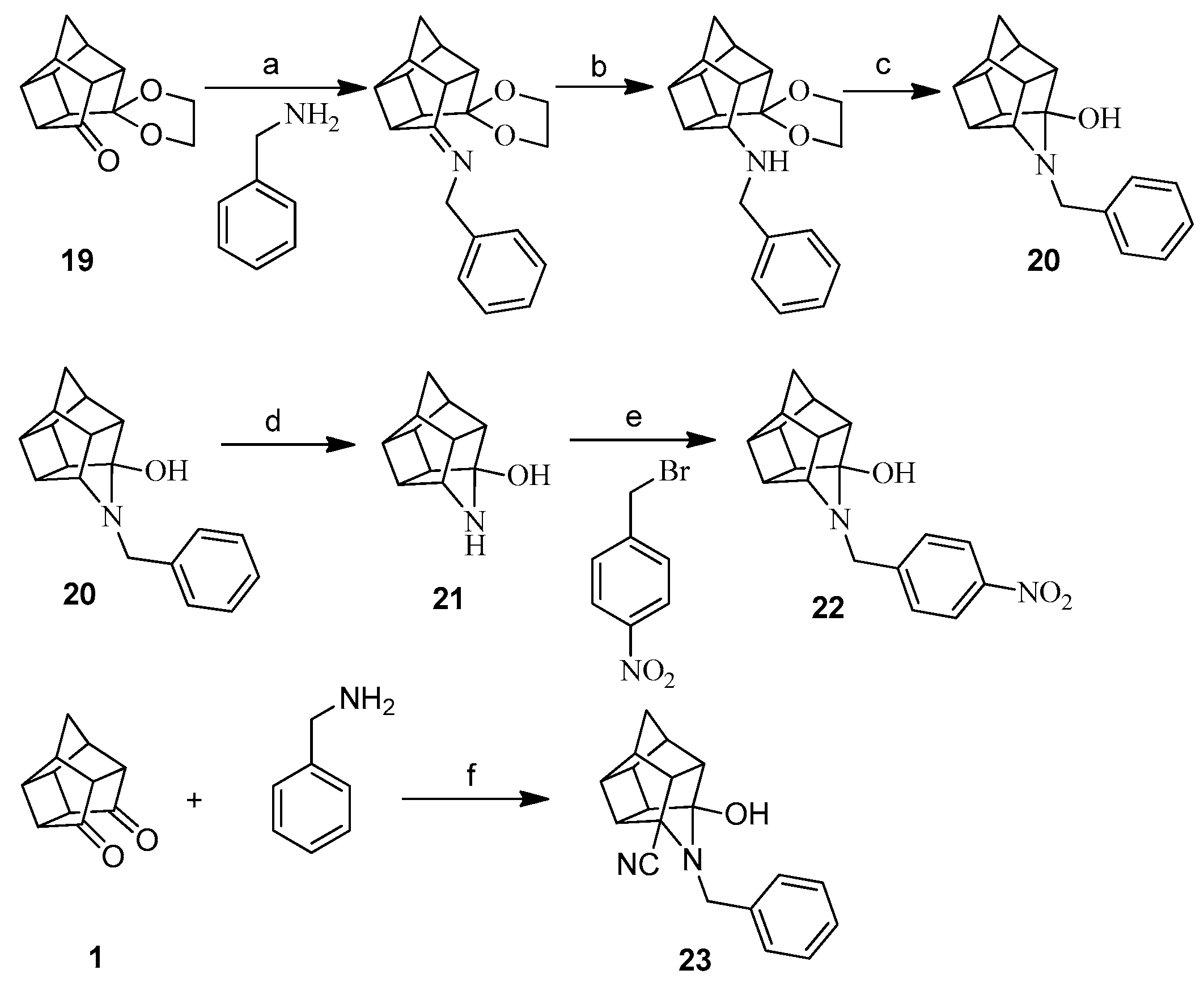
2.1. Synthesis
2.2. Biological Studies
2.2.1. Cytotoxicity Studies
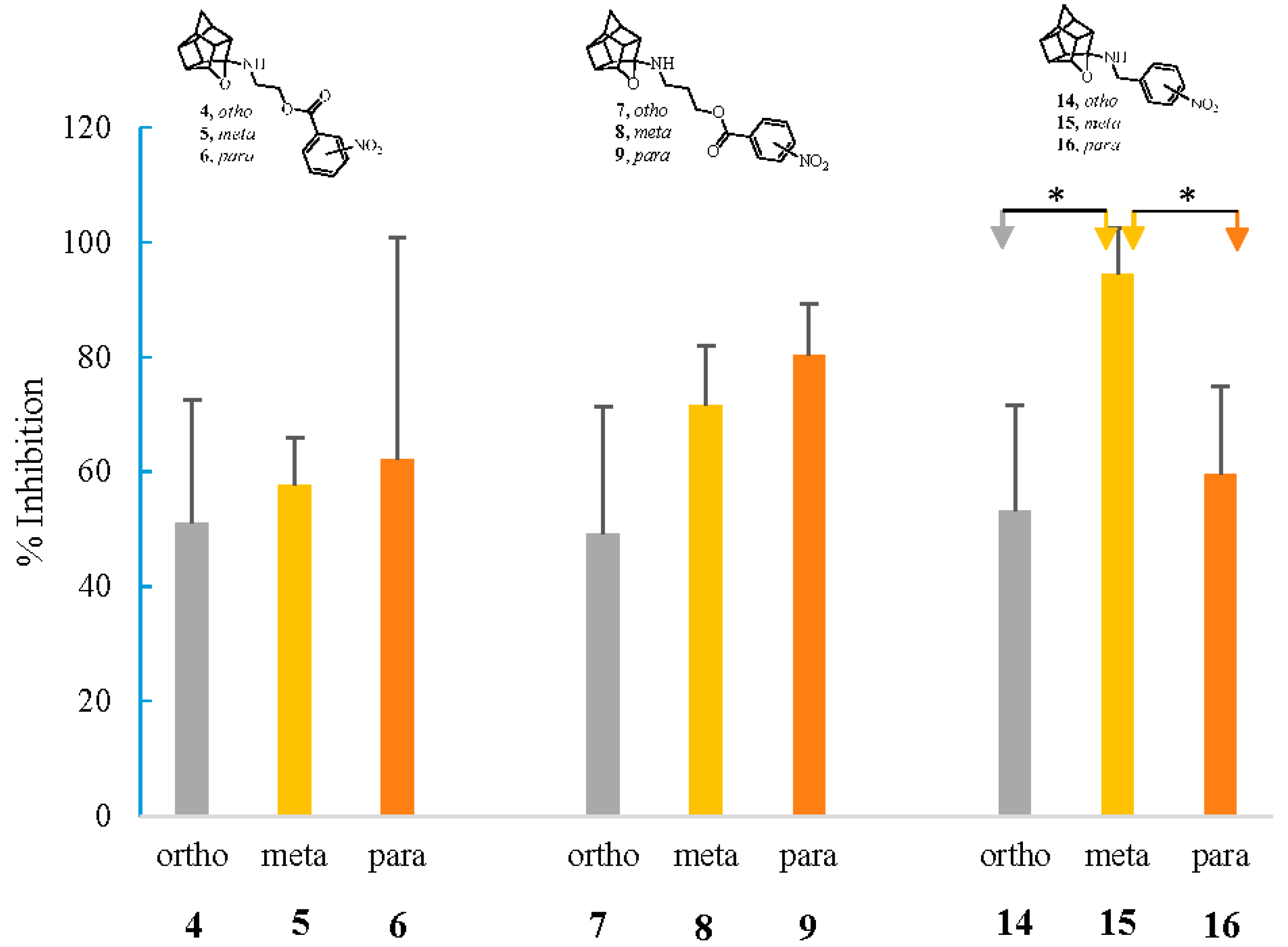
2.2.2. Neuroprotection Studies
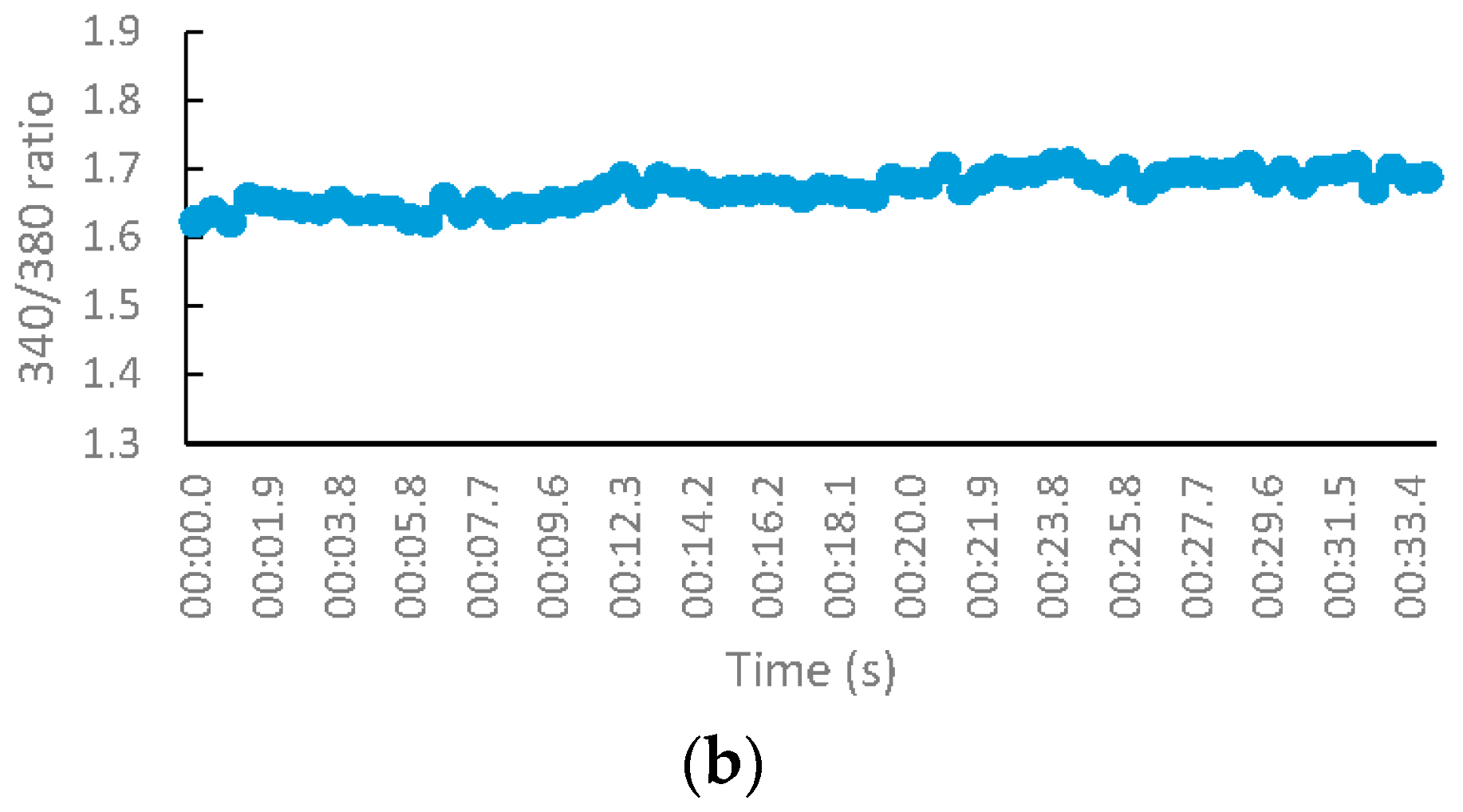
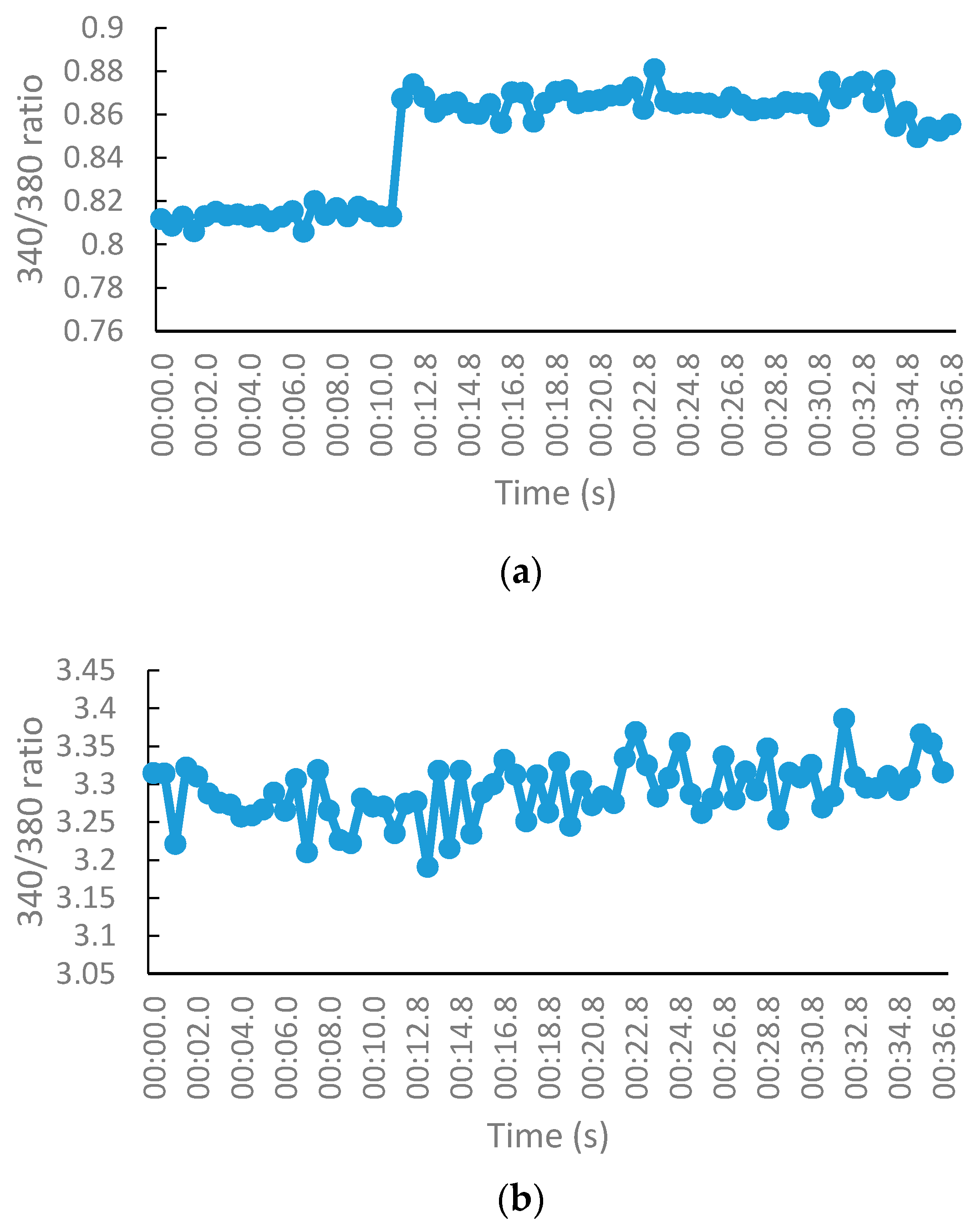
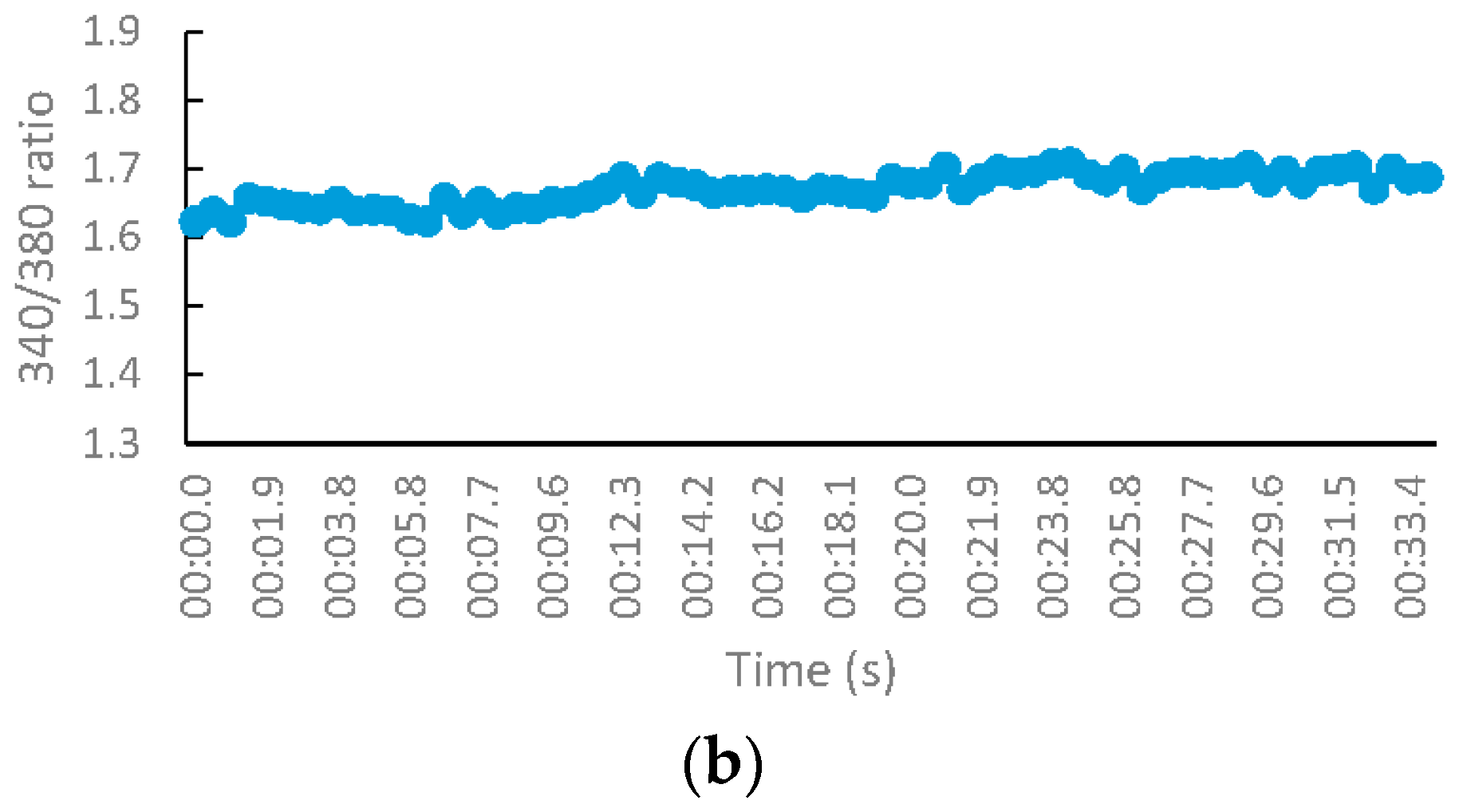
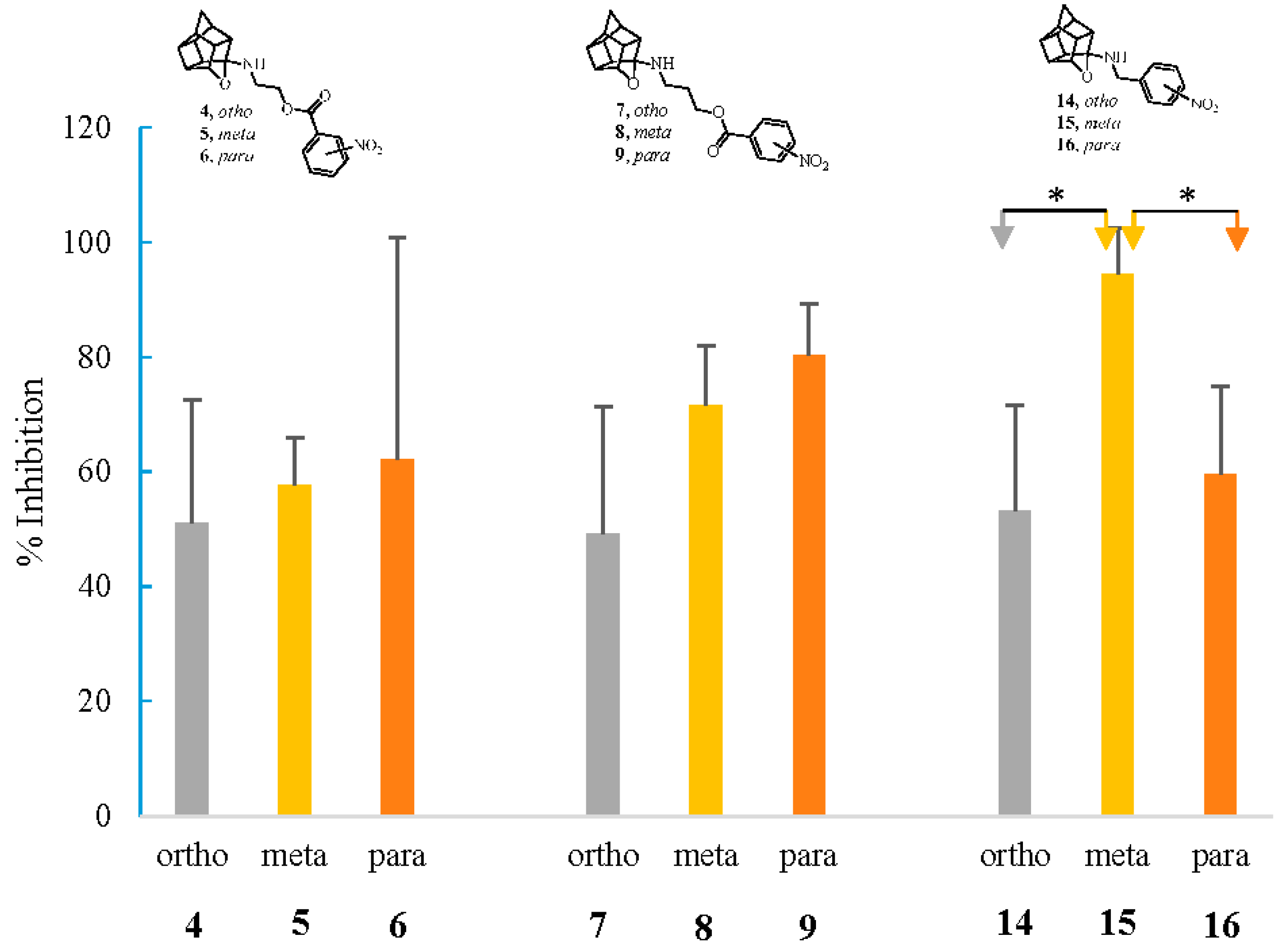
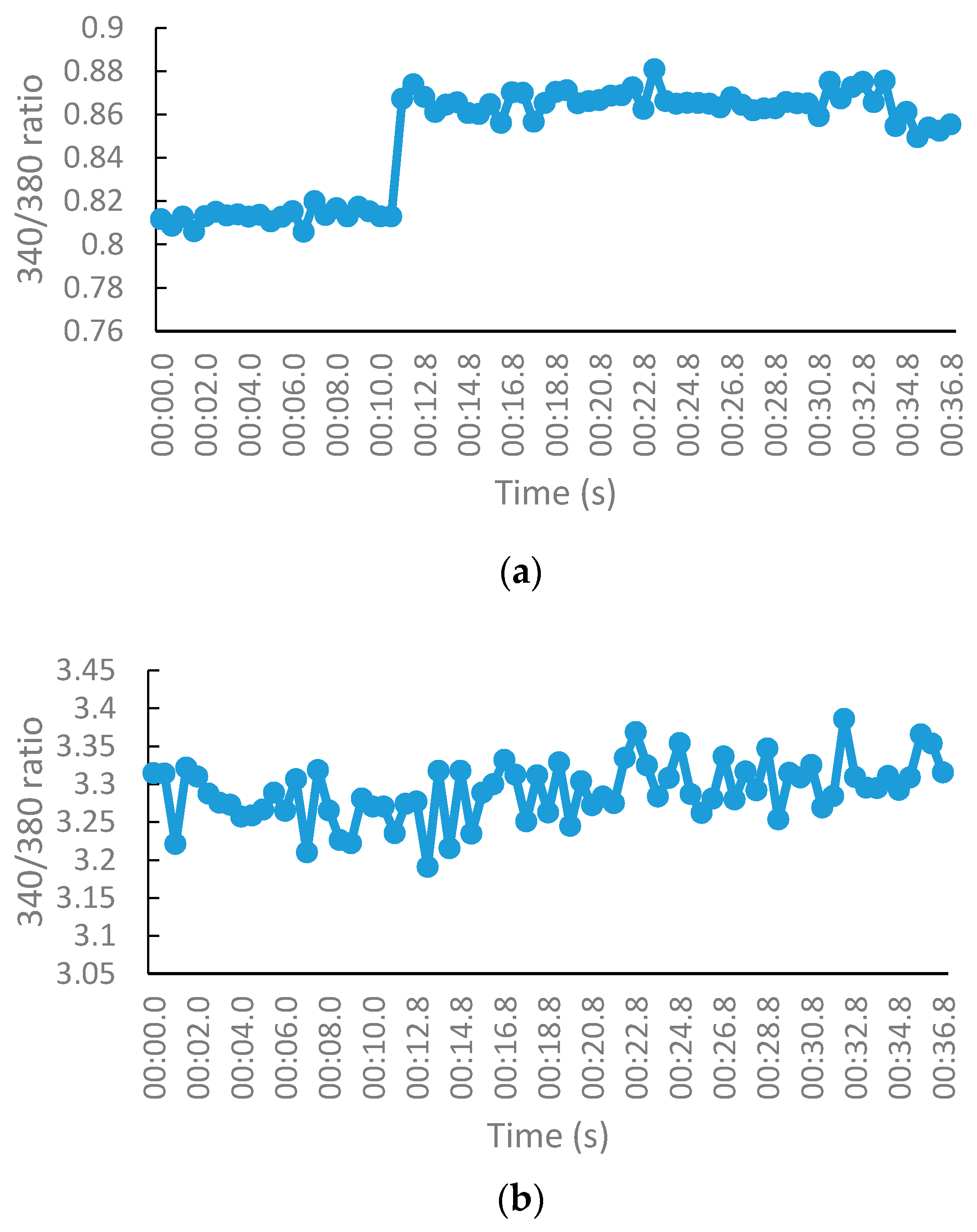
2.2.3. NMDA-Mediated Ca2+ Studies
2.2.4. Voltage-Gated Ca2+ Studies
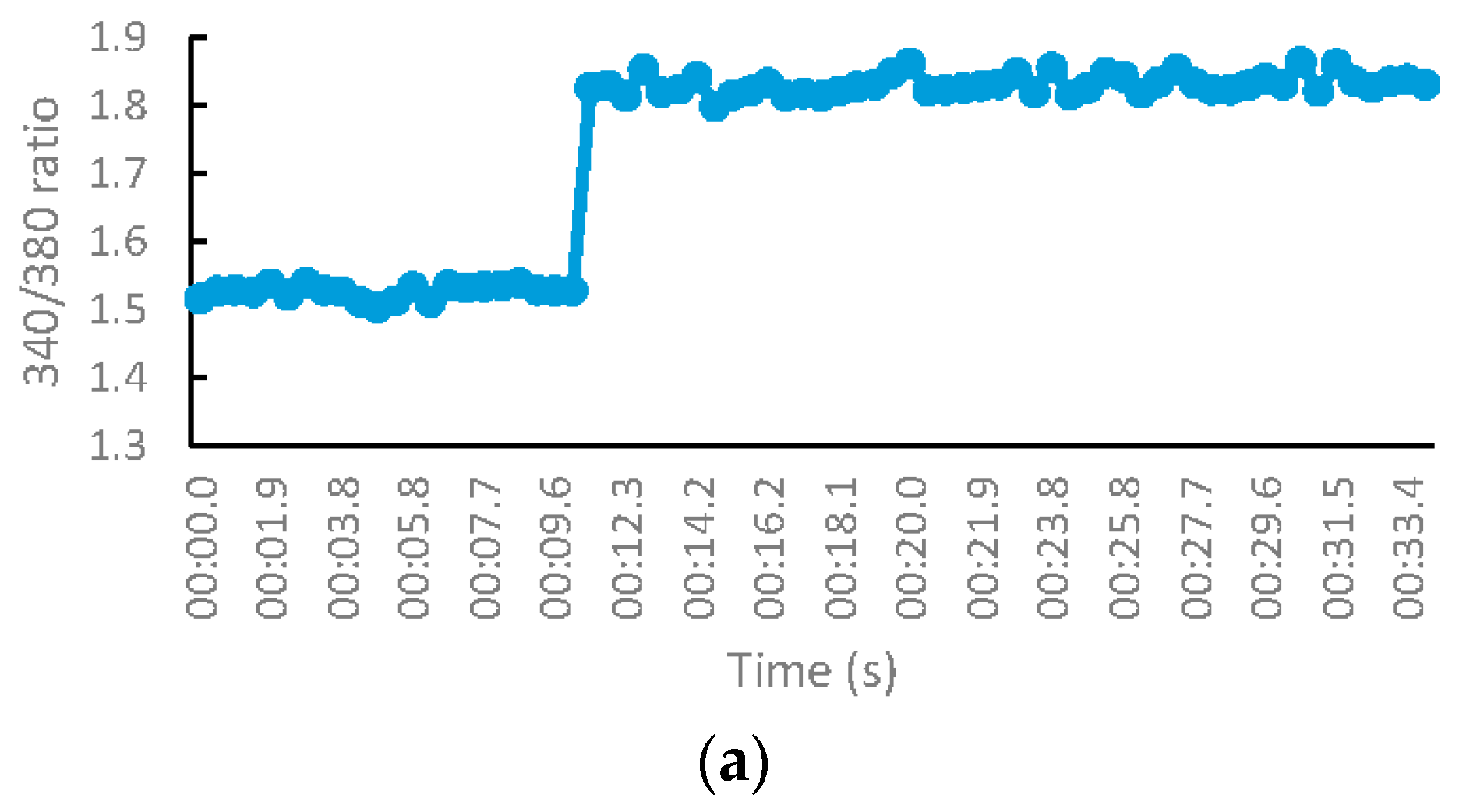
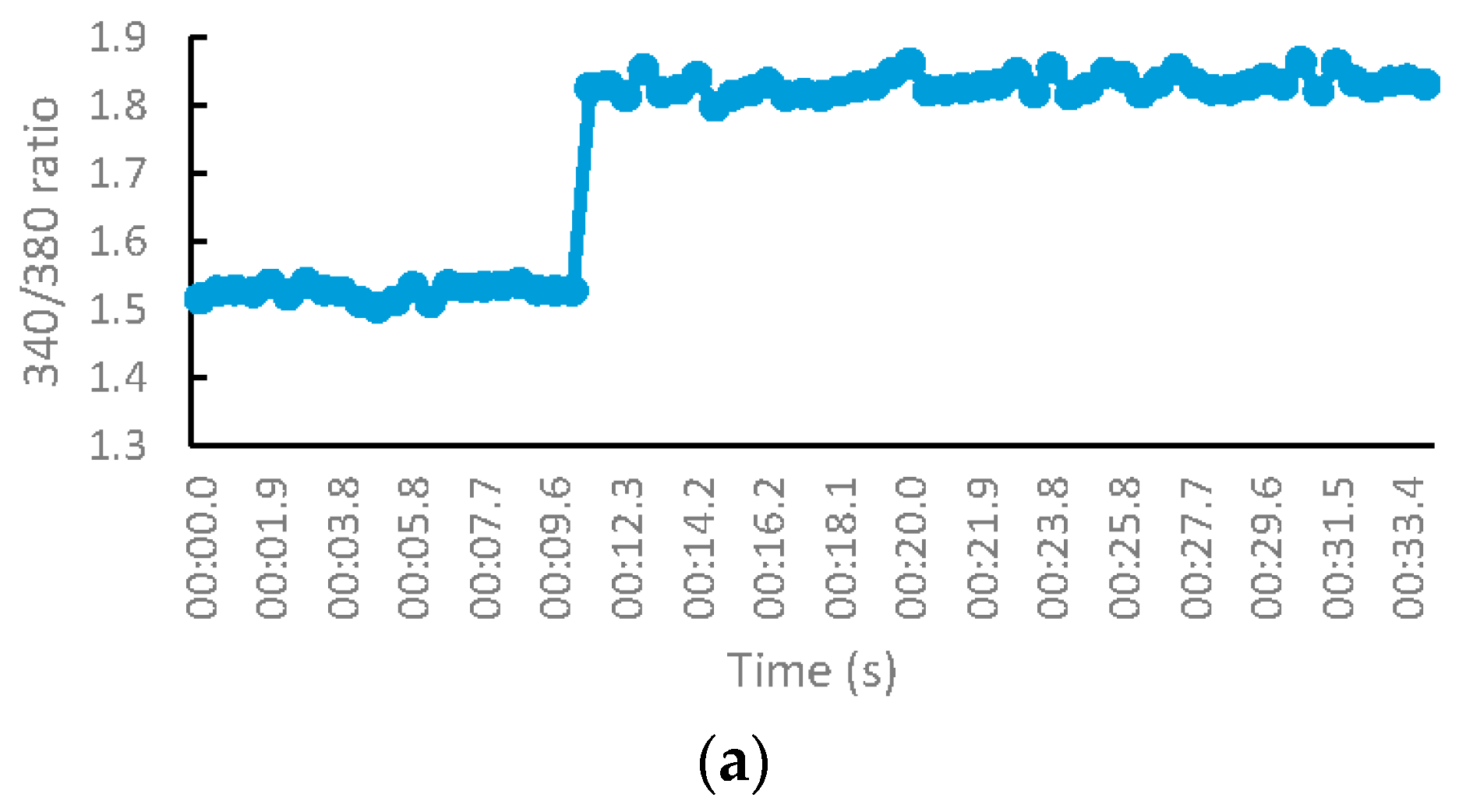
2.2.5. S-Nitsosylation Studies
3. Materials and Methods
3.1. Synthesis
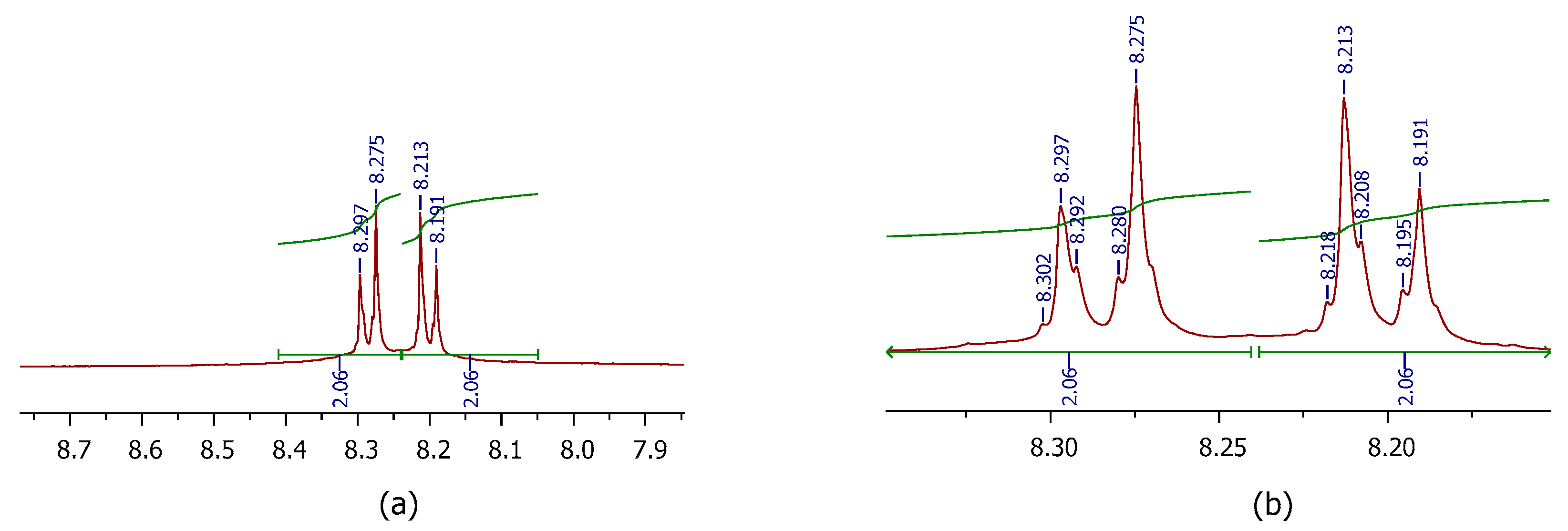
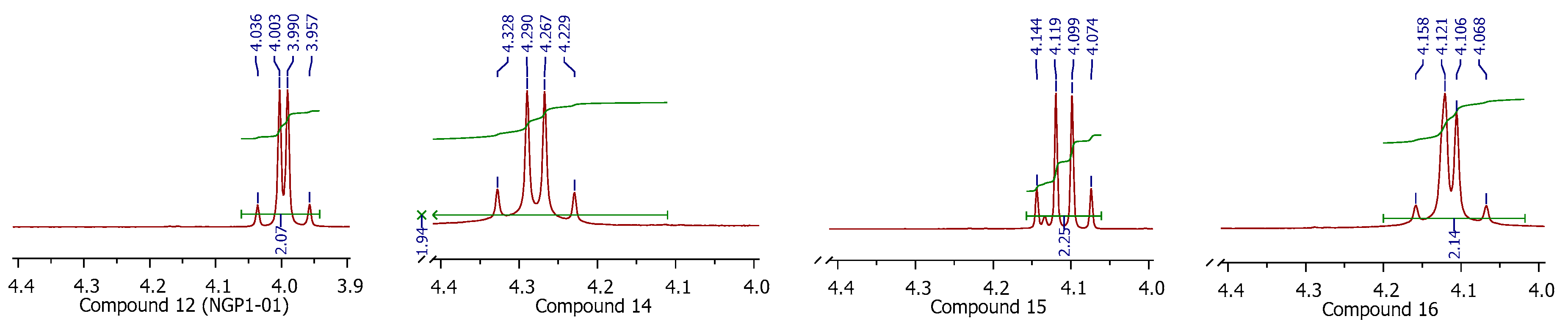
3.1.1. General Information
3.1.2. General Procedure for the Synthesis of Amino Alcohol Derivatives (2,3)
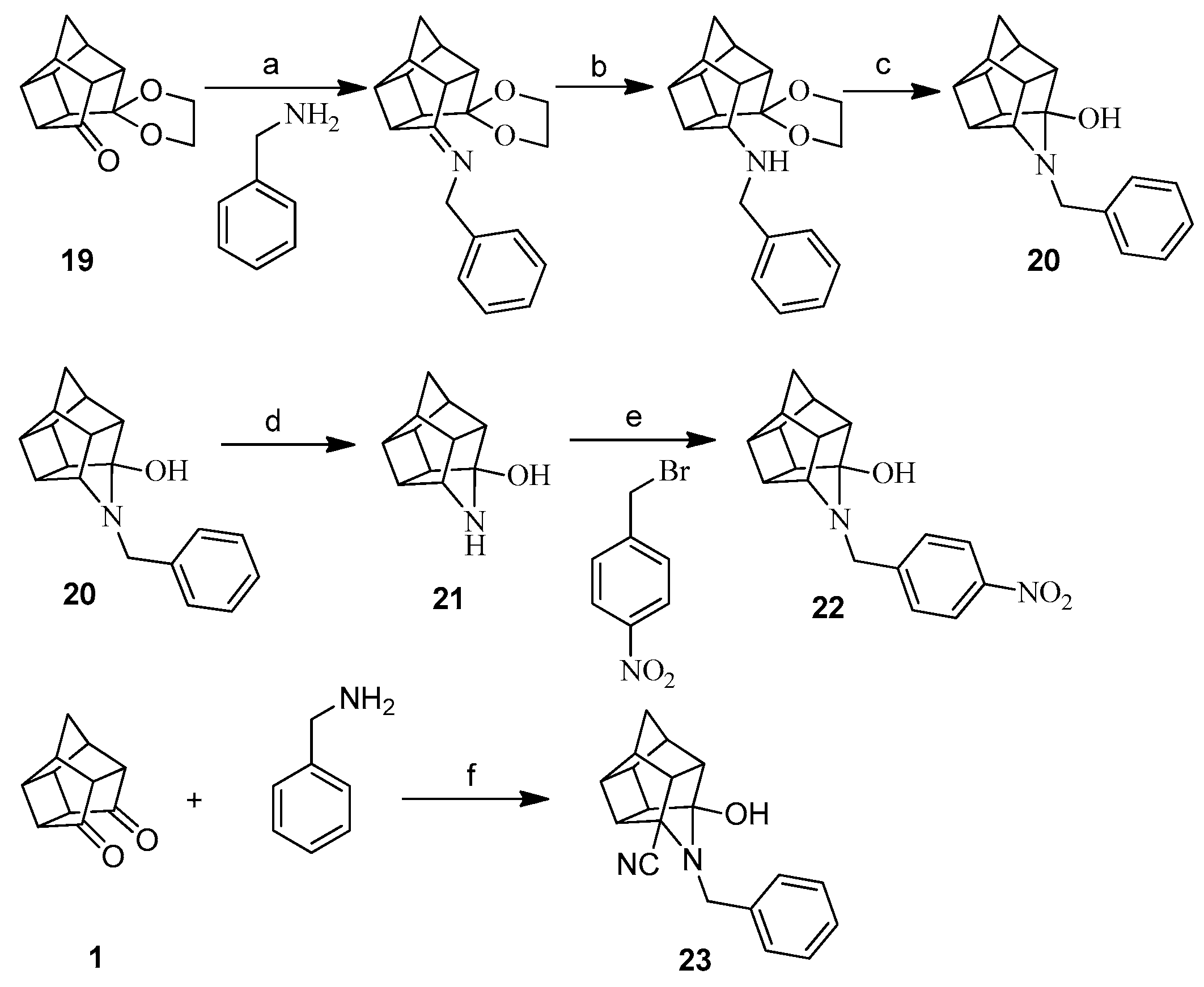
3.1.3. Procedure for Synthesis of Aza-Bridged Compounds 20
4-Benzyl-4-azahexacyclo[5.4.1.02,6.03,10.05,9.08,11]dodecan-3-ol (20)
3.1.4. Procedure for Synthesis of Oxa-Bridged Compounds (4–11)
2-(8,11-Oxapentacyclo[5.4.0.02,6.03,10.05,9]undecane-8-amino)ethyl 2-nitrobenzoate (4)
3.1.5. Procedure for Synthesis of 8-Benzylamino-8,11-oxapentacyclo[5.4.0.02,6.03,10.05,9]undecane (NGP 1-01) (12)
3.1.6. Procedure for Synthesis of Oxa-Bridged Monoamine Cage 13 and Aza-Bridged Monoamine Cage 21
8-Amino-8,11-oxapentacyclo[5.4.0.02,6.03,10.05,9]undecane (13)
4-Azahexacyclo[5.4.1.02,6.03,10.05,9.08,11]dodecan-3-ol (21)
3.1.7. Procedure for Synthesis of Compounds (14–18) and (22)
8-(2-Nitrobenzylamino)-8,11-oxapentacyclo[5.4.0.02,6.03,10.05,9]undecane (14)
3.1.8. Procedure for Synthesis of 3-Hydroxy-5-cyano-4-benzyl-4-azahexacyclo[5.4.1.02,6.03,10.05,9.08,11] Dodecane (23)
3.2. Biological Studies
3.2.1. Cytotoxicity Studies
3.2.2. Neuroprotection Studies
3.2.3. NMDA-Mediated and Voltage-Gated Ca2+ Influx Studies
Preparation of Synaptoneurosomes
Measurement of Intracellular Calcium for NMDA-Mediated Studies
Measurement of Intracellular Calcium for Voltage-Gated Studies
Calculation of Percentage Inhibition in NMDA and VG-Mediated Ca2+ Influx
- [R]control = Mean Ca2+ influx of control
- [R]test = Mean Ca2+ influx of test compound
3.2.4. S-Nitrosylation Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Population Aging: 1950–2050; Report Presented by Department of Economic and Social Affairs, Population Division, United Nations; United Nations: New York, NY, USA, 2002.
- World Health Organization. Neurological Disorders: Public Health Challenges; World Health Organization: Geneva, Switzerland, 2006; ISBN 92-4-156336-2. [Google Scholar]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rosenberg, R.A. Mechanisms of disease: Excitatory amino acids as a final pathway in neurologic disorders. N. Eng. J. Med. 1994, 330, 613–622. [Google Scholar]
- Majdi, M.; Chen, H.S.V. NMDA-gated ion channel research and its therapeutic potentials in neurodegenerative diseases: A review. J. Recept. Ligand Channel Res. 2009, 2, 59–73. [Google Scholar]
- Cataldi, M. The changing landscape of voltage-gated calcium channels in neurovascular disorders and in neurodegenerative diseases. Curr. Neuropharmacol. 2013, 11, 276–297. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Choi, Y.B.; Sucher, N.J.; Chen, H.S.V. Neuroprotective versus neurodestructive effects of NO-related species. BioFactors 1998, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Oliver, D.W.; Malan, S.F. Medicinal chemistry of polycyclic cage compounds in drug discovery research. Med. Chem. Res. 2008, 17, 137–151. [Google Scholar] [CrossRef]
- Joubert, J.; Geldenhuys, W.J.; Van der Schyf, C.J.; Oliver, D.W.; Kruger, H.G.; Govender, T.; Malan, S.F. Polycyclic Cage Structures as Lipophilic Scaffolds for Neuroactive Drugs. ChemMedChem 2012, 7, 375–384. [Google Scholar]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Fortuin, E.E.; Taylor, D.; Smith, P.J.; Malan, S.F. Pentacycloundecylamines and conjugates thereof as chemosensitizers and reversed chloroquine agents. Bioorg. Med. Chem. Lett. 2014, 24, 5516–5519. [Google Scholar] [CrossRef] [PubMed]
- Klimochkin, Y.N.; Shiryaev, V.A.; Leonova, M.V. Antiviral properties of cage compounds. New prospects. Russ. Chem. Bull. 2015, 64, 1473–1496. [Google Scholar] [CrossRef]
- Van der Schyf, C.J.; Geldenhuys, W.J. Polycyclic Compounds: Ideal drug scaffolds for the design of multiple mechanism drugs. Neurotherapeutics 2009, 6, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Malan, S.F.; Bloomquist, J.R.; Marchand, A.P.; Van der Schyf, C.J. Pharmacology and Structure-Activity Relationships of Bioactive Polycyclic Cage Compounds: A Focus on Pentacycloundecane derivatives. Med. Res. Rev. 2005, 25, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Cookson, R.C.; Crundwell, E.; Hudec, J. Synthesis of cage-like molecules by irradiation of Diels-Alder adducts. Chem. Ind. 1958, 32, 1003–1004. [Google Scholar]
- Marchand, A.P.; Arney, B.E., Jr.; Dave, P.R.; Satyanarayana, N.; Watson, W.H.; Nagl, A. Transannular cyclizations in the pentacyclo [5.4.0.02,6.03,10.05,9]undecane-8, 11-dione system. A reinvestigation. J. Org. Chem. 1988, 53, 2644–2647. [Google Scholar]
- Joubert, J.; Sharma, R.; Malan, S.F. Microwave-assisted methods for synthesis of pentacyclo[5.4.0.02,6.03,10.05,9]undecylamines. Tetrahedron Lett. 2013, 54, 6923–6927. [Google Scholar]
- Onajole, O.K.; Coovadia, Y.; Kruger, H.G.; Maguire, G.E.M.; Pillay, M.; Govender, T. Novel polycyclic cage’-1,2-diamines as potential anti-tuberculosis agents. Eur. J. Med. Chem. 2012, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fourie, T.G.; Snyckers, F.O.; Noristan Holdings Limited. 4-Azahexacyclododecane Compounds. U.S. Patent 5,137,908, 11 August 1992. [Google Scholar]
- Holst-Hansen, C.; Brünnner, N. MTT-cell proliferation assay. In Cell Biology: A Laboratory Handbook, 2nd ed.; Celis, J.E., Ed.; Academic Press: San Diego, CA, USA, 1998; pp. 16–18. [Google Scholar]
- Green, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef]
- Shahripour, B.R.; Alexandrov, A.V.; Harrigan, M.R. N-acetylcysteine (NAC) in neurological disorders: Mechanisms of action and therapeutic opportunities. Brain Behav. 2004, 4, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Malan, S.F.; Murugesan, T.; Van der Schyf, C.J.; Bloomquist, J.R. Synthesis and biological evaluation of pentacyclo[5.4.0.02,6.03,10.05,9]undecane derivatives as potential therapeutic agents in Parkinson’s disease. Bioorg. Med. Chem. 2004, 12, 1799–1806. [Google Scholar]
- Kiewert, C.; Hartmann, J.; Stoll, J.; Thekkumkara, T.J.; Schyf, C.J.; Klein, J. NGP1-01 is a Brain-permeable Dual Blocker of Neuronal Voltage- and Ligand-operated Calcium Channels. Neurochem. Res. 2006, 31, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Tyagi, N. Method and validation of synaptosomal preparation for isolation of synaptic membrane proteins from rat brain. MethodsX 2014, 1, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Westmark, P.R.; Westmark, C.J.; Jeevananthan, A.; Malter, J.S. Preparation of Synaptoneurosomes from Mouse Cortex using a Discontinuous Percoll-Sucrose Density Gradient. J. Vis. Exp. 2011, 55, e3196. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Malan, S.F.; Bloomquist, J.R.; Van der Schyf, C.J. Structure-activity relationships of pentacycloundecylamines at the N-methyl-D-aspartate receptor. Bioorg. Med. Chem. 2007, 15, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Forrester, M.T.; Foster, M.W.; Benhar, M.; Stamler, J.S. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 2009, 46, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Van der Schyf, C.J.; Liebenberg, W.; Bornman, R.; Dekker, T.G.; Van Rooyen, P.H.; Fourie, T.G.; Matthee, E.; Snyckers, F.O. The polycyclic calcium antagonist, NGP1-01, has an oxa rather than an aza bird-cage structure: Evidence from NMR spectroscopy and the x-ray crystal structure. S. Afr. J. Chem. 1989, 42, 46–48. [Google Scholar]
- Zah, J.; Terre’Blanche, G.; Erasmus, E.; Malan, S.F. Physicochemical prediction of a brain-blood distribution profile in polycyclic amines. Bioorg. Med. Chem. 2003, 11, 3569–3578. [Google Scholar] [CrossRef]
- Malan, S.F.; Van der Walt, J.J.; Van der Schyf, C.J. Structure-activity relationships of polycyclic aromatic amines with calcium channel blocking activity. Arch. Pharm. 2000, 333, 10–16. [Google Scholar] [CrossRef]
- Lemmer, H.J.R.; Joubert, J.; van Dyk, S.; van der Westhuizen, F.H.; Malan, S.F. S-Nitrosylation and attenuation of excessive calcium flux by pentacycloundecane derivatives. Med. Chem. 2012, 8, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Sanders, J.K.M. The nature of .pi.-.pi. Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |
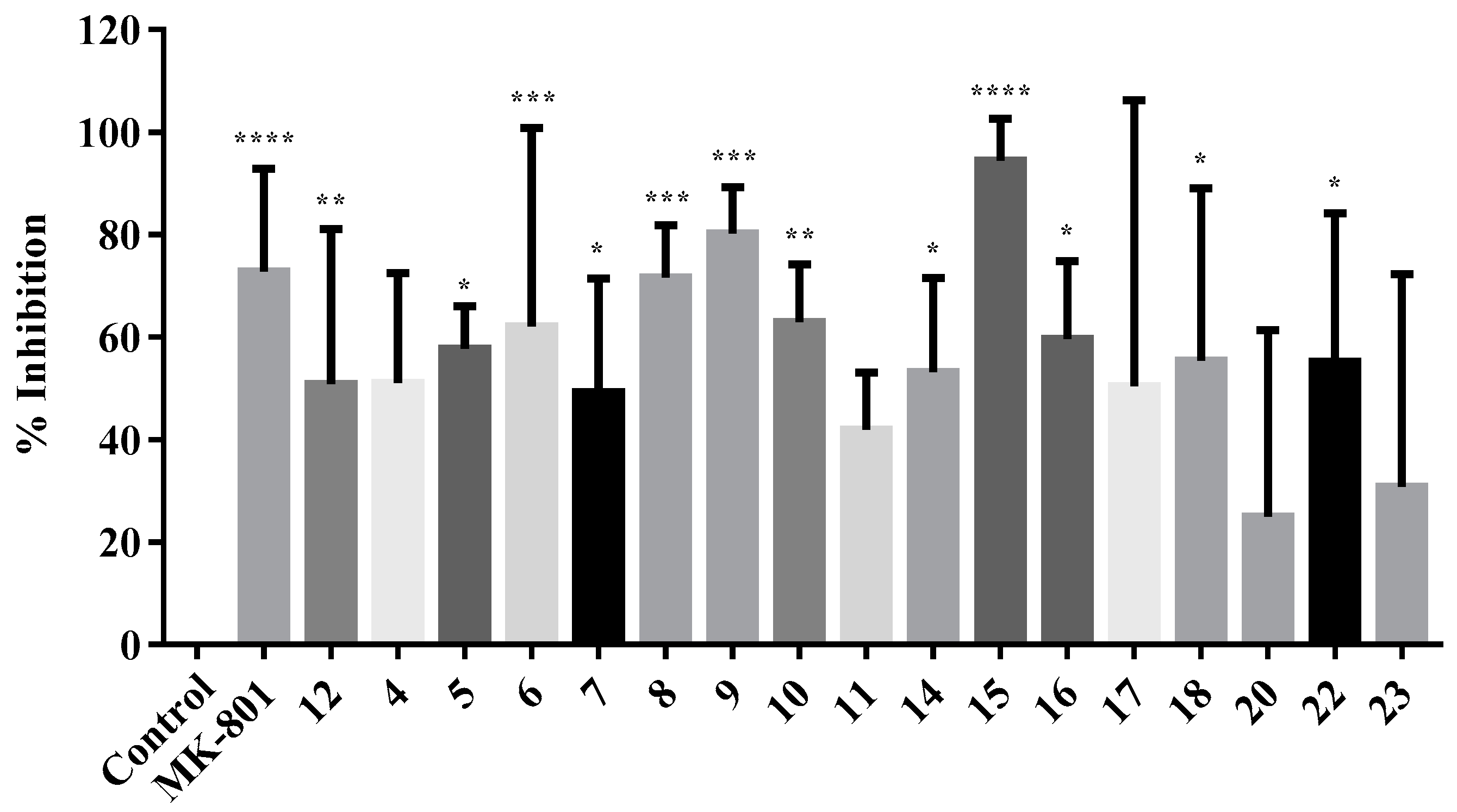
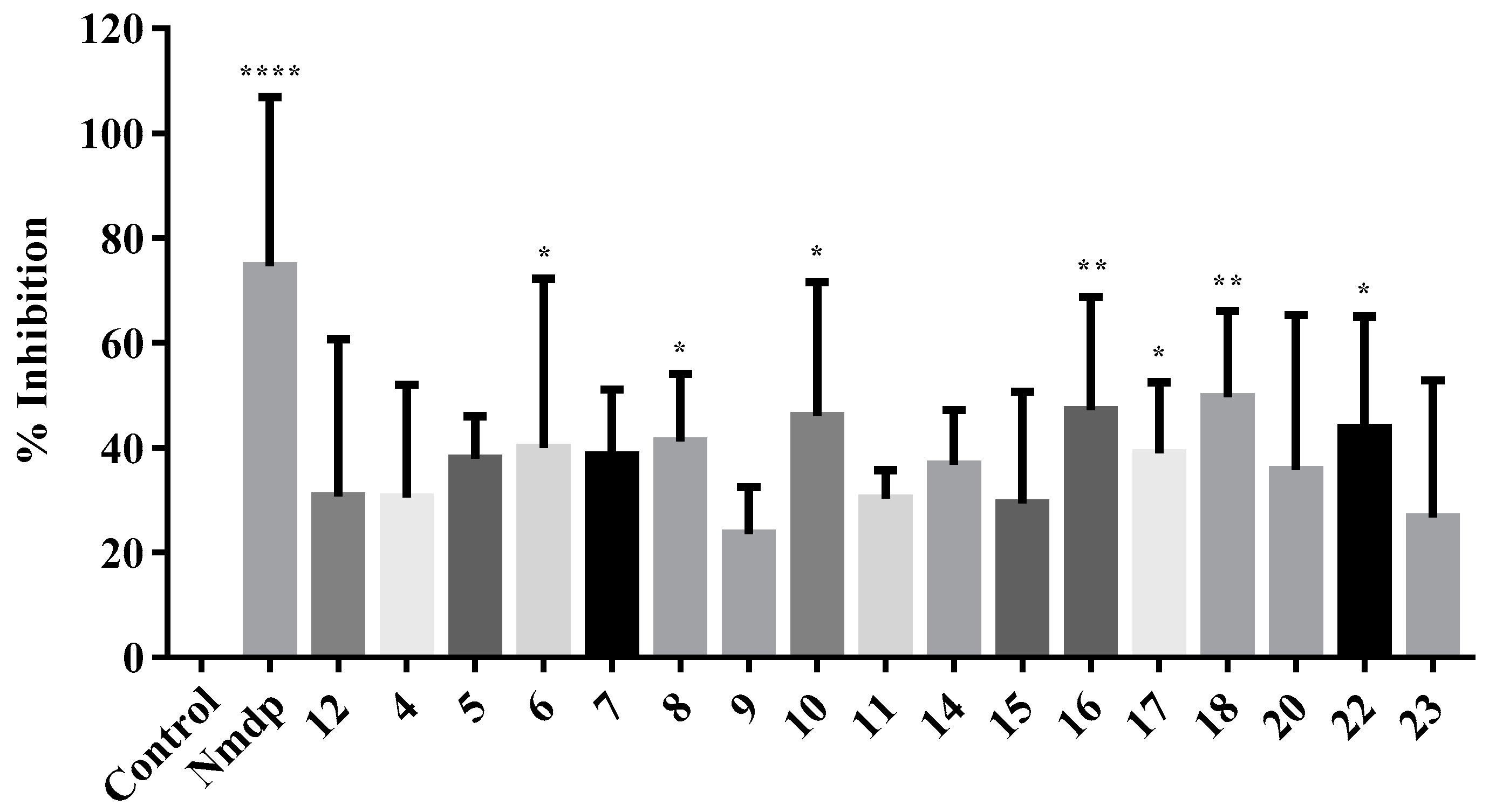
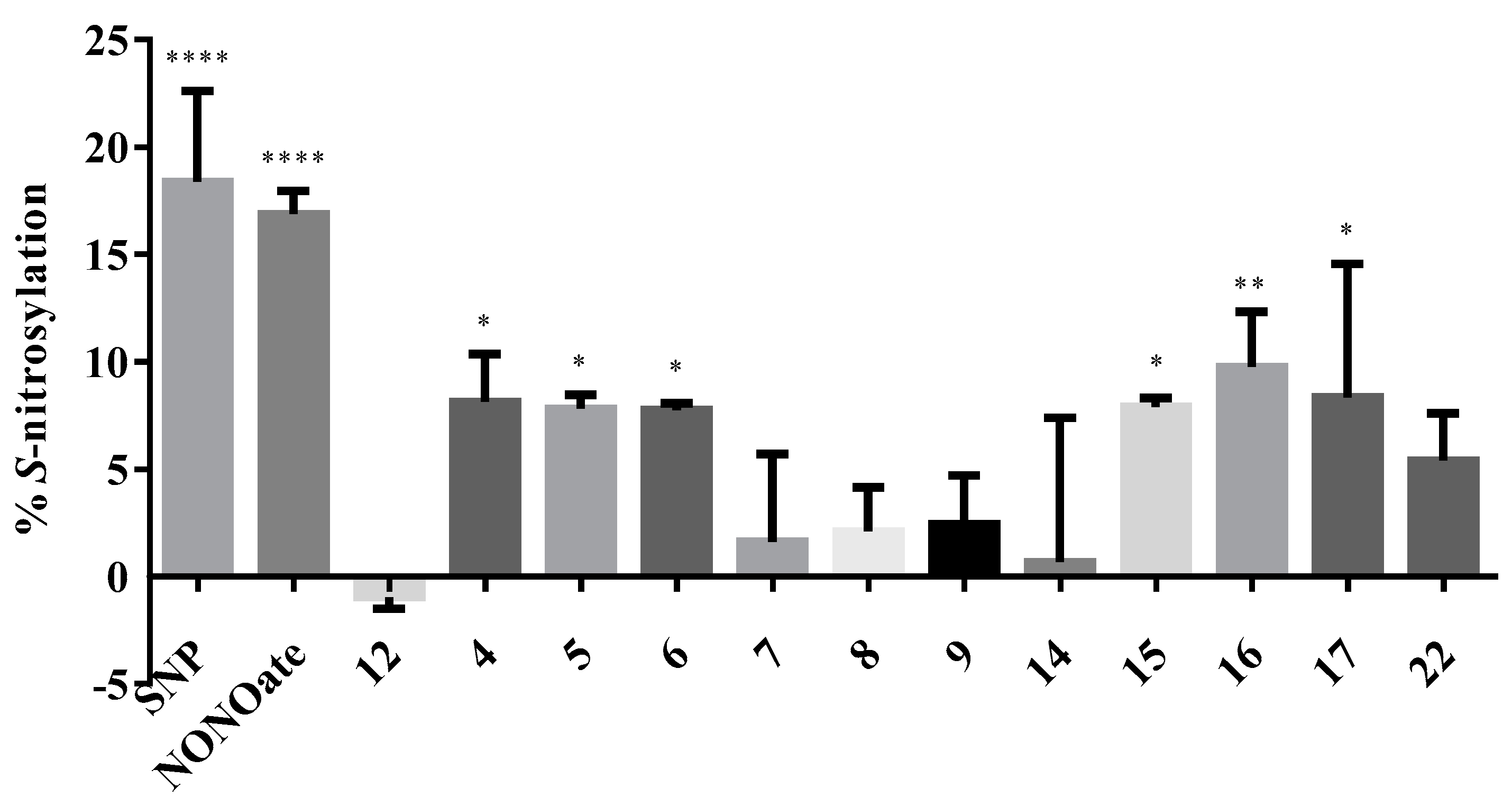

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
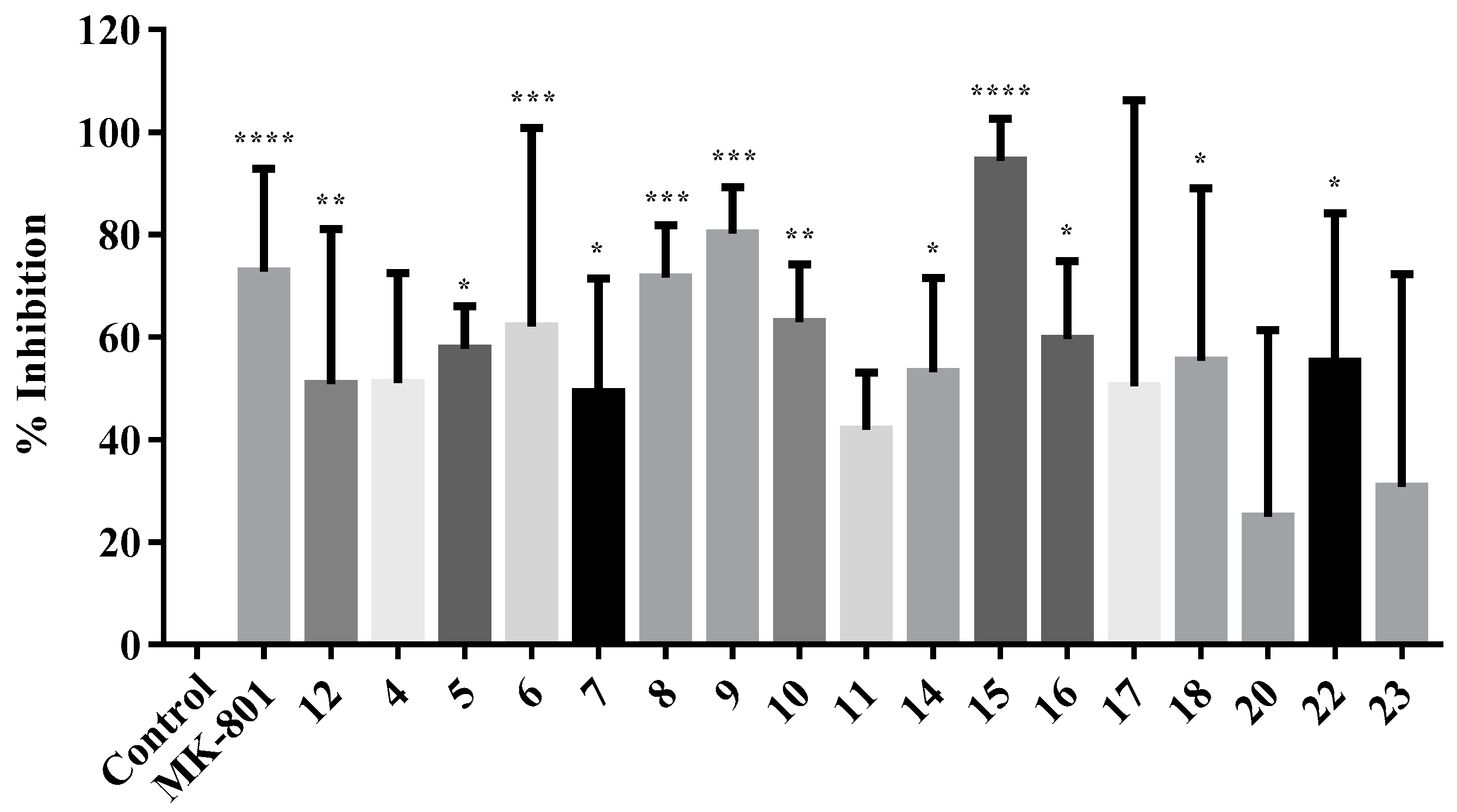{kind=link}
{kind=link}
{kind=link}
{kind=link}
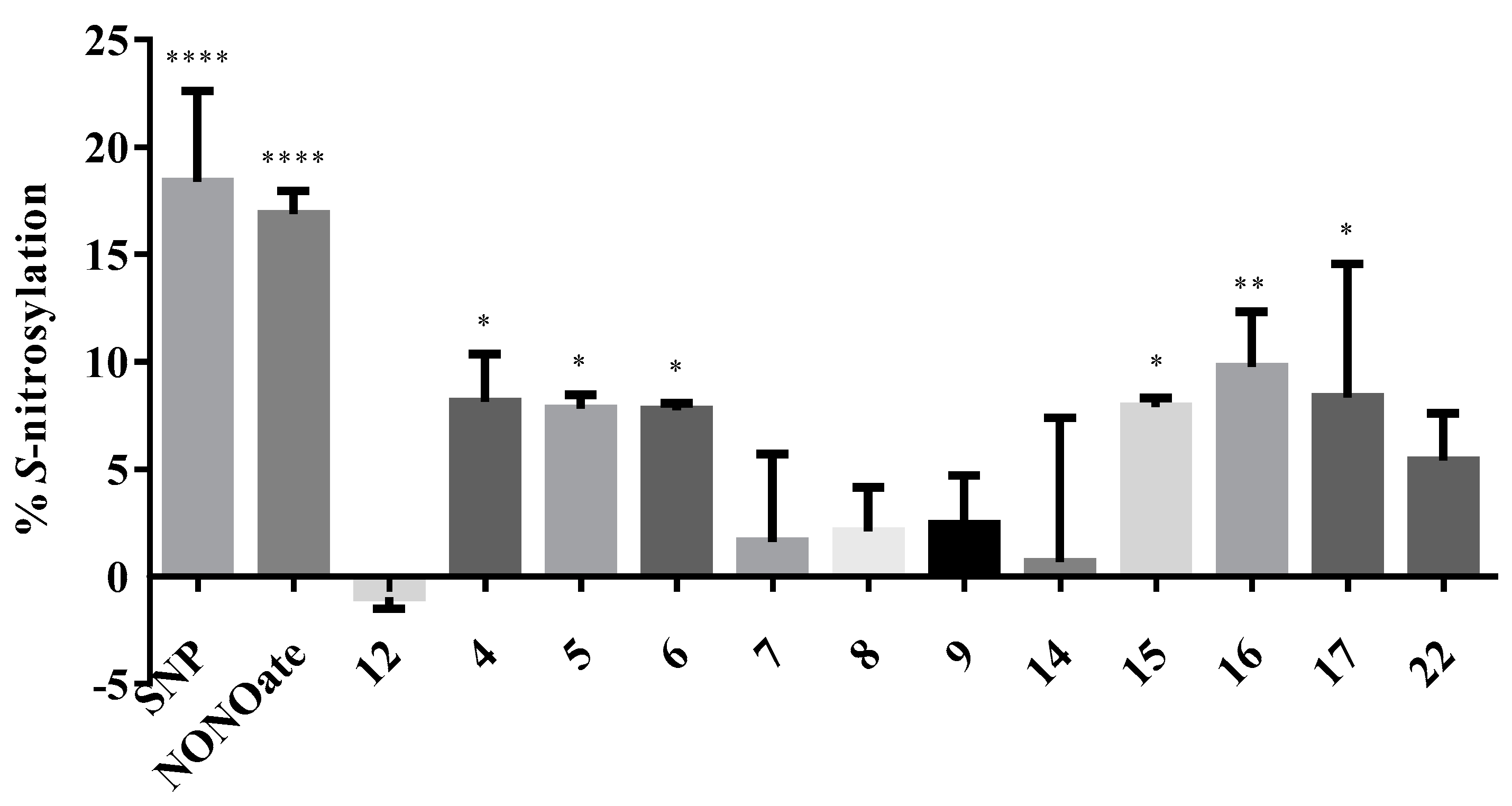{kind=link}
{kind=link}
{kind=link}
| Code | % Viability at Different Concentrations | |||||||
|---|---|---|---|---|---|---|---|---|
| 200 µM | 100 µM | 50 µM | 25 µM | 12.5 µM | 6.25 µM | 3.13 µM | 1.56 µM | |
| 4 | 84.55 | 97.24 | 93.68 | 95.46 | 96.02 | 98.67 | 98.85 | 95.85 |
| 5 | 85.90 | 91.47 | 84.30 | 92.58 | 100.18 | 90.40 | 95.10 | 98.68 |
| 6 | 75.47 | 98.77 | 98.27 | 103.91 | 99.17 | 97.12 | 94.12 | 106.21 |
| 7 | 102.42 | 105.10 | 109.14 | 116.15 | 110.57 | 117.52 | 106.64 | 110.51 |
| 8 | 96.43 | 108.25 | 111.05 | 112.36 | 109.39 | 114.19 | 111.45 | 116.13 |
| 9 | 121.08 | 122.17 | 113.29 | 111.37 | 109.14 | 113.08 | 113.76 | 115.58 |
| 10 | 117.90 | 120.65 | 117.10 | 112.69 | 116.29 | 111.34 | 116.34 | 117.53 |
| 11 | 98.53 | 101.52 | 97.62 | 104.37 | 101.06 | 104.19 | 103.87 | 101.56 |
| 12 | 90.55 | 103.71 | 100.13 | 109.19 | 98.65 | 86.40 | 92.88 | 99.39 |
| 14 | 68.33 | 108.98 | 90.37 | 98.13 | 100.56 | 96.09 | 109.83 | 104.97 |
| 15 | 79.46 | 93.89 | 109.08 | 115.43 | 102.87 | 105.63 | 101.42 | 107.42 |
| 16 | 81.07 | 85.38 | 93.21 | 100.70 | 96.06 | 106.68 | 107.63 | 109.98 |
| 17 | 61.16 | 102.19 | 102.91 | 107.85 | 97.14 | 103.35 | 102.25 | 110.06 |
| 18 | 96.82 | 94.19 | 92.45 | 105.69 | 94.99 | 90.97 | 95.88 | 102.41 |
| 20 | 63.05 | 80.70 | 88.05 | 90.08 | 90.57 | 93.91 | 90.68 | 99.67 |
| 22 | 72.98 | 112.89 | 130.70 | 128.28 | 118.47 | 94.80 | 105.84 | 115.89 |
| 23 | 127.86 | 126.76 | 128.73 | 123.01 | 132.12 | 102.78 | 110.27 | 99.59 |
| Code | %Neuroprotection 5 µM | %Neuroprotection 25 µM | %Neuroprotection 50 µM |
|---|---|---|---|
| 4 | −8.94 | 21.08 | 28.17 * |
| 5 | −13.47 | 24.48 | −23.05 |
| 6 | 6.15 | −0.66 | 26.55 * |
| 7 | 14.53 | 5.98 | −1.59 |
| 8 | −10.90 | 9.95 | 39.62 * |
| 9 | −9.58 | −7.78 | 3.42 |
| 10 | −12.54 | −3.92 | 9.52 |
| 11 | 14.25 | 9.37 | 1.38 |
| 12 | 2.88 | 19.88 * | 3.38 |
| 14 | 8.04 | 21.55 | 24.87 |
| 15 | 4.40 | 4.68 | 11.21 |
| 16 | 6.02 | 9.68 * | 18.71 ** |
| 17 | 5.95 | 5.72 | 16.69 |
| 18 | 12.72 | 5.52 | −8.01 |
| 20 | −1.58 | −15.46 | 0.29 |
| 22 | −5.36 | −6.19 | 24.92 * |
| 23 | 6.40 | 17.49* | 15.33 * |
| Code | %CYT 50 µM | %NP 50 µM | %NMDA 100 µM | %VG 100 µM | %S-NO |
|---|---|---|---|---|---|
| 4 | 93.68 | 28.17 * | 51.04 | 30.54 | 8.14 * |
| 5 | 84.3 | 24.48 at 25 μM | 57.74 * | 37.91 | 7.81 * |
| 6 | 98.27 | 26.55 * | 62.17 *** | 39.95 * | 7.76 * |
| 7 | 109.14 | 14.53 at 5 μM | 49.26 * | 38.51 | 1.61 |
| 8 | 111.05 | 39.62 * | 71.66 *** | 41.18 * | 2.09 |
| 9 | 113.29 | 3.42 | 80.29 *** | 23.61 | 2.42 |
| 10 | 117.1 | 9.52 | 62.92 ** | 46.03 * | n. d. |
| 11 | 97.62 | 14.25 at 5 μM | 41.88 | 30.33 | n. d. |
| 12 | 100.13 | 19.88 * at 25 μM | 50.84 ** | 30.71 | n. d. |
| 14 | 90.37 | 24.87 | 53.19 * | 36.79 | 0.68 |
| 15 | 109.08 | 11.21 | 94.48 **** | 29.35 | 7.89 * |
| 16 | 93.21 | 18.71 ** | 59.6* | 47.15 ** | 9.77 ** |
| 17 | 102.91 | 16.69 | 50.47 | 38.91 * | 8.34 * |
| 18 | 92.45 | 12.72 at 5 μM | 55.43 * | 49.69 ** | n. d. |
| 20 | 88.05 | 0.29 | 24.92 | 35.69 | n. d. |
| 22 | 130.70 | 24.92 * | 55.22 * | 43.76 * | 5.40 |
| 23 | 128.73 | 15.33 * | 30.78 | 26.68 | n. d. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, R.; Joubert, J.; Malan, S.F. Synthesis and Biological Evaluations of NO-Donating Oxa- and Aza-Pentacycloundecane Derivatives as Potential Neuroprotective Candidates. Molecules 2018, 23, 308. https://doi.org/10.3390/molecules23020308
Sharma R, Joubert J, Malan SF. Synthesis and Biological Evaluations of NO-Donating Oxa- and Aza-Pentacycloundecane Derivatives as Potential Neuroprotective Candidates. Molecules. 2018; 23(2):308. https://doi.org/10.3390/molecules23020308
Chicago/Turabian StyleSharma, Rajan, Jacques Joubert, and Sarel F. Malan. 2018. "Synthesis and Biological Evaluations of NO-Donating Oxa- and Aza-Pentacycloundecane Derivatives as Potential Neuroprotective Candidates" Molecules 23, no. 2: 308. https://doi.org/10.3390/molecules23020308
APA StyleSharma, R., Joubert, J., & Malan, S. F. (2018). Synthesis and Biological Evaluations of NO-Donating Oxa- and Aza-Pentacycloundecane Derivatives as Potential Neuroprotective Candidates. Molecules, 23(2), 308. https://doi.org/10.3390/molecules23020308