Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions


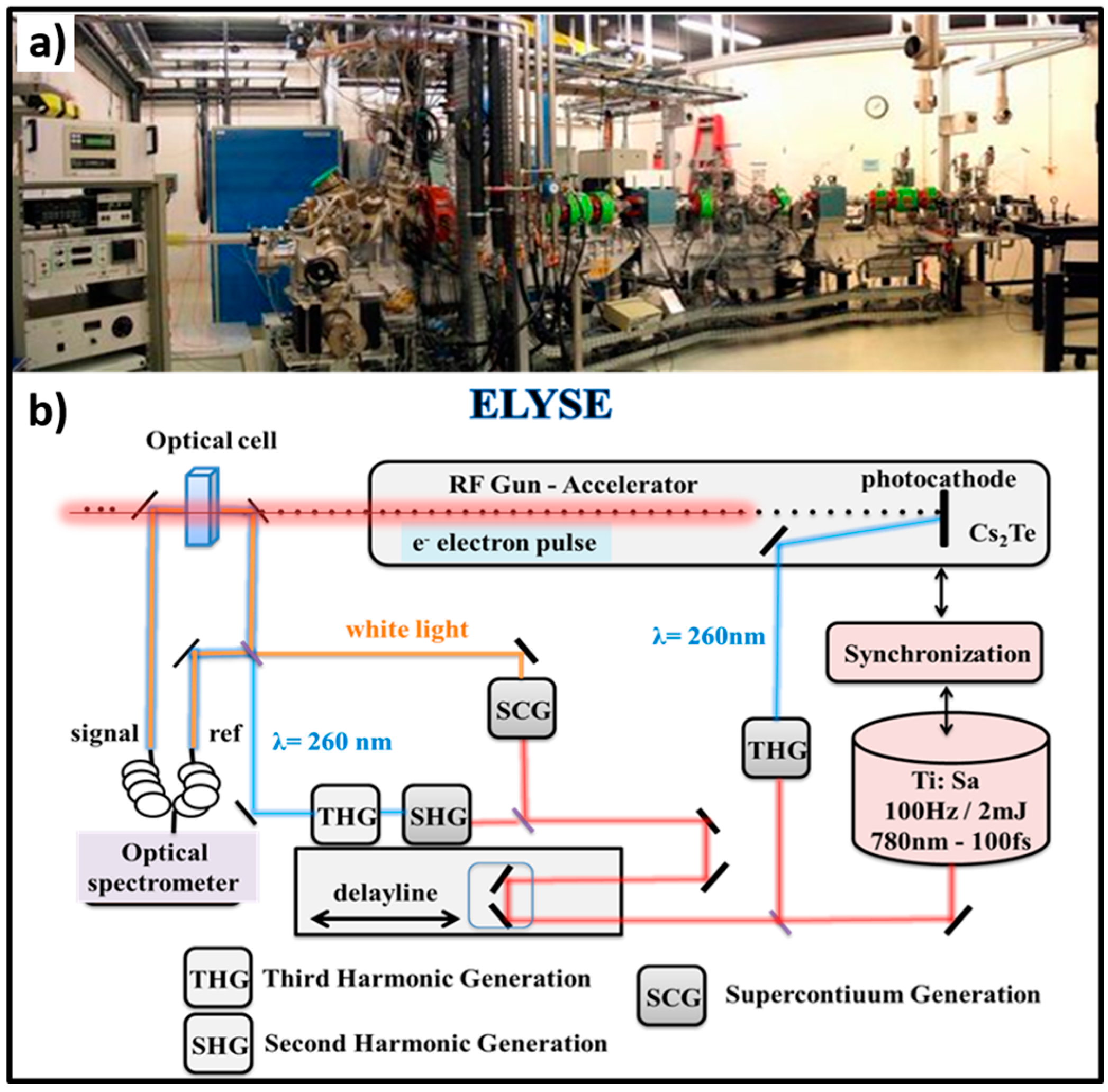{kind=link}
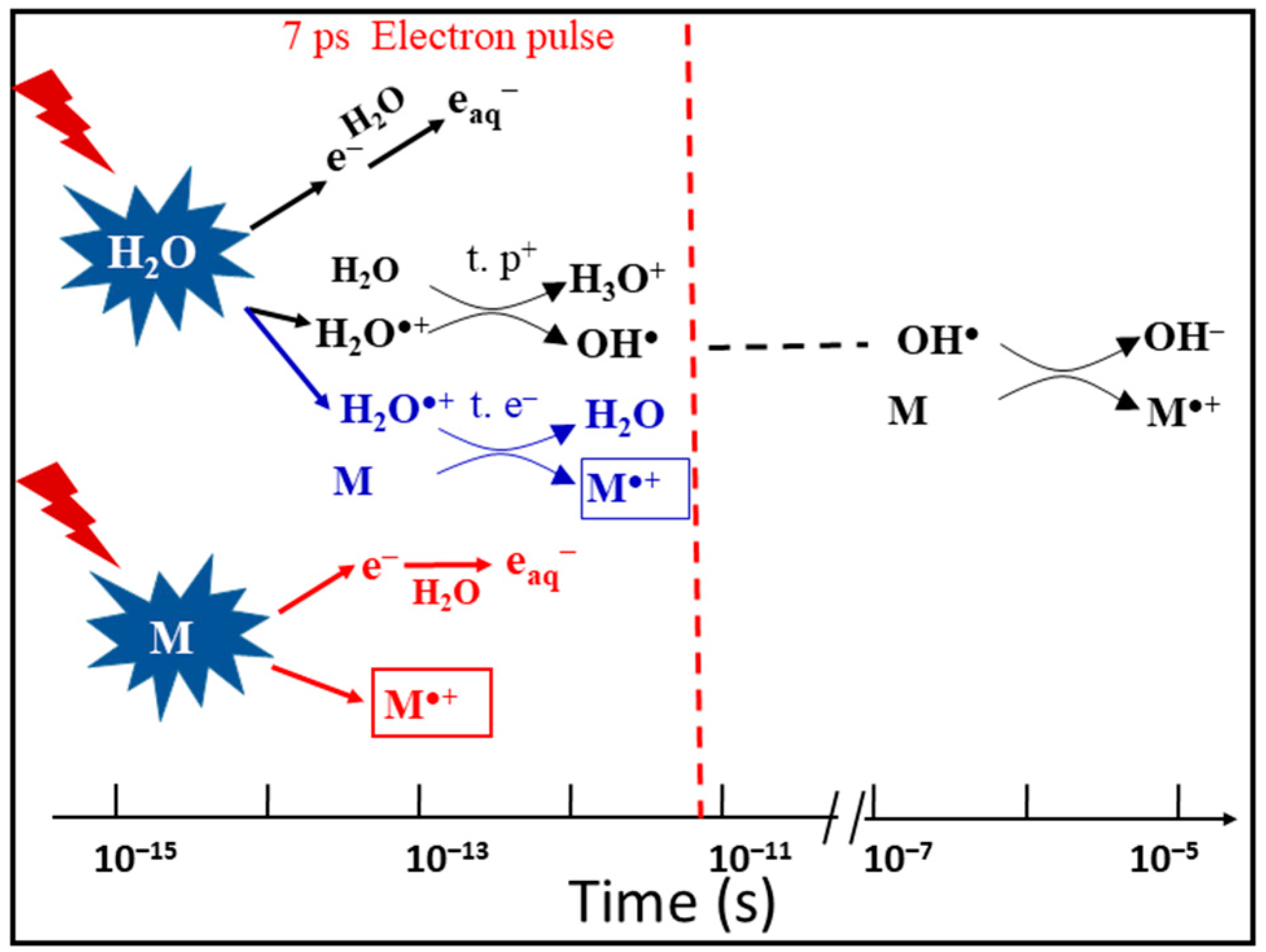{kind=link}
{kind=link}
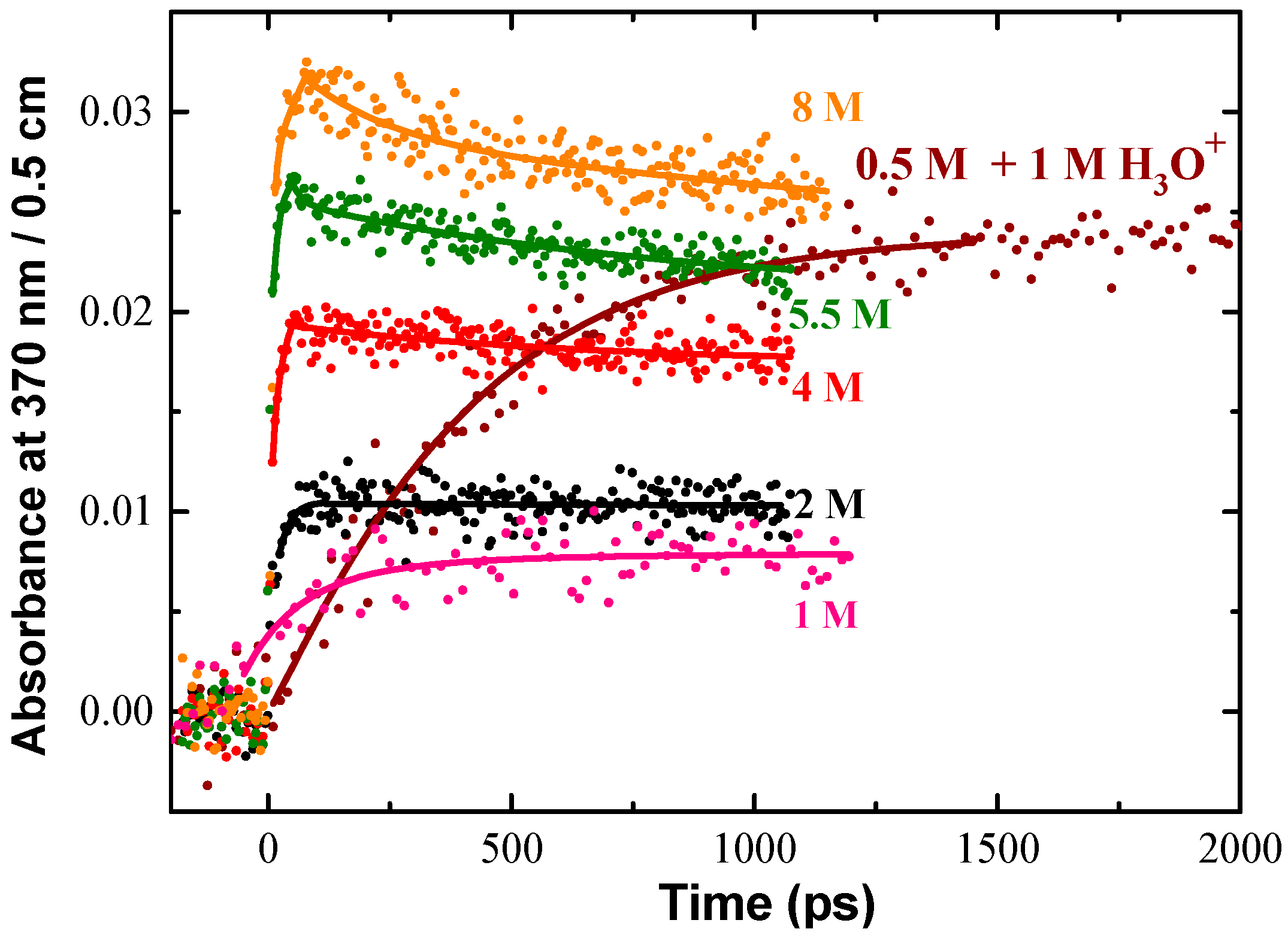{kind=link}
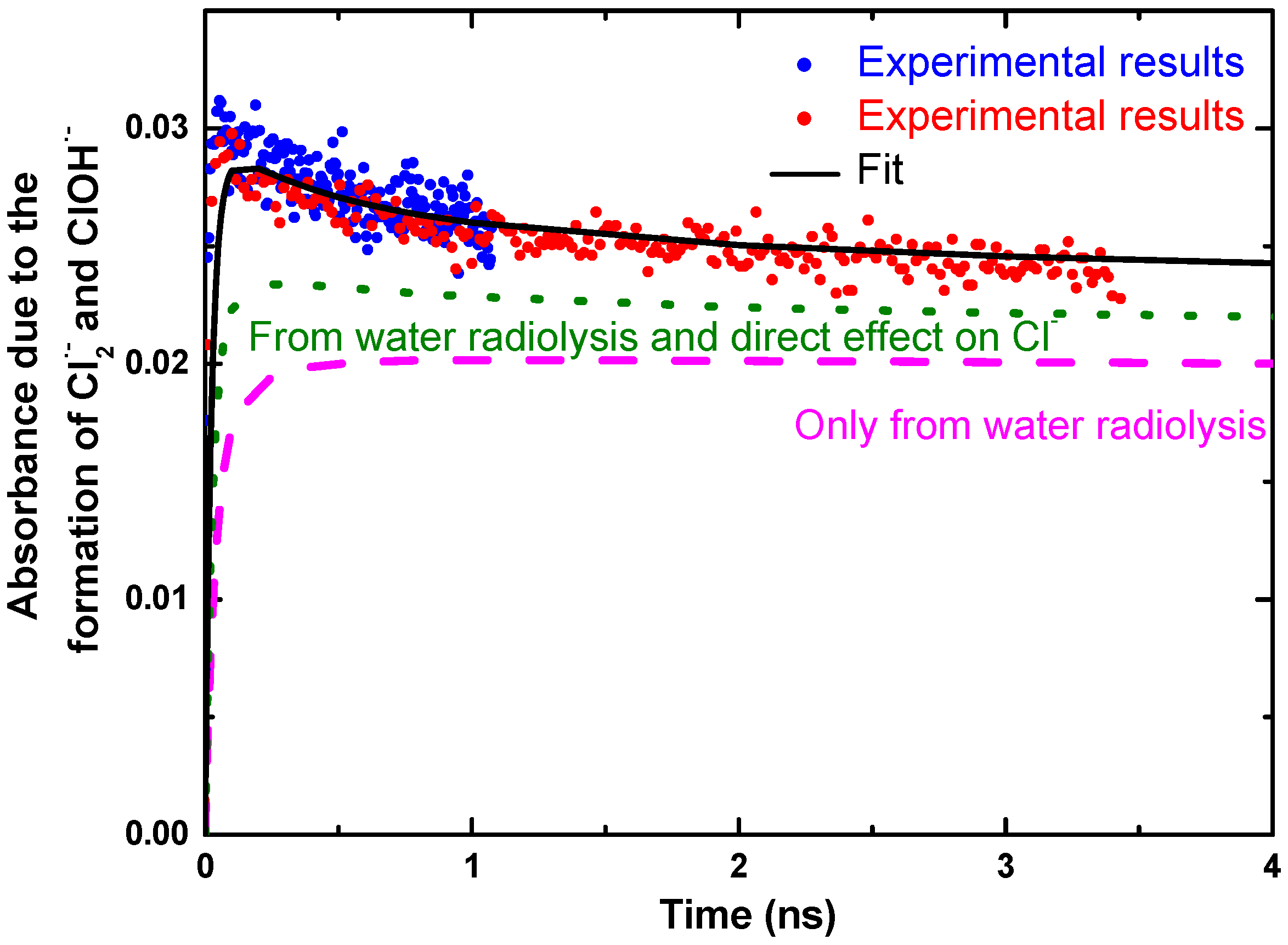{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxidation Reactions by Radicals
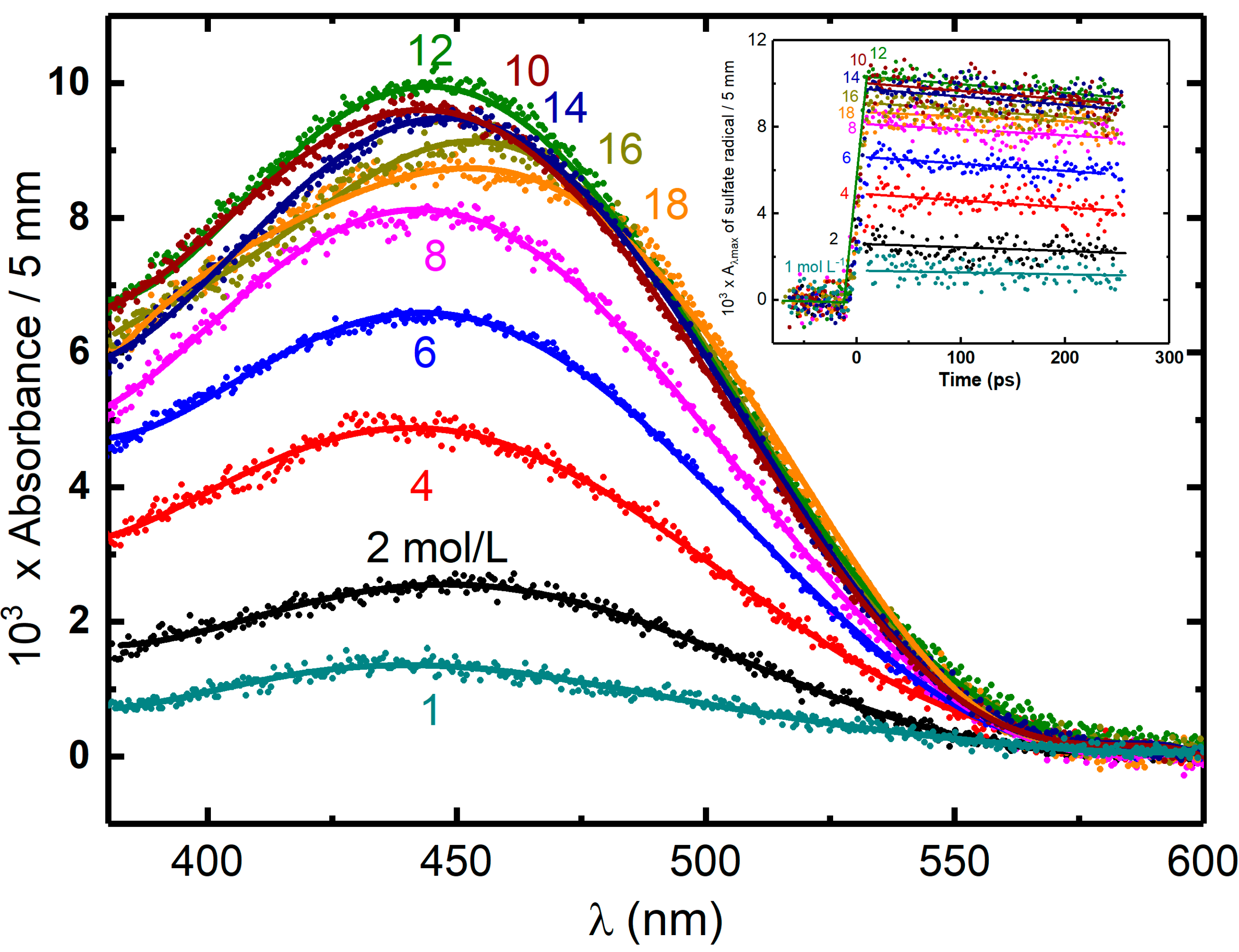
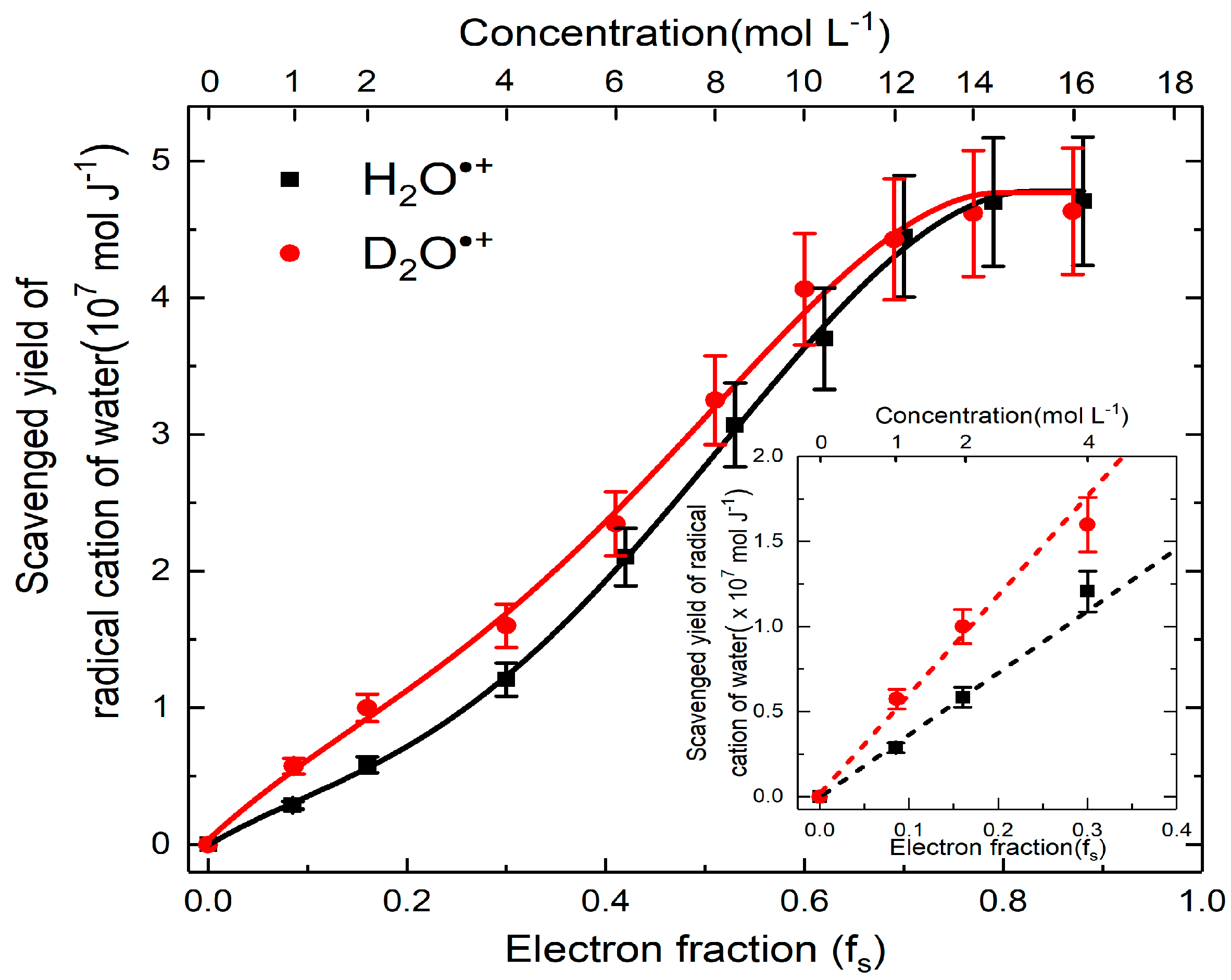
3. Reactivity of H2O•+ in Halide Solutions
4. Reactivity of H2O•+ in Highly Acidic Solutions
5. The Nature and Ultrafast Charge Migration of H2O•+
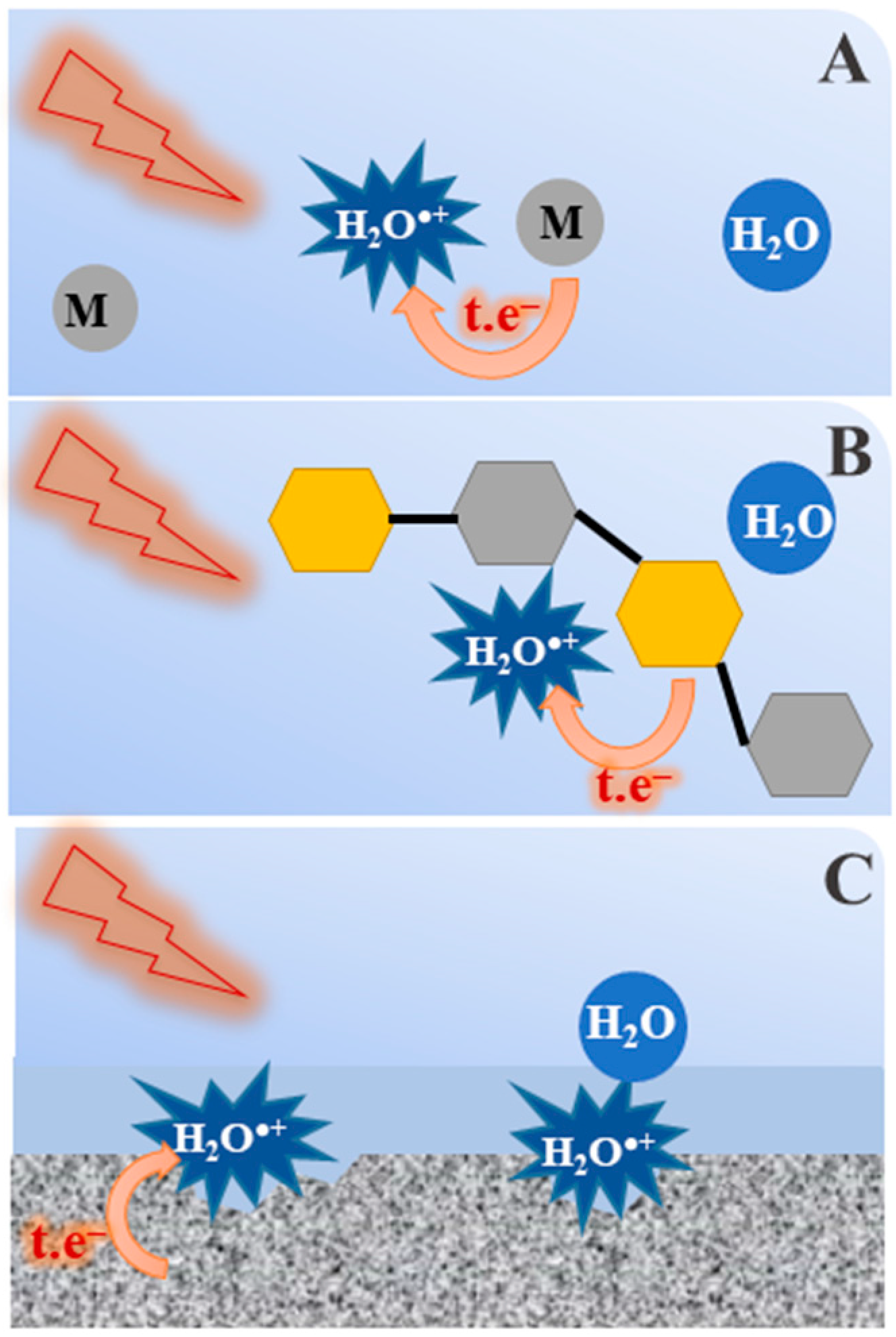
6. Perspectives on Reactivity of H2O•+ in Other Environments
- -
- Radiotherapy and radiobiology: It is known that large amounts of hydrating water molecules are in direct contact with biomolecules such as DNA or proteins [56]. When ionizing radiation is present, part of the radiation energy is absorbed directly by DNA and breaks the bonds of the sugars, phosphates and nucleobases, while some of the radiation is also absorbed by the water adjacent to the DNA. In fact, some amount of water is linked to the DNA and the local concentration of DNA is very high. In that case, the generated H2O•+ radical cation may induce a chemistry different from OH• radical and the consequence is producing the secondary biomolecule radicals which has not been yet considered before.
- -
- Treatment and storage of fuel in the nuclear industry: Spent nuclear fuel is processed in highly concentrated nitric acid aqueous solutions. In this case, the radical cation H2O•+ may react with nitrate ions to yield the NO3• radical, which is also a highly oxidizing species. Alternatively, when radioactive waste of low and medium level is coated by cement or any other porous material, the interface effect is extensive and the formation of radical cation H2O•+ and its oxidation reactions should be taken into account.
- -
- When the core of a nuclear power plant comes into contact with water, as happened during the Fukushima incident in Japan, the amount of radiation deposited at the interface of the exposed fuel/water is important. Although we do not provide any evidence, the present work suggests that, in this situation, metal corrosion by H2O•+ may be involved [57].
- -
- Is the positive charge distributed on several water molecules or is it localized mainly in one water molecule? H2O•+ symbol stands for the water radical cation, but is it possible that the positive charge is distributed on several water molecules (H2O•)+n?
- -
- The absorption band of the water radical cation is not known, but its deprotonated form, OH• radical, absorbs in the region of 200–300 nm. Thus, it is rationalized that ultrafast detection setup (˂30 fs) is needed to observe in deep UV region to confirm that the decay of this species is correlated to the formation of OH• radical in neat water.
- -
- What is the time constant for the decay of H2O•+? This value is important to consider for the competition between the electron transfer and the proton transfer reactions of H2O•+.
- -
- Finally, more precise estimation is needed to evaluate the oxidizing power of H2O•+. The redox potential and the pKa of this radical cation should get better estimation.
Acknowledgments
Conflicts of Interest
References
- Hatano, Y.; Mozumder, A. Introduction. In Charged Particle and Photon Interactions with Matter; CRC Press: Boca Raton, FL, USA, 2003; pp. 1–8. ISBN 9780824746230. [Google Scholar]
- Garrett, B.C.; Dixon, D.A.; Camaioni, D.M.; Chipman, D.M.; Johnson, M.A.; Jonah, C.D.; Kimmel, G.A.; Miller, J.H.; Rescigno, T.N.; Rossky, P.J.; et al. Role of Water in Electron-Initiated Processes and Radical Chemistry: Issues and Scientific Advances. Chem. Rev. 2005, 105, 355–390. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V. An overview of the radiation chemistry of liquids. In Radiation Chemistry—From Basics to Applications in Material and Life Sciences; Spotheim-Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Les Ulis, France, 2008; pp. 35–52. ISBN 9782759800247. [Google Scholar]
- Mozumder, A. Events in Radiation Chemistry: An Introduction. Int. J. Radiat. Appl. Instrum. Part C 1989, 34, 1–3. [Google Scholar] [CrossRef]
- Anicich, V.G. Evaluated Bimolecular Ion-Molecule Gas Phase Kinetics of Positive Ions for Use in Modeling Planetary Atmospheres, Cometary Comae, and Interstellar Clouds. J. Phys. Chem. Ref. Data 1993, 22, 1469–1569. [Google Scholar] [CrossRef]
- Marsalek, O.; Elles, C.G.; Pieniazek, P.A.; Pluhařová, E.; VandeVondele, J.; Bradforth, S.E.; Jungwirth, P. Chasing Charge Localization and Chemical Reactivity Following Photoionization in Liquid Water. J. Chem. Phys. 2011, 135, 224510. [Google Scholar] [CrossRef] [PubMed]
- Khorana, S.; Hamill, W.H. Electronic Processes in the Pulse Radiolysis of Aqueous Solutions of Halide Ions. J. Phys. Chem. 1971, 75, 3081–3088. [Google Scholar] [CrossRef]
- Kim, K.-J.; Hamill, W.H. Direct and Indirect Effects in Pulse Irradiated Concentrated Aqueous Solutions of Chloride and Sulfate Ions. J. Phys. Chem. 1976, 80, 2320–2325. [Google Scholar] [CrossRef]
- Musat, R.; Denisov, S.; Marignier, J.-L.; Mostafavi, M. Decoding the Three-Pronged Mechanism of NO3• Radical Formation in HNO3 Solutions at 22 °C and 80 °C Using Picosecond Pulse Radiolysis. J. Phys. Chem. B 2018. submitted. [Google Scholar] [CrossRef] [PubMed]
- La Vere, T.; Becker, D.; Sevilla, M.D. Yields of •OH in Gamma-irradiated DNA as A Function of DNA Hydration: Hole Transfer in Competition with •OH Formation. Radiat. Res. 1996, 145, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Janata, E.; Schuler, R.H. Rate Constant for Scavenging eaq- in Nitrous Oxide-saturated Solutions. J. Phys. Chem. 1982, 86, 2078–2084. [Google Scholar] [CrossRef]
- Roebke, W.; Renz, M.; Henglein, A. Pulsradiolyse der Anionen S2O82− und HSO5− in Wässriger Lösung. Int. J. Radiat. Phys. Chem. 1969, 1, 39–44. [Google Scholar] [CrossRef]
- Hentz, R.R.; Farhataziz; Hansen, E.M. Pulse Radiolysis of Liquids at High Pressures. II. Diffusion-Controlled Reactions of the Hydrated Electron. J. Chem. Phys. 1972, 56, 4485–4488. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tagawa, S. Direct Observation of Guanine Radical Cation Deprotonation in Duplex DNA Using Pulse Radiolysis. J. Am. Chem. Soc. 2003, 125, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Belloni, J.; Monard, H.; Gobert, F.; Larbre, J.-P.; Demarque, A.; De Waele, V.; Lampre, I.; Marignier, J.-L.; Mostafavi, M.; Bourdon, J.C.; et al. ELYSE—A Picosecond Electron Accelerator for Pulse Radiolysis Research. Nucl. Instrum. Methods Phys. Res. Sect. A 2005, 539, 527–539. [Google Scholar] [CrossRef]
- Schmidhammer, U.; Jeunesse, P.; Stresing, G.; Mostafavi, M. A Broadband Ultrafast Transient Absorption Spectrometer Covering the Range from Near-Infrared (NIR) down to Green. Appl. Spectrosc. 2014, 68, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Wishart, J.F.; Cook, A.R.; Miller, J.R. The LEAF Picosecond Pulse Radiolysis Facility at Brookhaven National Laboratory. Rev. Sci. Instrum. 2004, 75, 4359–4366. [Google Scholar] [CrossRef]
- Muroya, Y.; Lin, M.; Han, Z.; Kumagai, Y.; Sakumi, A.; Ueda, T.; Katsumura, Y. Ultra-fast Pulse Radiolysis: A Review of The Recent System Progress and Its Application to Study on Initial Yields and Solvation Processes of Solvated Electrons in Various Kinds of Alcohols. Radiat. Phys. Chem. 2008, 77, 1176–1182. [Google Scholar] [CrossRef]
- Saeki, A.; Kozawa, T.; Tagawa, S. Picosecond Pulse Radiolysis Using Femtosecond White Light with a High S/N Spectrum Acquisition System in One Beam Shot. Nucl. Instrum. Methods Phys. Res. Sect. A 2006, 556, 391–396. [Google Scholar] [CrossRef]
- Finkel, T.; Serrano, M.; Blasco, M.A. The Common Biology of Cancer and Ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein Oxidation and Aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.; Linn, S. Damage and Oxygen Radical. Science 1988, 240, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Mallard, W.G.; Ross, A.B.; Helman, W.P. NDRL/NIST Solution Kinetics; Notre Dame Radiation Laboratory and National Institute of Standards and Technology: Gaithersburg, MD, USA, 1998.
- Davidkova, M.; Spotheim-Maurizot, M. Predicting Radiation Damage Distribution in Biomolecules. In Radiation Chemistry—From Basics to Applications in Material and Life Sciences; Spotheim-Maurizot, M., Mostafavi, M., Douki, T., Belloni, J., Eds.; EDP Sciences: Les Ulis, France, 2008; pp. 265–276. [Google Scholar]
- Wishart, J.F.; Nocera, D.G. Photochemistry and Radiation Chemistry; Wishart, J.F., Nocera, D.G., Eds.; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1998; Volume 254, ISBN 0-8412-3499-X. [Google Scholar]
- Trasatti, S. The Absolute Electrode Potential: An Explanatory Note (Recommendations 1986). Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Reiss, H.; Heller, A. The Absolute Potential of the Standard Hydrogen Electrode: A New Estimate. J. Phys. Chem. 1985, 89, 4207–4213. [Google Scholar] [CrossRef]
- Wardman, P. Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef]
- Neta, P. Redox Properties of Free Radicals. J. Chem. Educ. 1981, 58, 110–113. [Google Scholar] [CrossRef]
- Mozumder, A. Ionization and Excitation Yields in Liquid Water due to the Primary Irradiation: Relationship of Radiolysis with Far UV-photolysisPresented at The Symposium on Recent Trends in Photochemical Sciences, Trivandrum, January 8–10, 2000. Phys. Chem. Chem. Phys. 2002, 4, 1451–1456. [Google Scholar] [CrossRef]
- Mizuse, K.; Kuo, J.-L.; Fujii, A. Structural Trends of Ionized Water Networks: Infrared Spectroscopy of Watercluster Radical Cations (H2O)n+ (n = 3–11). Chem. Sci. 2011, 2, 868–876. [Google Scholar] [CrossRef]
- Dorfman, L.M.; Adams, G.E. Reactivity of the Hydroxyl Radical in Aqueous Solutions; National Bureau of Standards: Gaithersburg, MD, USA, 1973.
- Balcerzyk, A.; LaVerne, J.; Mostafavi, M. Direct and Indirect Radiolytic Effects in Highly Concentrated Aqueous Solutions of Bromide. J. Phys. Chem. A 2011, 115, 4326–4333. [Google Scholar] [CrossRef] [PubMed]
- El Omar, A.K.; Schmidhammer, U.; Rousseau, B.; LaVerne, J.; Mostafavi, M. Competition Reactions of H2O•+ Radical in Concentrated Cl– Aqueous Solutions: Picosecond Pulse Radiolysis Study. J. Phys. Chem. A 2012, 116, 11509–11518. [Google Scholar] [CrossRef] [PubMed]
- Balcerzyk, A.; Schmidhammer, U.; El Omar, A.K.; Jeunesse, P.; Larbre, J.-P.; Mostafavi, M. Picosecond Pulse Radiolysis of Direct and Indirect Radiolytic Effects in Highly Concentrated Halide Aqueous Solutions. J. Phys. Chem. A 2011, 115, 9151–9159. [Google Scholar] [CrossRef] [PubMed]
- El Omar, A.K.; Schmidhammer, U.; Balcerzyk, A.; LaVerne, J.; Mostafavi, M. Spur Reactions Observed by Picosecond Pulse Radiolysis in Highly Concentrated Bromide Aqueous Solutions. J. Phys. Chem. A 2013, 117, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Hosoya, K.; Sakamoto, S.; Suzuki, H.; Usuki, T.; Uemura, O. Hydrogen-bonded Structure in Aqueous Sulfuric Acid Solutions. J. Mol. Liq. 1995, 65–66, 305–308. [Google Scholar] [CrossRef]
- Tang, Y.; Thorn, R.P.; Mauldin, R.L.; Wine, P.H. Kinetics and Spectroscopy of the SO4− Radical in Aqueous Solution. J. Photochem. Photobiol. A Chem. 1988, 44, 243–258. [Google Scholar] [CrossRef]
- Jiang, P.-Y.; Katsumura, Y.; Nagaishi, R.; Domae, M.; Ishikawa, K.; Ishigure, K.; Mechanism, F. Pulse Radiolysis Study of Concentrated Sulfuric Acid Solutions. J. Chem. Soc. Faraday Trans. 1992, 88, 1653–1658. [Google Scholar] [CrossRef]
- Ma, J.; Schmidhammer, U.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Sulfuric Acid Solutions: Evidence for the Oxidation Reactivity of Radical Cation H2O•+. J. Phys. Chem. A 2014, 118, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- El Omar, A.K.; Schmidhammer, U.; Jeunesse, P.; Larbre, J.-P.; Lin, M.; Muroya, Y.; Katsumura, Y.; Pernot, P.; Mostafavi, M. Time-Dependent Radiolytic Yield of OH• Radical Studied by Picosecond Pulse Radiolysis. J. Phys. Chem. A 2011, 115, 12212–12216. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Schmidhammer, U.; de La Lande, A.; Mostafavi, M. Ultra-fast Charge Migration Competes with Proton Transfer in The Early Chemistry of H2O+. Phys. Chem. Chem. Phys. 2017, 19, 2894–2899. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Schmidhammer, U.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Phosphoric Acid Solutions: Mechanism of Phosphate Radical Formation. J. Phys. Chem. B 2014, 119, 7180–7185. [Google Scholar] [CrossRef] [PubMed]
- Elmore, K.L.; Hatfield, J.D.; Dunn, R.L.; Jones, A.D. Dissociation of Phosphoric Acid Solutions at 25°1. J. Phys. Chem. 1965, 69, 3520–3525. [Google Scholar] [CrossRef]
- Caminiti, R.; Cucca, P.; Atzei, D. Phosphate-water Interactions in Concentrated Aqueous Phosphoric Acid Solutions. J. Phys. Chem. 1985, 89, 1457–1460. [Google Scholar] [CrossRef]
- Long, F.H.; Lu, H.; Eisenthal, K.B. Femtosecond Studies of The Presolvated Electron: An Excited State of The Solvated Electron? Phys. Rev. Lett. 1990, 64, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Gauduel, Y.; Pommeret, S.; Migus, A.; Antonetti, A. Some Evidence of Ultrafast H2O+-Water Molecule Reaction in Femtosecond Photoionization of Pure Liquid Water: Influence on Geminate Pair Recombination Dynamics. Chem. Phys. 1990, 149, 1–10. [Google Scholar] [CrossRef]
- Li, J.; Nie, Z.; Zheng, Y.Y.; Dong, S.; Loh, Z.-H. Elementary Electron and Ion Dynamics in Ionized Liquid Water. J. Phys. Chem. Lett. 2013, 4, 3698–3703. [Google Scholar] [CrossRef]
- Thürmer, S.; Ončák, M.; Ottosson, N.; Seidel, R.; Hergenhahn, U.; Bradforth, S.E.; Slavíček, P.; Winter, B. On the Nature and Origin of Dicationic, Charge-Separated Species Formed in Liquid Water on X-ray Irradiation. Nat. Chem. 2013, 5, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Crowell, R.A.; Bartels, D.M. Multiphoton Ionization of Liquid Water with 3.0−5.0 eV Photons. J. Phys. Chem. 1996, 100, 17940–17949. [Google Scholar] [CrossRef]
- Gauduel, Y.; Pommeret, S.; Migus, A.; Antonetti, A. Femtosecond Dynamics of Geminate Pair Recombination in Pure Liquid Water. J. Phys. Chem. 1989, 93, 3880–3882. [Google Scholar] [CrossRef]
- Jordan, I.; Huppert, M.; Brown, M.A.; van Bokhoven, J.A.; Wörner, H.J. Photoelectron Spectrometer for Attosecond Spectroscopy of Liquids and Gases. Rev. Sci. Instrum. 2015, 86, 123905. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.; Stevens, D.L.; Garman, E.F. Physical and Chemical Considerations of Damage Induced in Protein Crystals by Synchrotron Radiation: A Radiation Chemical Perspective. J. Synchrotron Radiat. 2002, 9, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Grambow, B.; Mostafavi, M. State of Fukushima Nuclear Fuel Debris Tracked by Cs137 in Cooling Water. Environ. Sci. Process. Impacts 2014, 16, 2472–2476. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Wang, F.; Mostafavi, M. Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions. Molecules 2018, 23, 244. https://doi.org/10.3390/molecules23020244
Ma J, Wang F, Mostafavi M. Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions. Molecules. 2018; 23(2):244. https://doi.org/10.3390/molecules23020244
Chicago/Turabian StyleMa, Jun, Furong Wang, and Mehran Mostafavi. 2018. "Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions" Molecules 23, no. 2: 244. https://doi.org/10.3390/molecules23020244
APA StyleMa, J., Wang, F., & Mostafavi, M. (2018). Ultrafast Chemistry of Water Radical Cation, H2O•+, in Aqueous Solutions. Molecules, 23(2), 244. https://doi.org/10.3390/molecules23020244