Highly Efficient One-Pot Synthesis of COS-Based Block Copolymers by Using Organic Lewis Pairs


Abstract
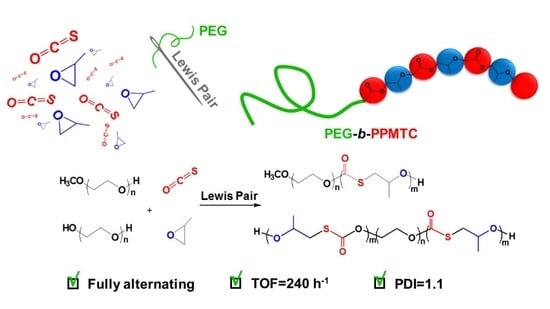
:
1. Introduction
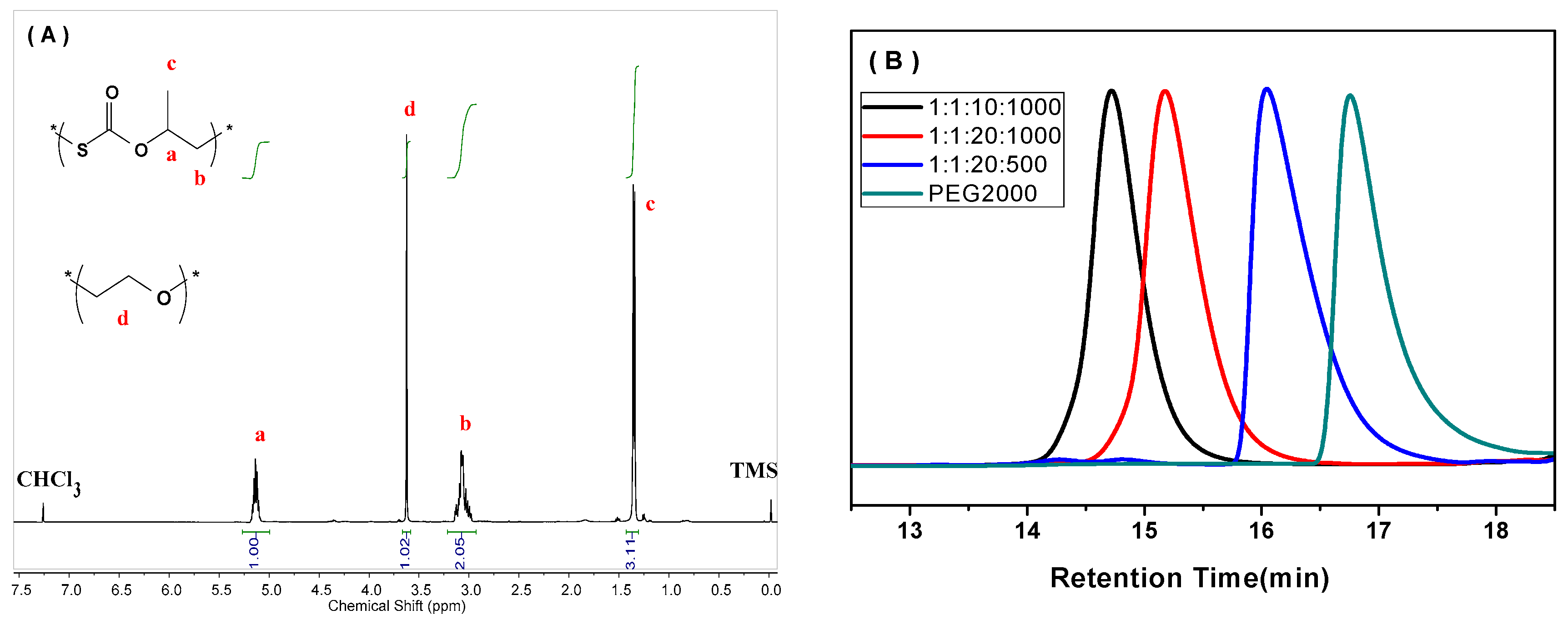
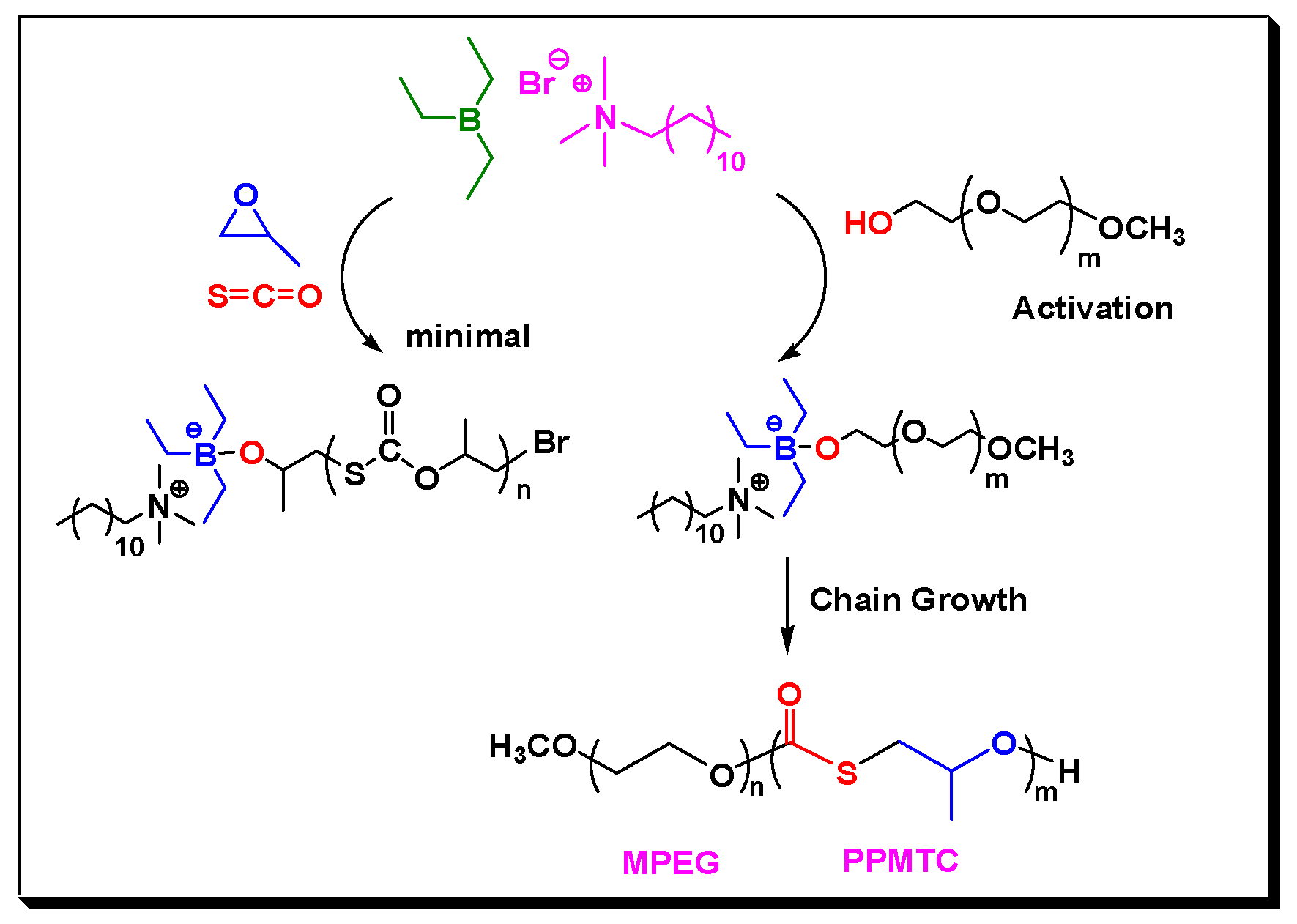
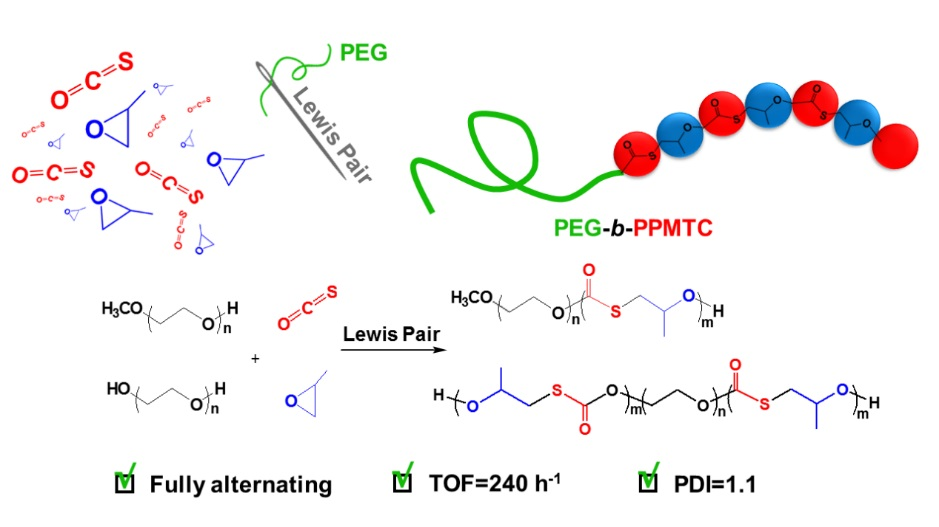
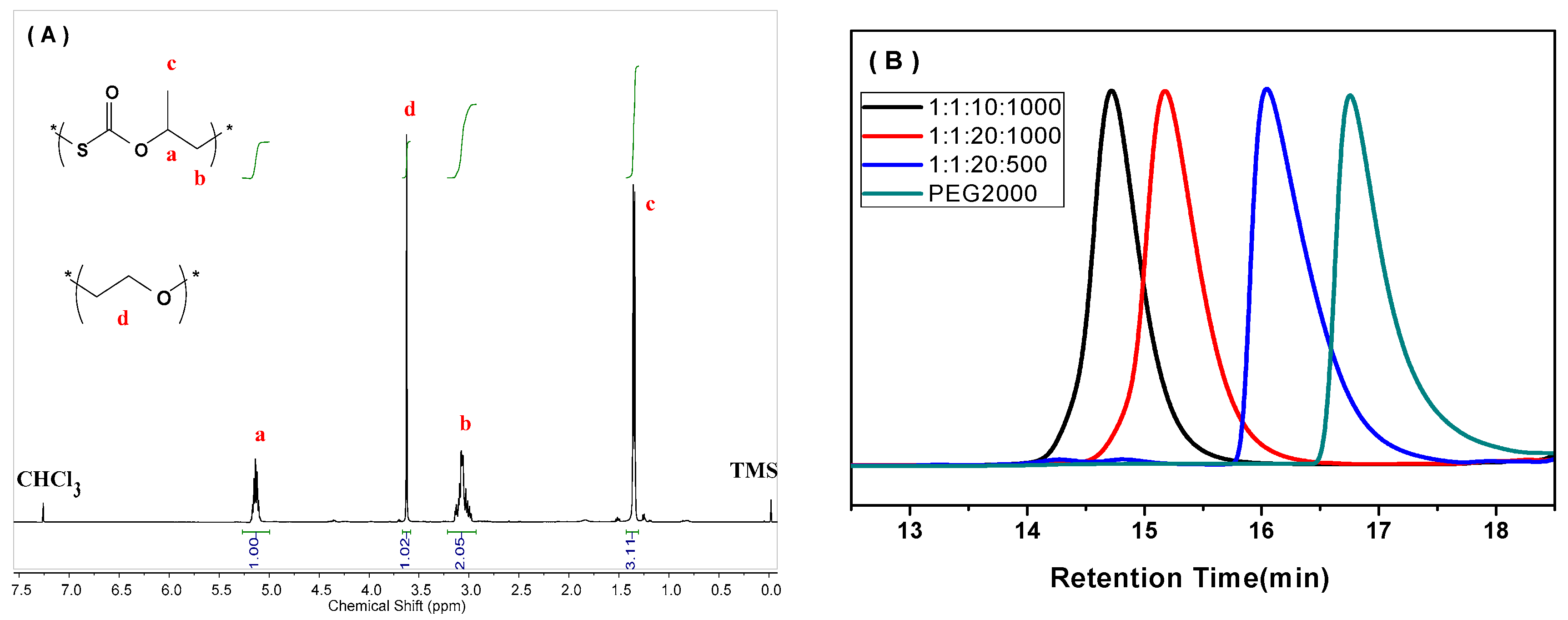
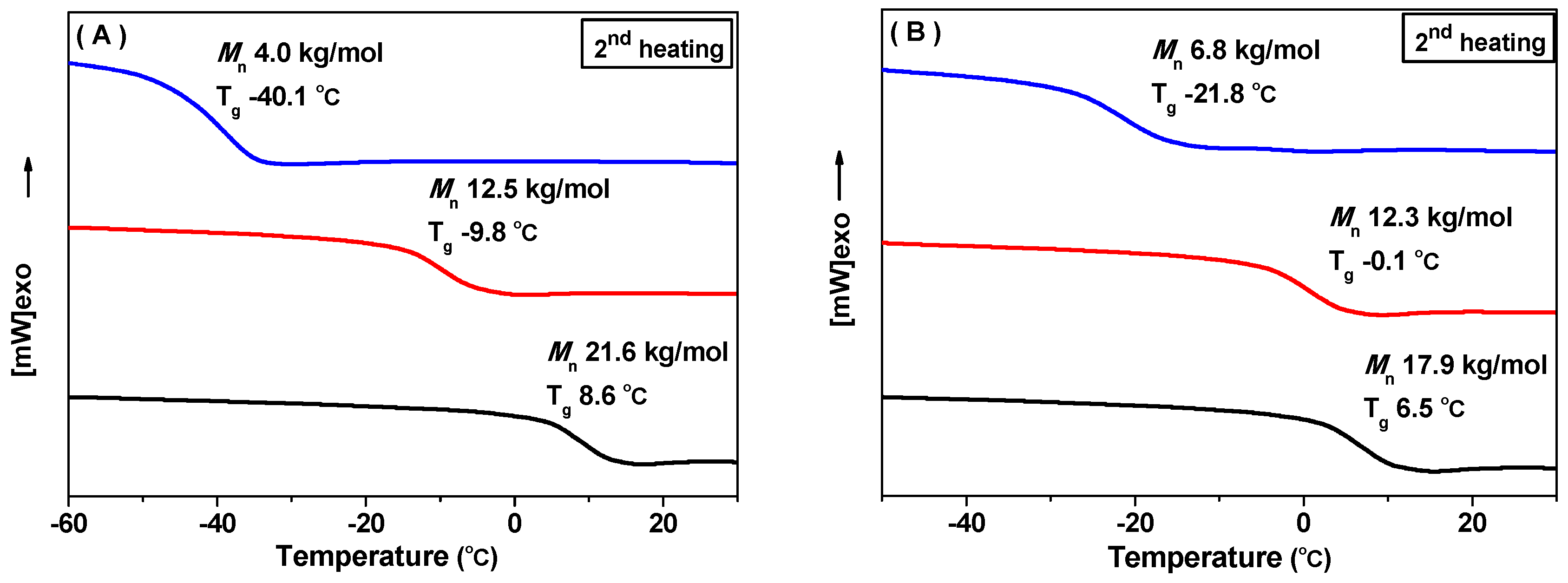
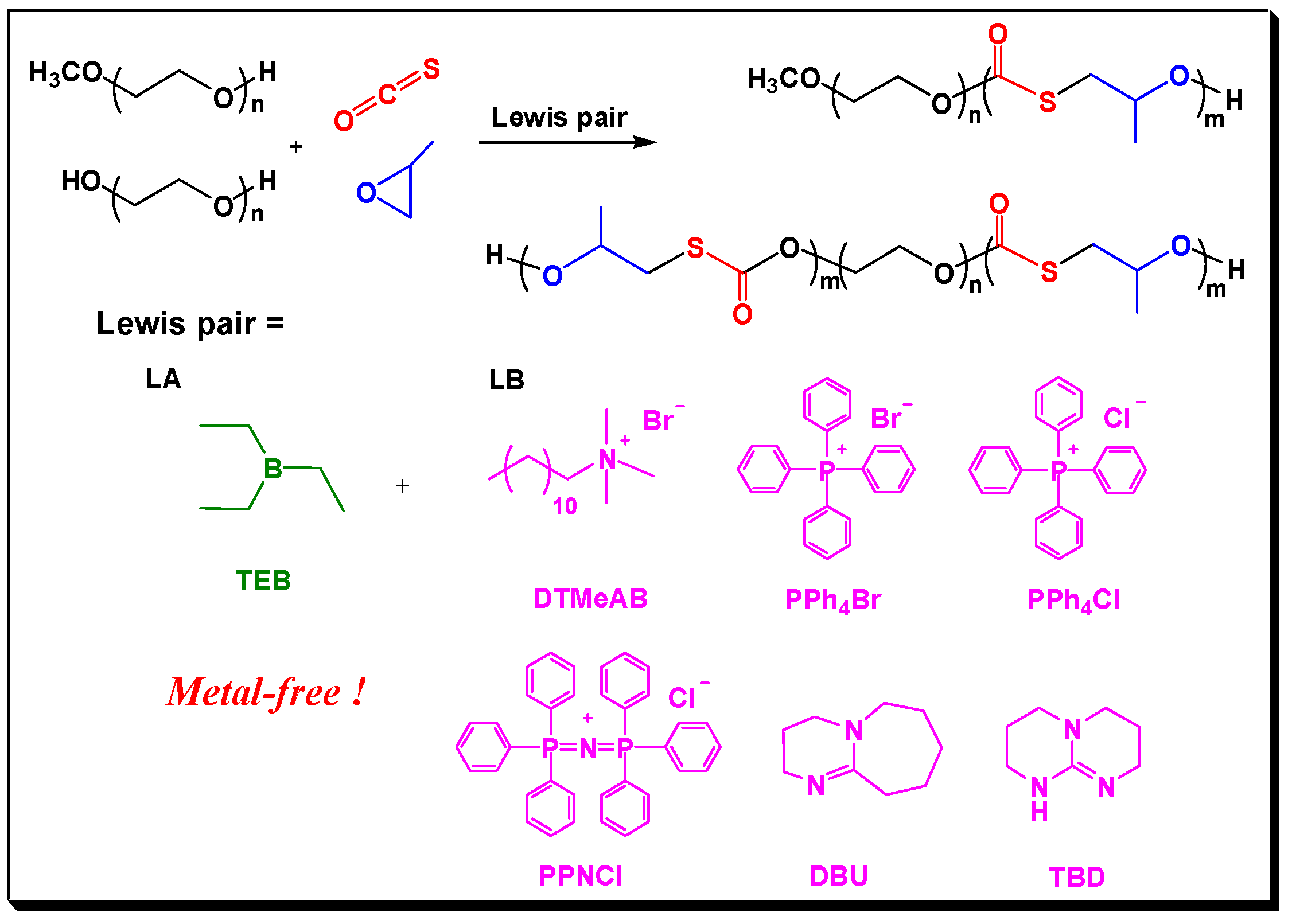
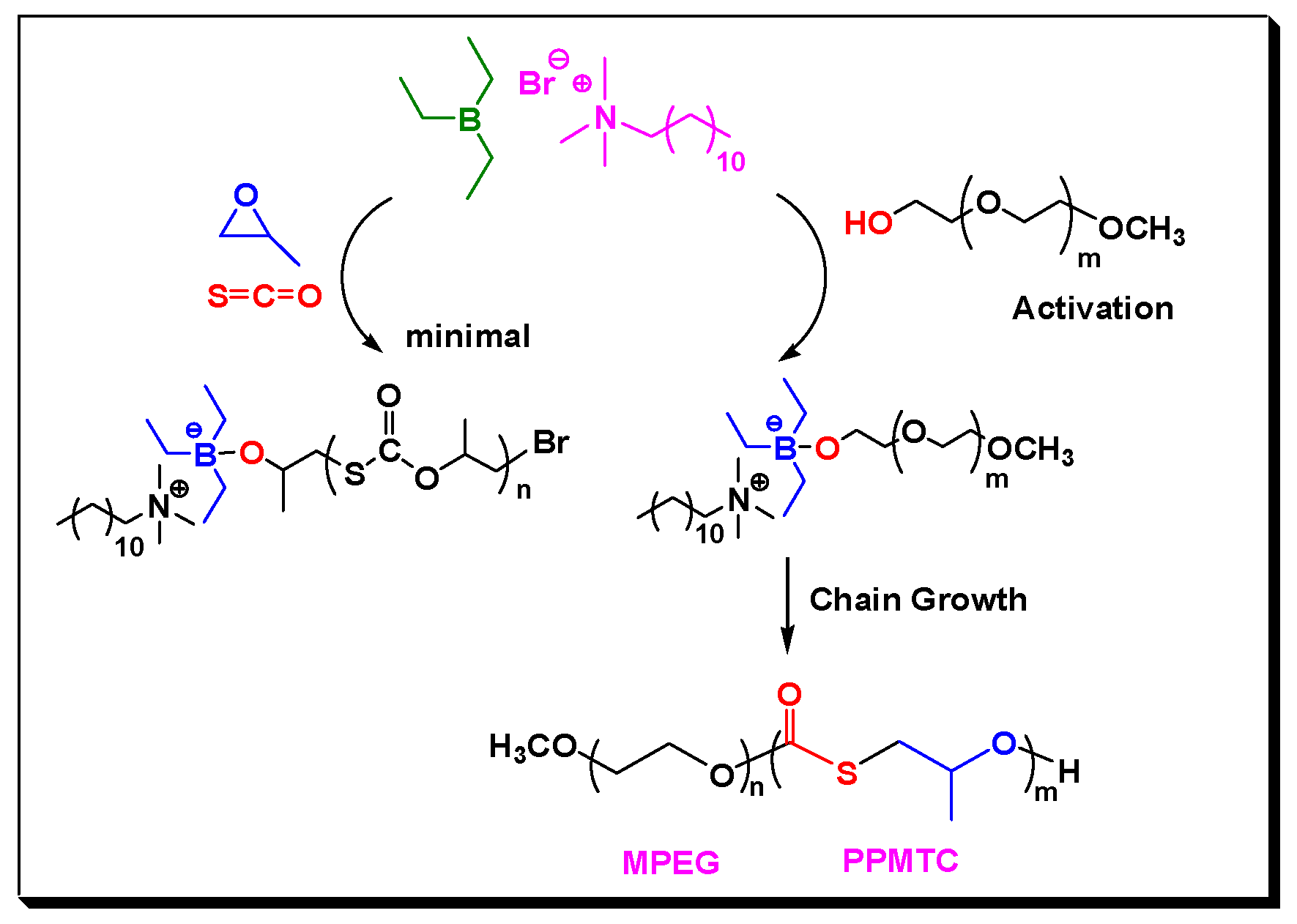
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Copolymerization of COS with Epoxides in the Presence of CTAs, Experimental Details
3.3. Characterization
3.3.1. Nuclear Magnetic Resonance (NMR)
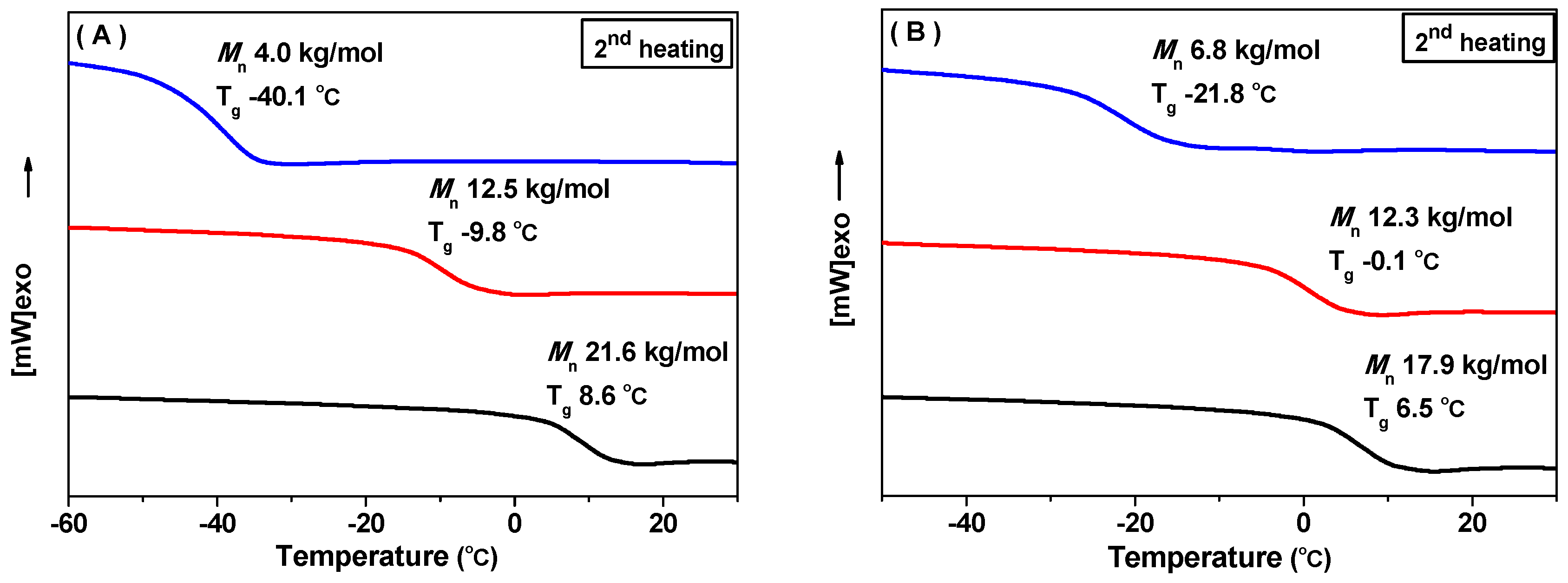
3.3.2. Differential Scanning Calorimetry (DSC)
3.3.3. Thermogravimetric Analysis (TGA)
3.4. Calculation of TOF, Conversion of CTAs and Copolymer Selectivity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

References
- Hong, M.; Chen, E.Y. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of gamma-butyrolactone. Nat. Chem. 2016, 8, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.J.; Tang, C.B. Controlled polymerization of next-generation renewable monomers and beyond. Macromolecules 2013, 46, 1689–1712. [Google Scholar] [CrossRef]
- Tian, H.Y.; Chen, X.S. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012, 37, 237–280. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Making plastics from carbon dioxide: Salen metal complexes as catalysts for the production of polycarbonates from epoxides and CO2. Chem. Rev. 2007, 107, 2388–2410. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Moore, D.R. Discrete metal-based catalysts for the copolymerization of CO2 and epoxides: Discovery, reactivity, optimization, and mechanism. Angew. Chem. Int. Ed. 2004, 43, 6618–6639. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, X.H. Using carbon dioxide and its sulfur analogues as monomers in polymer synthesis. Polymer 2016, 82, 406–431. [Google Scholar] [CrossRef]
- Lu, X.B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.S.; Wang, X.H. Recent advances in carbon dioxide based copolymers. J. CO2 Util. 2015, 11, 3–9. [Google Scholar] [CrossRef]
- Cox, R.A.; Sheppard, D. Reactions of OH radicals with gaseous sulfur compounds. Nature 1980, 284, 330–331. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, X.H. Regioselective and alternating copolymerization of carbonyl sulfide with racemic propylene oxide. Macromolecules 2013, 46, 5899–5904. [Google Scholar] [CrossRef]
- Nakano, R.; Nozaki, K. Copolymerization of carbon dioxide and butadiene via a lactone intermediate. Nat. Chem. 2014, 6, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Darensbourg, D.J.; Wilson, S.J. What’s new with CO2? Recent advances in its copolymerization with oxiranes. Green Chem. 2012, 14, 2665–2671. [Google Scholar] [CrossRef]
- Klaus, S.; Rieger, B. Recent advances in CO2/epoxide copolymerization-new strategies and cooperative mechanisms. Coord. Chem. Rev. 2011, 255, 1460–1479. [Google Scholar] [CrossRef]
- Kember, M.R.; Williams, C.K. Catalysts for CO2/epoxide copolymerisation. Chem. Commun. 2011, 47, 141–163. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.Q.; Coates, G.W. Cobalt-based complexes for the copolymerization of propylene oxide and CO2: Active and selective catalysts for polycarbonate synthesis. Angew. Chem. Int. Ed. 2003, 42, 5484–5487. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, X.H. Poly(monothiocarbonate)s from the alternating and regioselective copolymerization of carbonyl sulfide with epoxides. Acc. Chem. Res. 2016, 49, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhang, X.H. Well-defined high refractive index poly(monothiocarbonate) with tunable Abbe's numbers and glass-transition temperatures via terpolymerization. Polym. Chem. 2015, 6, 4978–4983. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, X.H. Highly regioselective and alternating copolymerization of carbonyl sulfide with phenyl glycidyl ether. Polym. Chem. 2015, 6, 6955–6958. [Google Scholar] [CrossRef]
- Wu, H.L.; Zhang, X.H. Poly(trimethylene monothiocarbonate) from the alternating copolymerization of COS and oxetane: A semicrystalline copolymer. Macromolecules 2016, 49, 8863–8868. [Google Scholar] [CrossRef]
- Ren, W.M.; Lu, X.B. Crystalline and elastomeric poly(monothiocarbonate)s prepared from copolymerization of COS and achiral epoxide. Macromolecules 2016, 50, 63–68. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, X.H. Alternating copolymerization of carbonyl sulfide and cyclohexene oxide catalyzed by zinc–cobalt double metal cyanide complex. Polymer 2014, 55, 3688–3695. [Google Scholar] [CrossRef]
- Luo, M.; Zhang, X.H. An examination of the steric and electronic effects in the copolymerization of carbonyl sulfide and styrene oxide. Macromolecules 2015, 48, 6057–6062. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.H. Mechanistic study of regio-defects in the copolymerization of propylene oxide/carbonyl sulfide catalyzed by (salen)CrX complexes. Macromolecules 2017, 50, 8426–8437. [Google Scholar] [CrossRef]
- Yang, J.L.; Zhang, X.H. Perfectly alternating and regioselective copolymerization of carbonyl sulfide and epoxides by metal-free Lewis pairs. Angew. Chem. Int. Ed. 2017, 56, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S. Immortal polymerization: The qutset, development, and application. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2861–2871. [Google Scholar] [CrossRef]
- Scharfenberg, M.; Frey, H. Multiarm polycarbonate star polymers with a hyperbranched polyether core from CO2 and common epoxides. Macromolecules 2017, 50, 6577–6585. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Switchable catalytic processes involving the copolymerization of epoxides and carbon dioxide for the preparation of block polymers. Inorg. Chem. Front. 2017, 4, 412–419. [Google Scholar] [CrossRef]
- Liu, S.J.; Wang, F.S. Construction of well-defined redox-responsive CO2-based polycarbonates: Combination of immortal copolymerization and prereaction approach. Macromol. Rapid Commun. 2017, 38, 1600754. [Google Scholar] [CrossRef] [PubMed]
- Reiter, M.; Rieger, B. In situ generated ABA block copolymers from CO2, cyclohexene oxide, and poly(dimethylsiloxane)s. ACS Macro Lett. 2016, 5, 419–423. [Google Scholar] [CrossRef]
- Cyriac, A.; Lee, B.Y. Immortal CO2/propylene oxide copolymerization: Precise control of molecular weight and architecture of various block copolymers. Macromolecules 2010, 43, 7398–7401. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Chen, E.Y. Alane-based classical and frustrated Lewis pairs in polymer synthesis: Rapid polymerization of MMA and naturally renewable methylene butyrolactones into high-molecular-weight polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.Q.; Chen, E.Y. Probing site cooperativity of frustrated phosphine/borane Lewis pairs by a polymerization study. J. Am. Chem. Soc. 2014, 136, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.B.; Li, B. Design of highly active binary catalyst systems for CO2/epoxide copolymerization: Polymer selectivity, enantioselectivity, and stereochemistry control. J. Am. Chem. Soc. 2006, 128, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.B.; Chen, E.Y. From meso-lactide to isotactic polylactide: Epimerization by B/N Lewis pairs and kinetic resolution by organic catalysts. J. Am. Chem. Soc. 2015, 137, 12506–12509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Liu, F. Atom-exchange coordination polymerization of carbon disulfide and propylene oxide by a highly effective double-metal cyanide complex. Macromolecules 2008, 41, 1587–1590. [Google Scholar] [CrossRef]
Sample Availability: Samples of the di-block and tri-block copolymers are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | CTA | LA/LB/CTA/PO | TOF (h−1) 2 | Conv. of CTA (%) 3 | Copolym. Select. (%) 4 | Mn (kg/mol) 5 | PDI (Mw/Mn) 5 |
|---|---|---|---|---|---|---|---|
| 1 | M750 | 1:1:5:1000 | 117 | >99 | 100 | 27.6 | 1.2 |
| 2 | M750 | 1:1:10:1000 | 115 | >99 | 97 | 14.7 | 1.2 |
| 3 | M750 | 1:1:20:1000 | 120 | 92 | 100 | 7.1 | 1.1 |
| 4 | M750 | 1:1:50:1000 | 123 | 95 | 100 | 3.8 | 1.1 |
| 5 | M750 | 1:1:20:500 | 60 | >99 | 100 | 5.9 | 1.1 |
| 6 | M750 | 1:1:20:2000 | 232 | >99 | 100 | 23.8 | 1.2 |
| 7 | P2000 | 1:1:5:1000 | 115 | >99 | 100 | 51.0 | 1.2 |
| 8 | P2000 | 1:1:10:1000 | 117 | 97 | 100 | 21.6 | 1.1 |
| 9 | P2000 | 1:1:20:1000 | 122 | >99 | 100 | 12.5 | 1.1 |
| 10 | P2000 | 1:1:50:1000 | 124 | 94 | 100 | 6.6 | 1.3 |
| 11 | P2000 | 1:1:20:500 | 59 | >99 | 100 | 4.0 | 1.1 |
| 12 | P2000 | 1:1:20:2000 | 240 | >99 | 100 | 21.9 | 1.1 |
| Entry | CTA | LA/LB/CTA/PO | TOF (h−1) 2 | Conv of CTA (%) 3 | Copolymer Selectivity (%) 4 | Mn (kg/mol) 5 | PDI (Mw/Mn) 5 |
|---|---|---|---|---|---|---|---|
| 1 6 | M750 | 1:1:20:1000 | 122 | 96 | 87 | 6.5 | 1.2 |
| 2 7 | M750 | 1:1:20:1000 | 123 | 98 | 84 | 7.1 | 1.1 |
| 3 6 | P2000 | 1:1:20:1000 | 119 | >99 | 100 | 11.5 | 1.1 |
| 4 7 | P2000 | 1:1:20:1000 | 124 | 99 | 86 | 10.9 | 1.2 |
| 5 | M2000 | 1:1:5:1000 | 121 | >99 | 95 | 17.9 | 1.3 |
| 6 | M2000 | 1:1:10:1000 | 117 | 99 | 100 | 12.3 | 1.2 |
| 7 | M2000 | 1:1:20:1000 | 122 | >99 | 100 | 6.8 | 1.1 |
| 8 | M5000 | 1:1:5:1000 | 98 | >99 | 100 | 21.6 | 1.1 |
| 9 | M5000 | 1:1:10:1000 | 68 | 32 | 100 | 23.3 | 1.7 |
| 10 | M5000 | 1:1:20:1000 | 56 | 16 | 100 | 33.5 | 1.3 |
| Entry | LB | TOF (h−1) 2 | Conv of CTA (%) 3 | Copolymer Selectivity (%) 4 | Mn (kg/mol) 5 | PDI (Mw/Mn) 5 |
|---|---|---|---|---|---|---|
| 1 | PPNCl | 237 | 99 | 84 | 17.9 | 1.3 |
| 2 | PPh4Cl | 234 | >99 | 95 | 16.1 | 1.2 |
| 3 | PPh4Br | 229 | 98 | 94 | 14.8 | 1.2 |
| 4 | DBU | 202 | 94 | 84 | 11.4 | 1.2 |
| 5 | TBD | 214 | 91 | 90 | 14.7 | 1.3 |
| 6 | DMAP | 0 | - | - | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.-L.; Cao, X.-H.; Zhang, C.-J.; Wu, H.-L.; Zhang, X.-H. Highly Efficient One-Pot Synthesis of COS-Based Block Copolymers by Using Organic Lewis Pairs. Molecules 2018, 23, 298. https://doi.org/10.3390/molecules23020298
Yang J-L, Cao X-H, Zhang C-J, Wu H-L, Zhang X-H. Highly Efficient One-Pot Synthesis of COS-Based Block Copolymers by Using Organic Lewis Pairs. Molecules. 2018; 23(2):298. https://doi.org/10.3390/molecules23020298
Chicago/Turabian StyleYang, Jia-Liang, Xiao-Han Cao, Cheng-Jian Zhang, Hai-Lin Wu, and Xing-Hong Zhang. 2018. "Highly Efficient One-Pot Synthesis of COS-Based Block Copolymers by Using Organic Lewis Pairs" Molecules 23, no. 2: 298. https://doi.org/10.3390/molecules23020298
APA StyleYang, J.-L., Cao, X.-H., Zhang, C.-J., Wu, H.-L., & Zhang, X.-H. (2018). Highly Efficient One-Pot Synthesis of COS-Based Block Copolymers by Using Organic Lewis Pairs. Molecules, 23(2), 298. https://doi.org/10.3390/molecules23020298